
Abstract
Disturbed circadian rhythms have been a risk factor for type 2 diabetes mellitus (T2DM). Melatonin is the major chronobiotic hormone regulating both circadian rhythm and glucose homeostasis. The rs10830963 (G allele) of the melatonin receptor 1B (MTNR1B) gene has the strongest genetic associations with T2DM according to several genome-wide association studies. The MTNR1B rs10830963 G allele is also associated with disturbed circadian phenotypes and altered melatonin secretion, both factors that can elevate the risk of diabetes. Furthermore, evolutionary studies implied the presence of selection pressure and ethnic diversity in MTNR1B, which was consistent with the “thrifty gene” hypothesis in T2DM. The rs10830963 G risk allele is associated with delayed melatonin secretion onset in dim-light and prolonged duration of peak melatonin. This delayed melatonin secretion may help human ancestors adapt to famine or food shortages during long nights and early mornings and avoid nocturnal hypoglycemia but confers susceptibility to T2DM due to adequate energy intake in modern society. We provide new insight into the role of MTNR1B variants in T2DM via disturbed circadian rhythms from the perspective of the “thrifty gene” hypothesis; these data indicate a novel target for the prevention and treatment of susceptible populations with the thrifty genotype.
1. Introduction
The International Diabetes Federation (IDF) estimated that diabetes affects 537 million adults (20-70 years old) across the world [Citation1]. Type 2 diabetes mellitus (T2DM) is a complex multifactorial metabolic disorder that develops interaction between lifestyle risk factors and genetic susceptibility [Citation2]. Accumulating evidence has demonstrated that the endogenous circadian rhythm, which is responsible for harmonizing the internal clock with the environment and regulating daily behavior (i.e. sleep-wake cycles, feeding and activity), is an additional risk factor for T2DM, as it impacts the diurnal rhythm of glucose metabolism [Citation3–5]. Large cross-sectional and prospective cohort studies have reported that chronic circadian misalignment, such as rotating night-shift work [Citation6–8], social jetlag [Citation9,Citation10], sleep disturbances [Citation6,Citation11] and an evening chronotype [Citation12], confer a higher risk of impaired glucose tolerance, insulin resistance, insufficient β-cell mass and T2DM [Citation13–15].
Melatonin is a critical circadian hormone and has been widely investigated for its complex contribution to sleep quality, circadian regulation and glucose homeostasis [Citation16,Citation17]. Circadian melatonin is mainly secreted from the pineal gland under the control of the hypothalamic suprachiasmatic nucleus (SCN) and environmental light exposure [Citation18,Citation19]. Despite substantial research, whether and how melatonin contributes to glucose metabolism through circadian regulation remains poorly understood. Several studies have mentioned that melatonin protects against glucose tolerance and diabetes mellitus. Large population-based studies within the Nurses’ Health cohort have suggested that lower melatonin release can increase insulin resistance and the incidence of type 2 diabetes [Citation19,Citation20]. Furthermore, a randomized, double-blind clinical trial among type 2 diabetes patients with insomnia also indicated that long-term administration of prolonged-release melatonin could improve glycemic control and inhibit hypertriglyceridemia as well as hyperinsulinemia [Citation21]. Conversely, two other placebo-controlled studies indicated a deleterious effect of acute melatonin administration in the morning or evening on glucose tolerance and insulin sensitivity both in young and older women [Citation22,Citation23].
The contradictory or even completely opposite role of melatonin in glucose metabolism is intriguing. Some researchers have proposed that melatonin receptors may partly explain this contradiction. Melatonin generally exerts its effects through specific, high-affinity G protein-coupled receptors with seven transmembrane domains; these receptors are widely expressed in central and peripheral tissues [Citation24,Citation25], including human islet cells [Citation26,Citation27]. The MTNR1B gene, which encodes melatonin receptor 1B, is robustly associated with various glycemic traits, such as increased fasting glucose, aggravated insulin resistance and attenuated β-cell function according to a genome-wide association (GWA) and replicated cohort studies, with T2DM risk most strongly linked to the rs10830963 G allele [Citation28–30]. Notably, this MTNR1B gene variant was associated with endogenous melatonin signaling, sleep status and circadian rhythms [Citation31]. Lane et al. previously confirmed that the MTNR1B rs10830963 G allele was linked with the prolongation of elevated melatonin levels and delayed offset of melatonin secretion in daytime [Citation32]. Therefore, rs10830963 G allele carriers might have an increased risk of impaired glucose tolerance or T2DM due to elevated melatonin secretion and food intake in the morning. In addition, these risks of rs10830963 allele carriers were more pronounced in individuals with early sleep time [Citation32]. Moreover, T2DM patients carrying rare variants in the MTNR1B coding region were more prone to behavioral circadian misalignment and irregular sleep [Citation33]. The most recent randomized crossover trial on this topic revealed that altered behavioral rhythms, such as late dinner time, were obviously associated with 3.5-fold higher melatonin levels; moreover, the effect of late dinners on insulin secretion was stronger in MTNR1B rs10830963 G risk allele carriers, which indicates further effects of MTNR1B variants on circadian rhythms in terms of β-cell function [Citation34]. These findings all indicate that variants of the MTNR1B gene mediate human circadian phenotypes. Additionally, the rs10830963 G allele was linked with the overexpression of MTNR1B in human islet cells [Citation35]. The highly expressed G-protein-coupled melatonin receptor was reported to enhance the inhibition of melatonin secretion to enable cyclic adenosine monophosphate (cAMP) synthesis; cAMP is a stimulator of insulin release [Citation18].
Nonetheless, whether and how the circadian rhythm and genetic variants of MTNR1B exert separate or joint effects on T2DM via the melatonin system are relatively unknown, and the possible mechanisms remain unclear. Circadian misalignment and MTNR1B variants appeared to facilitate the development of T2DM in a subset of the population [Citation36]. Supporting this finding, several evolutionary analyses indicated a prominent population difference implying selection pressures on MTNR1B in terms of T2DM [Citation37,Citation38]. The present review mainly focuses on the link between the MTNR1B gene and T2DM. We first briefly outline the interplay between the circadian and melatonin systems and then elucidate the role of melatonin and its receptor signaling in T2DM via the regulation of the circadian rhythm. We also provide an overview of the genetic contribution of MTNR1B to T2DM, thus discussing evolutionary selection on MTNR1B in terms of glucose homeostasis as well as T2DM, and hypothesize that the MTNR1B gene is a “thrifty gene” for humans.
2. Melatonin and circadian system
2.1. The interplay between melatonin synthesis and circadian rhythms
Various organisms, including humans, have evolved to adapt to the earth’s daily rotation and light/dark cycle. Human ancestors worked (were awake) during the daytime and rested (sleep) at night. Consequently, endogenous biological processes, including physiology, metabolism, and behaviors, accommodated this sleep-wake pattern, which is termed the circadian rhythm or circadian clock [Citation39,Citation40]. In essence, the circadian rhythm is an evolutionarily conserved endogenous autonomous timing system. The human circadian system consists of the central and peripheral clocks. The central clock is located in the SCN of the hypothalamus; it can reset intrinsic circadian rhythms every 24 h and synchronize the peripheral clocks in adipose tissue and the pancreas, liver and gut [Citation41,Citation42]. The accurate molecular mechanism underlying the circadian timing system is executed by transcriptional-translational feedback loops consisting of core clock genes, and the entrained timing signal is ultimately forwarded through neural and hormonal signals [Citation13].
Synthesis of the pineal hormone melatonin is regulated by the SCN master clock and synchronized to the environmental light-dark cycle. Melatonin secretion generally occurs in darkness (at night) and peaks at 00:00 and 4:00 am. Importantly, nighttime melatonin production is blocked by light, especially blue light at wavelengths of 460–480 nm and intensities < 200 lux [Citation43–45]. The biosynthetic precursor of melatonin is tryptophan, which is hydroxylated to 5-hydroxytryptophan and then decarboxylated to generate serotonin. Subsequently, serotonin is acetylated to N-acetylserotonin by arylalkylamine N-acetyltransferase (AANAT) and then converted to melatonin by acetylserotonin O-methyltransferase [Citation16]. When the environmental photoperiodic information reaches intrinsic photosensitive retinal ganglion cells (ipRGCs), it is conveyed to the SCN by the retinal hypothalamic tract. Afterward, the signal is projected to the pineal gland through a neuronal signaling cascade that promotes or inhibits melatonin secretion in pinealocytes () [Citation41,Citation46,Citation47].
Figure 1. Melatonin levels fluctuate across the 24-h light-dark cycle [Citation41,Citation46,Citation47]. Melatonin synthesis and release occur in dim light and are inhibited by daytime light. In the eyes, environmental light reaches intrinsic photosensitive retinal ganglion cells (ipRGCs) and is then transmitted to the SCN via the retinal hypothalamic tract. SCN signals are conveyed to the medial forebrain bundle by descending hypothalamic projections and then project to the spinal cord and superior cervical ganglia. Afterward, the sympathetic nerve from the superior cervical ganglion stimulates the pineal gland to secrete melatonin, thus entraining circadian rhythms to environmental light. was created with BioRender (https://biorender.com).
![Figure 1. Melatonin levels fluctuate across the 24-h light-dark cycle [Citation41,Citation46,Citation47]. Melatonin synthesis and release occur in dim light and are inhibited by daytime light. In the eyes, environmental light reaches intrinsic photosensitive retinal ganglion cells (ipRGCs) and is then transmitted to the SCN via the retinal hypothalamic tract. SCN signals are conveyed to the medial forebrain bundle by descending hypothalamic projections and then project to the spinal cord and superior cervical ganglia. Afterward, the sympathetic nerve from the superior cervical ganglion stimulates the pineal gland to secrete melatonin, thus entraining circadian rhythms to environmental light. Figure 1 was created with BioRender (https://biorender.com).](/cms/asset/fc0537a5-94b4-43c5-b244-b3b85d3c4bc7/iann_a_2191218_f0001_c.jpg)
The SCN-controlled pineal gland is the main source of melatonin production in humans. In turn, pineal melatonin is the key temporal molecule; through feedback, it orchestrates the internal clock to adapt to rhythmic changes in the external light-dark cycle. Once secreted, the neuromodulator melatonin is conveyed to the SCN through the cerebrospinal fluid of the third ventricle or blood circulation system and then binds to specific high-affinity receptors expressed in the SCN [Citation48,Citation49]. It is well established that the binding sites of melatonin in the SCN are essential for transmitting the oscillating signals of the environmental light/dark cycle to the biological clock, subsequently modulating various physiological processes and behaviors [Citation16,Citation49,Citation50]. Hence, melatonin receptors are efficient therapeutic targets for treating circadian abnormalities or sleep disorders [Citation25]. However, a few studies have speculated that melatonin may directly (i.e. not through its receptors) mediate the action of the SCN [Citation50–52]. Moreover, the fluctuating levels of melatonin in the bloodstream affect the circadian rhythm of peripheral tissues. Melatonin shows a rapid pharmacokinetic profile with a half-life of 20–30 min [Citation53]. Therefore, the chronobiotic properties of melatonin in the SCN and peripheral tissues contribute to whole-body synchronization of circadian rhythms.
In summary, melatonin is primarily and indirectly generated by the SCN, consistent with the circadian rhythm, but it is also the key temporal feedback received by the SCN; the feedback loop between the SCN and the neuromodulator melatonin efficiently synchronizes the endogenous circadian rhythm to the photoperiod in humans.
2.2. The physiological function of melatonin
Melatonin is a pleiotropic molecule and has multiorgan effects in humans [Citation16,Citation47,Citation54–73] (). The physiological functions and ensuing effects of melatonin vary in different systems. We primarily focused on its role in metabolic functions in terms of immediate effects, consecutive effects and chronobiotic effects in a previous study [Citation45]. Pineal melatonin was characterized by dim-light melatonin onset (DLMO) and a nocturnal daily peak; melatonin was released in the CSF and blood via this classic hormonal pathway and immediately mediated biological function by melatonin signaling [Citation45]. After the cessation of melatonin signaling in the light phase, the intracellular adenylyl cyclase/cAMP/PKA/CREB transduction pathway became hypersensitive, which was inhibited by melatonin signaling in the dark phase. In particular, melatonin facilitates insulin secretion of islet β-cells by directly sensitizing glucagon-like peptide-1 (GLP-1)/AMP/PKA/CREB signaling, attenuating apoptosis of β-cells, diminishing oxidative stress in cells exposed to hyperglycemia and maintaining the glucose-stimulated insulin response [Citation74–77]. Additionally, the chronobiotic property of melatonin allows targeted regulation of peripheral clock gene expression in pancreatic β-cells, adipose tissue, skeletal muscle, liver tissue and other tissues, which are directly involved in multiple metabolic functions [Citation63,Citation78–80]. The melatonin-mediated circadian timing process, including the central and peripheral oscillators, is one of the key regulatory patterns of metabolism and includes several different physiological effects of melatonin.
Table 1. The physiological functions of melatonin.
3. Melatonin system influences T2DM via circadian rhythms
3.1. Circadian rhythms in glucose homeostasis
The circadian rhythm plays an important role in glucose metabolism by sustaining daily fluctuations in physiological, hormonal, and behavioral processes [Citation3,Citation81]. Especially, the central circadian clock in the SCN is a pacemaker which aligns our daily sleep-wake, activity-rest as well as feeding-fasting cycles and synchronizes the metabolic activity of peripheral clocks [Citation82–85]. Therefore, the central and peripheral circadian clocks jointly regulate endogenous diurnal rhythm in glucose metabolism (). The sleep-wake cycle is the main manifestation of the endogenous circadian clock and is also the metabolic master switch [Citation81]. A normal sleep-wake pattern aligned with the circadian rhythm supports normal metabolism; for example, energy expenditure during sleep is just two-thirds of that during wakefulness [Citation81,Citation86]. Moreover, the rhythm of the sleep-wake cycle corresponds to the fasting and feeding rhythm and thereby influences glucose metabolism synergistically [Citation81]. The glucose tolerance in response to the same meal is relatively impaired in the evening/night compared with the morning in healthy individuals [Citation4]. Thus, glucose metabolism oscillates with the circadian rhythm such that decreased glucose tolerance and a lower response of β-cells occur at night. The progressive impairment of glucose tolerance and insulin sensitivity due to long-term shift work or circadian misalignment might be due to the joint action of a destroyed endogenous rhythm and altered feeding pattern [Citation87].
Figure 2. Circadian rhythms maintains glucose homeostasis. Circadian signals are conveyed from the SCN to the adjacent subparaventricular zone (SPZ), and the input is then integrated and amplified in the dorsomedial nucleus of the hypothalamus (DMH). Neurons in the DMH relay information to the ventrolateral preoptic nucleus (VLPO), the lateral hypothalamic area (LHA), orexin receptors and the paraventricular nucleus (PVN), which drive the circadian cycles of sleep, activity, feeding and corticosteroid secretion, respectively. The SCN could directly exerts excitatory-inhibitory effects on the neuronal response of the arcuate nucleus (ARC) to hypoglycemia and modulate food intake. The central circadian clock synchronizes peripheral clocks, and jointly regulate glucose metabolism. was created with BioRender (https://biorender.com).
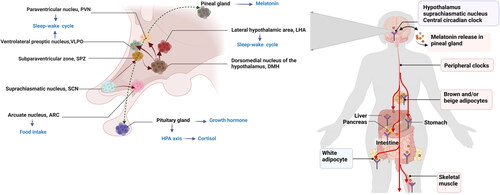
Several hormones affecting the fluctuation of glucose tolerance and insulin secretion, such as corticosteroids, melatonin and growth hormone, were also found to be controlled by the daily rhythm [Citation88]. Growth hormone is an anabolic hormone with levels that increase during the night and peak immediately after sleep onset [Citation88]; this hormone can rapidly elevate glucose levels by inhibiting muscle glucose uptake and suppressing glucose oxidation [Citation89]. Cortisol release depends on the interaction between the SCN and the PVN. The nadir of human endogenous cortisol occurs at midnight, and the levels rise for 2-3 h after sleep onset, followed an increasing trend until 9 o’clock in the morning (the peak); a lower level is then maintained during the day, with a decline usually during the later afternoon [Citation90]. Cortisol plays a critical role in the wakening response in the early morning and in glucose homeostasis. Circadian disruption disturbs the daily rhythm of cortisol secretion and induces hypercortisolemia, which leads to hepatic and peripheral insulin resistance [Citation91].
3.2. Melatonin as the “circadian zeitgeber” for glucose metabolism
Melatonin is a major chronobiotic hormone that provides feedback to the central and peripheral circadian clocks to regulate metabolic function by binding to its receptors (MT1 and MT2), which are expressed in central and peripheral sites, and initiating several specific signal transduction mechanisms. In SCN cells, rhythmic melatonin production inhibits the phosphorylation of cAMP responsive element binding protein (CREB) induced by the retinohypothalamic transmitter pituitary adenylate cyclase-activating polypeptide (PACAP); melatonin production thus regulates neuronal sensitivity to endogenous clock phase shifts [Citation92] (). In vitro studies have shown that melatonin receptor signaling increases the activity of Kir3 ion channels (G-protein-coupled potassium channels) in the SCN and hypothalamus, which inhibits the firing rate of neurons and the regulation of the circadian system [Citation93]. Melatonin can also facilitate circadian clock resetting by activating the protein kinase C (PKC) pathway via MT2 signaling [Citation94]. Intriguingly, melatonin acts as a circadian modulator that alters the expression of clock genes (per1, per2, cry1, bmal1 and clock) via PKC signaling in the SCN [Citation95–97], which entrains neuronal activity rhythms. Melatonin, as the main signaling molecule of the central circadian clock, can coordinate human physiological metabolism and behavioral activities with the circadian temporal structure.
Figure 3. Melatonin acts as a circadian pacemaker and advances the SCN phase [Citation93–97]. The retinohypothalamic tract mediates cAMP responsive element binding protein (CREB) phosphorylation via pituitary adenylate cyclase-activating polypeptide (PACAP) release under light stimulation in SCN cells; PACAP release is responsible for light-induced phase shifts. The binding of melatonin to MT1 inhibits PACAP-induced CREB phosphorylation in the SCN. Melatonin receptors activate G-protein-coupled Kir3 ion channels, inhibit the rhythmic firing of SCN neurons and regulate circadian rhythms. Melatonin activates PKC in the SCN and induces phase resetting, and through this signaling, the expression of core clock genes, Period 1 (Per1) and Period 2 (Per2), increases within the SCN. was created with BioRender (https://biorender.com).
![Figure 3. Melatonin acts as a circadian pacemaker and advances the SCN phase [Citation93–97]. The retinohypothalamic tract mediates cAMP responsive element binding protein (CREB) phosphorylation via pituitary adenylate cyclase-activating polypeptide (PACAP) release under light stimulation in SCN cells; PACAP release is responsible for light-induced phase shifts. The binding of melatonin to MT1 inhibits PACAP-induced CREB phosphorylation in the SCN. Melatonin receptors activate G-protein-coupled Kir3 ion channels, inhibit the rhythmic firing of SCN neurons and regulate circadian rhythms. Melatonin activates PKC in the SCN and induces phase resetting, and through this signaling, the expression of core clock genes, Period 1 (Per1) and Period 2 (Per2), increases within the SCN. Figure 2 was created with BioRender (https://biorender.com).](/cms/asset/ba748284-ce38-4546-af14-d78d748d74bc/iann_a_2191218_f0003_c.jpg)
Furthermore, melatonin receptors are expressed in peripheral tissues and mediate peripheral clocks in the pancreas, muscle, liver, adipose tissue and gut; consequently, these receptors affect glucose homeostasis through various intracellular mechanisms [Citation13,Citation16]. Previous in vitro and in vivo studies have demonstrated that insulin secretion in pancreatic β-cells is mediated by melatonin via cAMP, cyclic guanosine monophosphate (cGMP) and inositol triphosphate (IP3) signaling cascades through the melatonin receptor subtypes MT1 and MT2 [Citation98–100] (). Glucose transport to muscle cells was also found to be controlled by melatonin through the activation of insulin receptor substrate-1 (IRS-1)/phosphoinositide 3-kinase (PI-3-kinase) signaling and downstream PKC-ζ via MT2 in rats [Citation101] () . Melatonin also has the ability to ameliorate incremental glycogen increases and improve glucose utilization in HepG2 cells through a PKCζ-Akt-glycogen synthase kinase 3 beta (GSK3β)-related pathway via melatonin-specific receptors [Citation102] (). Hence, melatonin signaling is a potent ‘internal zeitgeber’ that entrains the endogenous circadian rhythm and synchronizes various organs and tissues to control glucose metabolism.
Figure 4. Melatonin regulates insulin secretion in pancreatic β-cells via the cAMP, cGMP and IP3 signaling pathways [Citation98,Citation236]. The binding of melatonin to MT1 inhibits cyclic adenosine monophosphate (cAMP) signaling and decreases insulin secretion in pancreatic β-cells. The binding of melatonin to MT2 inhibits cyclic guanosine monophosphate (cGMP) signaling and decreases insulin secretion in pancreatic β-cells. Melatonin stimulates IP3 release accompanied by a transient increase in Ca2+ concentrations and leads to Ca2+-dependent insulin secretion. was created with BioRender (https://biorender.com).
![Figure 4. Melatonin regulates insulin secretion in pancreatic β-cells via the cAMP, cGMP and IP3 signaling pathways [Citation98,Citation236]. The binding of melatonin to MT1 inhibits cyclic adenosine monophosphate (cAMP) signaling and decreases insulin secretion in pancreatic β-cells. The binding of melatonin to MT2 inhibits cyclic guanosine monophosphate (cGMP) signaling and decreases insulin secretion in pancreatic β-cells. Melatonin stimulates IP3 release accompanied by a transient increase in Ca2+ concentrations and leads to Ca2+-dependent insulin secretion. Figure 3 was created with BioRender (https://biorender.com).](/cms/asset/ff548456-86c5-42b5-8923-ba9001a00596/iann_a_2191218_f0004_c.jpg)
Figure 5. The binding of melatonin to MT2 stimulates glucose transport to skeletal muscle cells via the IRS-1/PI-3-kinase pathway [Citation101]. Melatonin increases the phosphorylation level of insulin receptor substrate-1 (IRS-1) and the activity of phosphoinositide 3-kinase (PI-3-kinase), activates downstream protein kinase C (PKC)-ζ and stimulates glucose transport to muscle cells. was created with BioRender (https://biorender.com).
![Figure 5. The binding of melatonin to MT2 stimulates glucose transport to skeletal muscle cells via the IRS-1/PI-3-kinase pathway [Citation101]. Melatonin increases the phosphorylation level of insulin receptor substrate-1 (IRS-1) and the activity of phosphoinositide 3-kinase (PI-3-kinase), activates downstream protein kinase C (PKC)-ζ and stimulates glucose transport to muscle cells. Figure 4 was created with BioRender (https://biorender.com).](/cms/asset/1229f3fc-6a0e-4f09-8723-4c451e8e8eb6/iann_a_2191218_f0005_c.jpg)
Figure 6. The binding of melatonin to MT1 stimulates glycogen synthesis in hepatic cells via the protein kinase Cζ (PKCζ)-Akt-glycogen synthase kinase 3B (GSK3β) pathway [Citation102]. Melatonin increases the phosphorylation of PKCζ, Akt, and GSK3β and stimulates glycogen synthesis in hepatic cells. was created with BioRender (https://biorender.com).
![Figure 6. The binding of melatonin to MT1 stimulates glycogen synthesis in hepatic cells via the protein kinase Cζ (PKCζ)-Akt-glycogen synthase kinase 3B (GSK3β) pathway [Citation102]. Melatonin increases the phosphorylation of PKCζ, Akt, and GSK3β and stimulates glycogen synthesis in hepatic cells. Figure 5 was created with BioRender (https://biorender.com).](/cms/asset/e50ad619-8b0e-4903-9cbf-12535276a696/iann_a_2191218_f0006_c.jpg)
Given the critical role of melatonin in glucose homeostasis, the effect of melatonin signaling on T2DM has been widely studied. Rats lacking melatonin showed marked glucose intolerance and insulin resistance, which may result from a widespread reduction in glucose transporter-4 (GLUT-4) in many insulin-targeted adipose tissues, such as adipose and hepatic cells [Citation103,Citation104]. Rats with experimentally induced diabetes displayed lower peak melatonin levels, pancreatic melatonin receptor overexpression and higher insulin levels during glucose metabolism impairment [Citation105,Citation106]. In diabetic rats, combined treatment with low-dose melatonin and insulin ameliorated insulin sensitivity in adipose tissue and improved glycemic control [Citation107], supporting a dynamic interaction between melatonin and insulin signaling [Citation107]. In insulin-resistant mice, the administration of melatonin enhanced insulin function related to vasodilatation, which facilitated glucose transport in skeletal muscle and subsequently improved glucose utilization [Citation108]. Population-based studies have also reported the beneficial effect of melatonin regarding glucose homeostasis. Case–control and prospective cohort studies confirmed a higher risk of glucose tolerance, insulin sensitivity and T2DM in participants with decreased melatonin [Citation19,Citation23,Citation109]; treatment with melatonin elevated the levels of adiponectin, leptin, and ghrelin; reduced insulin resistance; and improved glycemic traits and diabetic complications [Citation5,Citation110,Citation111]. Although the studies above provide evidence of the beneficial role of melatonin in the circadian rhythm and glycemic control, recent studies have only partially elucidated the causal relationship between melatonin signaling and T2DM risk. It is possible that genetic effects may also be critical in the development of T2DM.
4. MTNR1B gene and T2DM
4.1. Genetic variants of MTNR1B affect glucose traits and T2DM
The physiological functions of melatonin are mainly mediated by the specific high-affinity receptors MT1 and MT2. MTNR1B encodes melatonin receptor MT2, which is expressed in the human brain, pancreatic islets and β-cells [Citation28], and is a prime candidate gene for glycemic and metabolic traits, especially the common variant rs10830963 within the single 11.5-kb intron of MTNR1B. A previous meta-analysis combined data from 10 GWASs of individuals of European ancestry free of diabetes that validated the appreciable associations between several variant loci (rs1387153, rs11020107 and rs10830963) of MTNR1B and fasting glucose levels as well as reduced β-cell function [Citation28]. The strongest signal consistently occurred at rs10830963, and the fasting glucose concentration was elevated by an average of 0.07 mmol/l per risk (G) allele [Citation28]. Moreover, the findings from another extensive meta-analysis including 21 GWASs of 46,186 nondiabetic participants of European descent were consistent with the aforementioned evidence and indicate the effects of MTNR1B rs10830963 on fasting glucose and β-cell function [Citation30]. Subsequent replication studies in large-scale case–control and cohort research provided robust evidence that the glucose-elevating allele (MTNR1B rs10830963) was associated with glycemic traits such as impaired glucose tolerance, inhibited β-cell function, increased fasting glucose levels and T2DM in Europeans [Citation30,Citation112–114], Asians [Citation115–118], North Americans [Citation119] and African Americans, among others [Citation120,Citation121]. In addition, GWASs in different populations, including lean adults, lean adolescents, obese adults and obese adolescents, as well as subsequent replication analyses all identified that rs1387153 (in the 5′ region of MTNR1B) T allele was linked with elevated concentrations of fasting plasma glucose and glycosylated hemoglobin (HbA1c) as well as elevated T2DM risk [Citation28,Citation113]. Furthermore, a similar study confirmed that rs10830963 was in linkage disequilibrium (LD) with rs1387153, supporting the strongest genetic role of rs10830963 at MTNR1B in T2DM [Citation29,Citation112]. summarizes the GWASs investigating glycemic traits and T2DM since 2008 [Citation28–30, Citation113,Citation114,Citation119,Citation122–170].
Table 2. Common variants of MTNR1B significantly associated with glycemic traits or T2DM in GWASs.
4.2. MTNR1B is associated with therapeutic and intervention targets for T2DM
A study also reported that T2DM patients with the MTNR1B rs10830963 G risk allele experienced attenuated therapeutic efficacy of hypoglycemic treatment, such as repaglinide and nateglinide [Citation171,Citation172]. This attenuation is possibly due to the increased expression of MTNR1B in pancreatic β-cells; MTNR1B has an antagonistic effect on the triggering of insulin release by antidiabetic pharmacotherapy. This finding also provides a personalized therapy target for specific diabetes patients. Another study found that a high rate of insulin treatment efficacy could not be predicted in rs10830963 G risk allele carriers with T2DM [Citation173]. Remarkably, in women with normal glycemic levels, the MTNR1B rs10830963 risk variant predicted approximately 26% of individual variation in the curative effect of melatonin on glucose tolerance [Citation173]. In healthy participants, after the administration of 5 mg of melatonin in the morning (9:00 am), the deleterious effect of melatonin on glucose tolerance was elevated six-fold in MTNR1B G allele carriers compared with noncarriers [Citation174]. Another clinical trial found that in healthy participants, the administration of a supraphysiological dose of melatonin led to reduced insulin sensitivity in homozygous rs10830963 CC carriers [Citation175]. Therefore, we suspect that MTNR1B acts as a genetic bridge between circadian rhythms and glucose metabolism, and the common variant (rs10830963) could provide predictive biomarkers and therapeutic targets for T2DM.
Curiously, a large-scale population-based study reported an inverse correlation of the risk allele of MTNR1B with the hepatic insulin resistance (IR) index [Citation113] in nondiabetic participants. IR in the liver generally contributes to impaired hepatic glucose synthesis and enhanced gluconeogenesis and glycogenolysis, ultimately leading to hyperglycemia or T2DM [Citation176,Citation177]. Researchers hypothesized that the increased hepatic insulin sensitivity of MTNR1B risk allele carriers might be a compensatory mechanism that balances impaired insulin secretion and the maintenance of a normal range of glycemia levels in nondiabetic participants [Citation178,Citation179]. Studies have consistently demonstrated that MTNR1B is a true causal gene for diabetes, but the underlying molecular mechanisms remain unclear.
Functional data has shown that individuals with the risk genotype have higher expression of MTNR1B in human pancreatic β-cells and that MTNR1B expression is positively correlated with impaired insulin secretion [Citation18,Citation180,Citation181]. As previously mentioned, melatonin signaling modulates insulin gene expression via MT2, and MTNR1B overexpression aggravates the inhibitory effect of melatonin on the insulin gene; however, silencing MTNR1B diminishes the above effect in pancreatic β-cells. Thus, melatonin-MT2 binding may inactivate the mitogen-activated protein kinase (MAPK) pathway and eventually reduce insulin gene expression in β-cells [Citation182]. Genomic annotation and functional assays in individuals of European ancestry have indicated that the MTNR1B variant region overlaps with the forkhead box transcription factor A2 (FOXA2)-bound site and subsequently reinforces FOXA2-bound enhancement of activity in human islet- or liver-derived cells [Citation183]. The risk allele of rs10830963 also has a specific binding site that matches with NEUROD1 in human islet cells [Citation183]. Thus, the common variants of rs10830963 drive signaling by inducing islet MTNR1B expression by enhancing FOXA2 expression and binding to NEUROD1. However, large-scale exon sequencing revealed 40 nonsynonymous MTNR1B variants and four very rare variants (with an MAF < 0.1%), which contributed to the total deficits in MT2 function and T2DM risk [Citation184]. The findings from this research suggest that the increased expression of MTNR1B was not the cause of T2DM but rather a phenomenon occurring under reduced negative feedback due to the MTNR1B mutation and impaired G protein signaling; in other words, the increased expression of MTNR1B was a compensatory and adaptive activation via intracellular regulatory mechanisms. The attenuated effect of the melatonin system on insulin release regulation and circadian rhythms appeared to be a key cause of T2DM. Overall, more evidence is needed to clarify the effects of MTNR1B variants on the risk of T2DM and to provide definite therapeutic targets for the disease.
MTNR1B is also expressed in other human islet cell types, including α- and δ-cells, which determine the stimulatory impact of melatonin on glucagon and somatostatin secretion, respectively [Citation100,Citation185]. Physiological concentrations of melatonin elevate somatostatin and decrease somatostatin release both in vitro and in vivo; these two hormones are important regulators of blood glucose homeostasis [Citation186,Citation187]. However, MTNR1B density is lower in human pancreatic islet α-cells than in β-cells, and MTNR1A is the major transmitter of the inhibitory effect of melatonin in human δ-cells [Citation185]. Scant evidence on the influence of genetic variants of MTNR1B on the function of α-cells in humans is available; however, Anna Jonsso et al. found that the strongest correlation between glucose levels and glucagon occurred in individuals with the MTNR1B rs10830963 variant [Citation188]. Future studies are needed to determine whether MTNR1B variants impair insulin secretion and sensitivity in pancreatic islet β-cells and promote glucagon release in α-cells.
4.3. MTNR1B contributes to the common genetic background for GDM and T2DM
Many studies have assumed that gestational diabetes mellitus (GDM) shares genetic roots with T2DM, and women with GDM may have 8.9-folds increased risk of T2DM [Citation189]. A multiancestry genome-wide association study found four risk loci (MTNR1B, TCF7L2, CDKAL1, and CDKN2A-CDKN2B) for GDM, which were all linked with the risk of T2DM [Citation190]; these findings also indicate that GDM and T2DM have shared pathophysiology. Three variants of MTNR1B (rs10830963, rs1387153 and 10830962) exhibited genome-wide significant associations with GDM [Citation191]. The MTNR1B risk allele of rs10830963 was also associated with alterations in gestational glucose tolerance, including impaired insulin sensitivity, increased early-phase insulin release and fasting insulin conversion in women without GDM [Citation192]. Similarly, the MTNR1B rs7936247 T allele was strongly associated with elevated fasting plasma glucose (FPG) levels in pregnant women [Citation134]. Moreover, another study also showed that MTNR1B rs10830962 and rs1387153 were genetic risk loci for GDM in both Asian and Caucasian cohorts [Citation130,Citation193–196].
Common variants of MTNR1B are linked to substantial heterogeneity in glycemic control during pregnancy. Among pregnant women with prior GDM, the effectiveness of lifestyle intervention was attenuated by the rs10830963 risk allele (G); only women homozygous for the rs10830963 C allele benefitted from the lifestyle intervention and showed a decrease in GDM risk [Citation197]. The MTNR1B rs10830962 G allele also increased the risk of GDM, and in women homozygous for the rs10830962 G allele, physical activity decreased maternal fasting insulin and insulin resistance (according to the homeostatic model assessment of insulin resistance, HOMA-IR) [Citation198]. Thus, it seems that genetic susceptibility loci of MTNR1B could be candidate pharmacogenetic markers and prevention targets for GDM in risk population subgroups. Recent evidence further suggested that the maternal G allele at MTNR1B rs10830962 increased the risk of childhood obesity and metabolic abnormalities in offspring given observed interactions with gestational weight gain in a GDM mother-child paired cohort [Citation199]. A cohort study followed women with a GDM history during postpartum years 1-5; the results showed that women with the MTNR1B rs10830963 GG genotype had higher postpartum fasting glucose levels [Citation200]. Thus, MTNR1B may be part of a common genetic predisposition for GDM and T2DM.
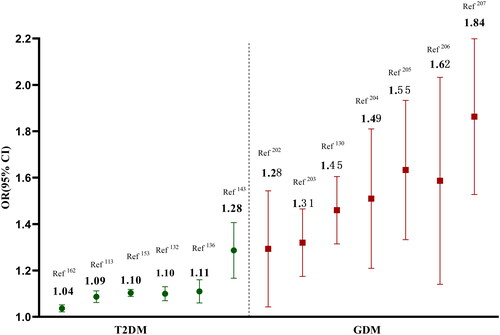
Although, the common variants of MTNR1B provided a shared genetic basis between GDM and T2DM, but the genetic effect sizes were pronounced difference [Citation201]. Especially, the MTNR1B rs10830963 presented a higher genetic effect size for GDM when compared to T2DM, and the odds ratios (ORs) of rs10830963 risk allele were generally ranged from 1.04 to 1.28 for T2DM, but from1.28 to1.84 for GDM [Citation113,Citation132,Citation136,Citation143,Citation153,Citation162,Citation202–207] (). Pregnancy is a special physiological state which is accompanied by progressive insulin resistance, but just a small proportion of pregnancy women developed as GDM. Previous study had reported increased nighttime serum melatonin levels after 24 weeks of gestation [Citation208], and this hormone could aggravate insulin desensitization. Concurrently, the expression levels of MTNR1B in placenta seem to up-regulate among GDM women, especially in participants with rs10830963 GG and GC genotypes [Citation209]. The pregnant women of concurrent rising levels of melatonin and MTNR1B gene contribute to increased insulin resistance, which may be genetically predisposed to GDM. Moreover, several specific environmental factors altered during pregnancy, such as maternal adiposity, placenta hormones and paternal or fetal genotypes, may modulate the genetic effects of MTNR1B on maternal GDM via gene-environment or gene-gene interactions [Citation202].
Figure 7. The different genetic effect sizes of MTNR1B in T2DM and GDM.
4.4. The different genetic effect sizes of MTNR1B in Sub-populations
Notably, several other factors, including body mass index (BMI), age and gender should be taken into account when assessed the genetic effect of MTNR1B on glycemic traits. The genetic effects of MTNR1B on glycemic traits appeared to be more preponderant in overweight and obese individuals [Citation203,Citation210–214] (). A meta-analysis detected the effects of an interaction between the MTNR1B rs10830963 genetic variant and prepregnancy BMI on the risk of GDM; specifically, rs10830963 G carriers with a pre-pregnancy BMI ≥25 kg/m2 were more susceptible to GDM [Citation203]. Among GDM patients with a prepregnancy BMI ≥29 kg/m2, carriers of the MTNR1B rs10830963 G risk allele presented a five-fold higher rate of antenatal insulin therapy (AIT) initiation [Citation214]. Studies with overweight/obese children and adolescents also confirmed an allele-dosage effect of the rs10830963 G allele on impaired fasting glucose and B-cell functions [Citation210,Citation212,Citation213]. Furthermore, several MTNR1B variants, such as rs76371840, rs8192552, rs6177139, rs6483208, rs4388843, rs4601728, and rs12804291, are associated with obesity traits, and may also increase the risk of T2DM indirectly [Citation171]. Therefore, the interaction between MTNR1B and BMI could provide critical candidate targets for glucose metabolism.
Table 3. The different genetic effects of MTNR1B on glycemic traits in overweight/obesity individuals.
Cohort studies indicated that MTNR1B rs10830963 was associated with elevated fasting glucose in early life among nondiabetic individuals [Citation112,Citation180]. The common genetic variants contributed to a stable age-related rise of fasting glucose, but a more pronounced age-related increase of 2-h postload glucose [Citation180,Citation215]. These results were consisted with the impaired insulin secretion in carriers with risk MTNR1B genotype over the lifespan [Citation180]. Another longitudinal observation indicated that the MTNR1B rs10830963 risk allele led to a faster rate of progression from normal fasting glucose to impaired fasting glucose (IFG) than the rate of progression from IFG to T2DM [Citation216]. Therefore, age may modify the heterogenetic effects of MTNR1B risk alleles on glycaemic phenotype via progressively impaired fasting glucose and insulin response
Gender differences in the genetic effects of MTNR1B on T2DM remain elusive. A population-based analysis presented similar trajectories of glucose, insulin and C-peptide in rs10830963 CC, CG, and GG genotypes among young healthy volunteers, but the glycemic curves exhibited gender differences [Citation217]. Specifically, the glycemic curves were monophasic, biphasic, triphasic, or more complex in healthy volunteers. The proportion of biphasic glycemic curves in men was twice as high as that in women, while the ratio of triphasic glycemic curves was significantly higher in women than in men [Citation217]. A joint analysis of nine diabetes-related genes (MTNR1B, TCF7L2, KCNJ11, HHEX, SLC30A8, WFS1, KCNQ1, FTO, and PPARG) indicated that the weighted risk score of these diabetes risk alleles predicted impaired glucose tolerance (IGT) in females after adjustment for age, BMI and insulin sensitivity; no such association was found in males [Citation218]. However, another large cohort study of 5,327 nondiabetic men found that 8 T2DM-related genes (MTNR1B, TCF7L2, KCNJ11, HHEX, SLC30A8, CDKN2B, CDKAL1, and IGF2BP2) were associated with impaired early-phase insulin release, individually or in combination [Citation219]. A randomized crossover study in healthy men aged 20-40 years reported that the effects of acute-term melatonin treatment on glucose metabolism were modified by MTNR1B genotypes and that deteriorated insulin sensitivity seemed to be driven by the MTNR1B rs10830963 C allele; however, the glucose levels were unchanged on melatonin days compared to placebo days [Citation175]. A similar clinical trial in young women (mean age: 24 ± 6 years) confirmed a melatonin-induced impairment of glucose tolerance in carriers of the MTNR1B rs10830963 G allele [Citation174]. The complicated interactions between MTNR1B and gender and their effects on circadian rhythm and T2DM have yet to be fully elucidated.
4.5. Is MTNR1B the genetic overlap of T1DM and T2DM?
T2DM and type 1 diabetes mellitus (T1DM) are etiologically different diabetes subtypes. Genetic overlap between the two forms of diabetes mellitus has rarely been reported. A previous study in children found no association of the T2DM susceptibility gene MTNR1B with islet autoimmunity and T1DM [Citation220]. However, a Finnish cohort study found that carriers of the MTNR1B rs10830963 G allele were predisposed to T1DM and latent autoimmune diabetes in adulthood (LADA) among participants older than 35 years [Citation221]. Another study used data-driven cluster analysis to divide individuals with new-onset diabetes into five subgroups; they found that rs10830963 was associated with severe autoimmune diabetes (SAID), which is characterized by relatively low BMI, insulin deficiency, metabolic disturbance and positivity for glutamic acid decarboxylase antibodies (GADAs) [Citation222]. However, the available data are still limited to clarifying the role of MTNR1B in T1DM.
5. Evolutionary selection pressures on MTNR1B in T2DM
T2DM is a complex metabolic disease with high heritability. The common variants of MTNR1B were unequivocally classified as genetic determinants of energy expenditure and metabolic abnormalities, including glycemic and adiposity traits, which are implicated in T2DM [Citation140]. Recent evolutionary evidence suggests positive genetic selection at MTNR1B for energy expenditure, glucose regulation and the circadian rhythm, which may make MTNR1B a thrifty gene. The “thrifty gene hypothesis” may partially resolve the evolutionary puzzle of MTNR1B and provide a potential explanation for T2DM.
5.1. The thrifty gene hypothesis and T2DM
The thrifty genotype of T2DM was first proposed by Neel in 1962; he speculated that thrifty genes promoted survival advantage during periods of famine via enhanced energy storage, fat deposition and glucose regulation [Citation223]. However, in modern times with plentiful food supply, the thrifty gene for efficient intake and utilization of food favors the occurrence of T2DM. Subsequent research even distinguished “thrifty early” and “thrifty late” genes [Citation224]. Human ancestors exhibited a hunter-gatherer lifestyle with feast and famine during the day and night, respectively. This lifestyle did not change until the arrival of the Neolithic Age approximately 10 thousand years ago, when humans began to domesticate animals and grow plant crops as food sources. Since then, humans have had a regular diet, although with insufficient nutrients; this regular diet gradually became the template of diet habits in modern society. The “thrifty early” gene hypothesis assumes that the coalescence of autosomes of humans was completed approximately 1 to 2 million years ago; thus, the genetic variants (thrifty alleles) occurring in ancestral genes before 2 million years ago were shared in the whole population. This perspective seems to be consistent with Neel’s assumption [Citation224]. “Thrifty late” genes are theorized to have arisen much later in human evolution, after the differentiation of living environments and evolutionary selection after the out-of-Africa migration [Citation224,Citation225].
The thrifty gene hypothesis has sparked great interest in genetic risks for T2D among different ethnic groups over the years. Previous studies provided genetic evidence for the role of the transcription factor 7-like 2 (TCF7L2) gene in the relationship between T2DM and adaptive evolution [Citation226,Citation227]; the researchers observed potential positive selection for HapA (a cluster of homogeneous haplotypes in TCF7L2), which was linked with T2DM, as well as several phenotypic traits related to energy metabolism among East Asian, European and West African populations [Citation227]. Another study in 2009 found high population differentiation for rs7901695 at TCF7L2 but failed to acquire a general signal of positive selection for 17 obesity and T2DM risk loci [Citation228]. In addition, 58 T2DM- or obesity-associated loci from GWASs were examined in 53 different populations by geographical region; the findings implied that different populations experienced substantial variation in selection pressures on obesity and T2DM risk alleles, and no evidence was able to distinguish the ancestral or derived thirty genotypes for T2DM/obesity [Citation229]. The specific variants tended to appear in specific populations; for example, the region containing T2DM loci exhibited predominantly group-specific differentiation between East Asians and Sub-Saharan Africans [Citation229]. A large genetic analysis of thrifty genes in 65 T2DM index SNPs failed to identify a global signal for positive selection but detected several positively selected loci in one or more specific populations; for example, five SNPs in PROX1, GRB14, UEB2E2, IGF2BP2 and ARAP1 in African populations; three SNPs in PROX1, HMGA2 and PRC1 in European populations; and nine SNPs in NOTCH2, THADA, GRB14, WFS1, TP53INP1, TCF7L2 and PRC1 in East Asian populations [Citation224]. Some studies have provided a potential explanation for this phenomenon: the extreme disparities in T2DM risk allele frequencies across diverse populations from Sub-Saharan Africa and from Europe to East Asia may have been caused by an adaptation to different climates, agricultural revolutions or dietary components after human mass migration [Citation230,Citation231]. Neel also emphasized the influences of the complex interplay of genetic and environmental factors on T2DM 36 years after proposing the thrifty gene concept; he supported the role of detrimental lifestyle changes in industrialized societies in genetic homeostasis among populations at high risk of T2DM [Citation232].
5.2. Selective pressure on MTNR1B is linked with T2DM
Early evolutionary studies of the MTNR1B gene implied positive selection on the lineage leading to human adaptation [Citation37,Citation38]. Human G-protein-coupled MTNR1B consists of 362 amino acids, and its predicted membrane topology comprises four extracellular domains, seven transmembrane domains, four cytoplasmic domains, an extracellular N-terminal domain and an intracellular C-terminal domain [Citation233]. The MTNR1B gene is on chromosome 11q21-22 and has two exons separated by an intron [Citation234]. It is encoded by the first exon from the N-terminal extracellular domain to the first intracellular loop; the other six transmembrane domains and C-terminal domain are coded by the second exon [Citation234]. Phylogenetic analysis revealed a positional difference in the evolutionary rate of the MTNR1B amino acid sequence. For instance, the transmembrane domains were relatively conserved, while the N-terminal domain and an intracellular C-terminal domain have several positively selected sites. Moreover, in primates, an additional 4 sites of positive selection were specifically detected in extracellular domains (2 sites), cytoplasmic domains (1 site) and the intracellular C-terminal domain (1 site) [Citation234].
Population genetic analyses based on HapMap SNP data suggest evolutionary selective pressure on MTNR1B alleles [Citation37]. Several reported variant loci in MTNR1B, such as rs10830963, rs1280429 and rs7951037, were mapped in introns, but rs10830962 and rs4753426 were mapped in the 5′ region. The MTNR1B rs1830963 G allele and rs4753426 C allele, which have a detrimental impact on β-cell function as well as glucose levels, exhibited ethnic diversity of risk allele frequencies between Central Europeans and Asians (Chinese Han population and Japanese population) [Citation37]. The frequency of the rs10830963 G allele was also higher in Europe compared to Africa [Citation235]. The different risk allele frequencies of MTNR1B loci may be due to a combination of a genetic drift effect and evolving adaptation. The MTNR1B variants generated from evolutionary pressures may act as intermediates that link the circadian rhythm with glucose homeostasis. MTNR1B rs10830963 G carriers had a higher risk of T2DM due to its mediation of melatonin secretion, the circadian rhythm and other physiological activities [Citation32]. The risk allele (rs1830963 G) was associated with later melatonin secretion onset in the dim-light phase as well as prolongation of these elevated melatonin levels to the morning; risk allele carriers who woke up earlier were at higher risk of T2DM [Citation32]. Perhaps the common variant at rs1830963 helped human ancestors adapt to a lack of food intake during the long night and even in the morning during the hunter-gatherer stage, and the delayed onset and prolonged duration of melatonin secretion prevented nocturnal and early-morning hypoglycemia. However, in modern society, carriers of this genetic variant maintain a relatively elevated fasting glucose level as well as increased insulin resistance from night to morning, which especially aggravates glucose tolerance in the morning after a substantial breakfast. Hence, we hypothesize that in individuals with the rs1830963 risk allele who wake up early in the morning, the effects of the risk variants on T2DM risk, such as impaired fasting glucose and glucose tolerance, are magnified. Moreover, the frequency of the MTNR1B rs4753426 C allele was negatively correlated with environmental sunshine duration among populations originating from different geographic regions [Citation38], which indicates that this disease-predisposition gene in the melatonin system is a candidate gene subject to ambient light and circadian rhythms. MTNR1B rs16918190, located 105 kb upstream of the gene, was identified as having experienced positive selection in East Asia [Citation235]. Although recent reports were unable to find an association between rs16918190 and glucose regulation, the SNP may act as a gene transcriptional control in the regulatory region.
Nevertheless, another meta-analysis showed a consistent genetic effect of MTNR1B rs10830963 on glycemic traits in Europeans and African Americans but no positive selection in the locus [Citation121]. Indeed, recent phylogenetic analyses have indicated a conserved evolutionary relationship of MTNR1B between humans and vertebrates both in terms of structure and function [Citation1,Citation234,Citation235]; specifically, a synteny analysis found a conserved suite consisting of seven neighboring genes in different species that varied upstream and downstream of MTNR1B, which may enhance the conservation of the gene [Citation1]. Thus, it is difficult to determine the evolutionary selection of the “thrifty gene” for T2DM.
T2DM is a disease attributed to polygenic inheritance, environmental factors and behaviors. Although the genetic variants of MTNR1B appear to explain only small effect sizes regarding the risk of T2DM, the rs1830963 G allele consistently provides a robust signal for T2DM risk across multiple populations. MTNR1B is a critical mediator in melatonin signaling that links circadian rhythms to T2DM, and a common variant of MTNR1B rs10830963 seems to further alter the nocturnal release of melatonin, inhibiting its release from β-cells; this alteration leads to a delayed rhythm and elevated glucose levels during the night as well as in the early morning. In terms of the “thrifty gene” hypothesis, it is plausible that harsh survival stressors might have triggered the evolution of adaptive variants of MTNR1B rs10830963 that maintained elevated fasting glucose levels to prevent overnight starvation. While this finding of a possible link between the selective pressure on MTNRIB and T2DM risk is of great interest, the thrifty mechanisms of MTNR1B need further elucidation.
6. Conclusion and future perspectives
A great deal of evidence supports the key role of the circadian rhythm in glucose homeostasis. Pineal melatonin is a critical chronobiotic hormone that synchronizes the endogenous circadian rhythm to the exogenous photoperiod. This review highlights the role of melatonin and its receptors in glucose metabolism and T2DM via the circadian rhythm. The possible mechanisms include the binding of melatonin to its receptors (MT1 and MT2) in the SCN and peripheral tissues (pancreatic, muscle, liver, adipose and gut), which control glucose metabolism via multiple downstream signaling cascades, such as the Kir3 ion channel, PKC, cGMP, and IP3. Furthermore, the findings from GWASs and replicated cohort studies indicate a robust genetic effect of MTNR1B, which codes melatonin receptor MT2, on glycemic traits and T2DM risk, especially the rs10830963 G allele. However, there is currently not insufficient functional evidence to determine the effects of MTNR1B variants on the risk of T2DM. Evolutionary studies have implied selective pressure on MTNR1B and supported its importance in glucose homeostasis. A common variant at MTNR1B rs10830963 may lead to later melatonin secretion onset in the dim-light cycle and an extended duration of elevated melatonin levels, further affecting the circadian rhythm and elevating fasting glucose levels as well as increasing insulin resistance. Accordingly, we propose that genetic variants in MTNR1B resulted from evolutionary selection to facilitate adaptation to famine or food shortage by maintaining fasting glucose for survival; unfortunately, these variants now confer susceptibility to T2DM. In conclusion, MTNR1B may be a “thrifty gene” that is experienced in positive selection to link T2DM via circadian misalignment in modern society. The “thrifty gene” hypothesis might indicate a possible mechanism underlying T2DM and provide a novel target for the prevention and treatment of T2DM.
Ethics statement
Not applicable.
Author contributions
Conception and design: Jin Xu and Lin-dan Ji; writing- original draft preparation: Hui Zhu; preparing of figures: Zhi-jia Zhao and Hong-yi Liu; editing review: Jie Cai and Qin-kang Lu; Revising: Jin Xu and Lin-dan Ji; Final approval: Jin Xu and Lin-dan Ji. All authors agree to be accountable for all aspects of the work.
Disclosure statement
The authors declare that they have no conflict of interest.
Data availability statement
The authors confirm that the data supporting the findings of this study are available within the review.
Additional information
Funding
References
- International Diabetes Federation (IDF) 2021. Available from: https://diabetesatlasorg/.
- Smushkin G, Vella A. Genetics of type 2 diabetes. Curr Opin Clin Nutr Metab Care. 2010;13(4):471–477.
- Panda S. Circadian physiology of metabolism. Science. 2016;354(6315):1008–1015.
- Qian J, Scheer F. Circadian system and glucose metabolism: implications for physiology and disease. Trends Endocrinol Metab. 2016;27(5):282–293.
- Mason IC, Qian J, Adler GK, et al. Impact of circadian disruption on glucose metabolism: implications for type 2 diabetes. Diabetologia. 2020;63(3):462–472.
- Shan Z, Li Y, Zong G, et al. Rotating night shift work and adherence to unhealthy lifestyle in predicting risk of type 2 diabetes: results from two large US cohorts of female nurses. BMJ. 2018;363:k4641.
- Pan A, Schernhammer ES, Sun Q, et al. Rotating night shift work and risk of type 2 diabetes: two prospective cohort studies in women. PLoS Med. 2011;8(12):e1001141.
- Suwazono Y, Sakata K, Okubo Y, et al. Long-term longitudinal study on the relationship between alternating shift work and the onset of diabetes mellitus in male japanese workers. J Occup Environ Med. 2006;48(5):455–461.
- Koopman ADM, Rauh SP, van ‘t Riet E, et al. The association between social jetlag, the metabolic syndrome, and type 2 diabetes mellitus in the general population: the new hoorn study. J Biol Rhythms. 2017;32(4):359–368.
- Mota MC, Silva CM, Balieiro LCT, et al. Social jetlag and metabolic control in non-communicable chronic diseases: a study addressing different obesity statuses. Sci Rep. 2017;7(1):6358.
- Mokhlesi B, Temple KA, Tjaden AH, et al. Association of self-reported sleep and circadian measures with glycemia in adults with prediabetes or recently diagnosed untreated type 2 diabetes. Diabetes Care. 2019;42(7):1326–1332.
- Merikanto I, Lahti T, Puolijoki H, et al. Associations of chronotype and sleep with cardiovascular diseases and type 2 diabetes. Chronobiol Int. 2013;30(4):470–477.
- Stenvers DJ, Scheer F, Schrauwen P, et al. Circadian clocks and insulin resistance. Nat Rev Endocrinol. 2019;15(2):75–89.
- Gale JE, Cox HI, Qian J, et al. Disruption of circadian rhythms accelerates development of diabetes through pancreatic beta-cell loss and dysfunction. J Biol Rhythms. 2011;26(5):423–433.
- Scheer FA, Hilton MF, Mantzoros CS, et al. Adverse metabolic and cardiovascular consequences of circadian misalignment. Proc Natl Acad Sci U S A. 2009;106(11):4453–4458.
- Karamitri A, Jockers R. Melatonin in type 2 diabetes mellitus and obesity. Nat Rev Endocrinol. 2019;15(2):105–125.
- Peschke E. Melatonin, endocrine pancreas and diabetes. J Pineal Res. 2008;44(1):26–40.
- Tuomi T, Nagorny CLF, Singh P, et al. Increased melatonin signaling is a risk factor for type 2 diabetes. Cell Metab. 2016;23(6):1067–1077.
- McMullan CJ, Schernhammer ES, Rimm EB, et al. Melatonin secretion and the incidence of type 2 diabetes. JAMA. 2013;309(13):1388–1396.
- McMullan CJ, Curhan GC, Schernhammer ES, et al. Association of nurnal melatonin secretion with insulin resistance in nondiabetic young women. Am J Epidemiol. 2013;178(2):231–238.
- Garfinkel D, Zorin M, Wainstein J, et al. Efficacy and safety of prolonged-release melatonin in insomnia patients with diabetes: a randomized, double-blind, crossover study. Diabetes Metab Syndr Obes. 2011;4:307–313.
- Rubio-Sastre P, Scheer FA, Gomez-Abellan P, et al. Acute melatonin administration in humans impairs glucose tolerance in both the morning and evening. Sleep. 2014;37(10):1715–1719.
- Cagnacci A, Arangino S, Renzi A, et al. Influence of melatonin administration on glucose tolerance and insulin sensitivity of postmenopausal women. Clin Endocrinol (Oxf). 2001;54(3):339–346.
- Dubocovich ML. Melatonin receptors: are there multiple subtypes? Trends Pharmacol Sci. 1995;16(2):50–56.
- Stein RM, Kang HJ, McCorvy JD, et al. Virtual discovery of melatonin receptor ligands to modulate circadian rhythms. Nature. 2020;579(7800):609–614
- Ramracheya RD, Muller DS, Squires PE, et al. Function and expression of melatonin receptors on human pancreatic islets. J Pineal Res. 2008;44(3):273–279.
- Blodgett DM, Nowosielska A, Afik S, et al. el observations from Next-Generation RNA sequencing of highly purified human adult and fetal islet cell subsets. Diabetes. 2015;64(9):3172–3181.
- Bouatia-Naji N, Bonnefond A, Cavalcanti-Proenca C, et al. A variant near MTNR1B is associated with increased fasting plasma glucose levels and type 2 diabetes risk. Nat Genet. 2009;41(1):89–94.
- Prokopenko I, Langenberg C, Florez JC, et al. Variants in MTNR1B influence fasting glucose levels. Nat Genet. 2009;41(1):77–81.
- Dupuis J, Langenberg C, Prokopenko I, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010;42(2):105–116.
- Hu C, Jia W. Linking MTNR1B variants to diabetes: the role of circadian rhythms. Diabetes. 2016;65(6):1490–1492.
- Lane JM, Chang AM, Bjonnes AC, et al. Impact of common diabetes risk variant in MTNR1B on sleep, circadian, and melatonin physiology. Diabetes. 2016;65(6):1741–1751.
- Imam A, Winnebeck EC, Buchholz N, et al. Circadian, sleep and caloric intake phenotyping in type 2 diabetes patients with rare melatonin receptor 2 mutations and controls: a pilot study. Front Physiol. 2020;11:564140.
- Garaulet M, Lopez-Minguez J, Dashti HS, et al. Interplay of dinner timing and MTNR1B type 2 diabetes risk variant on glucose tolerance and insulin secretion: a randomized crossover trial. Diabetes Care. 2022;45(3):512–519.
- Mulder H. Melatonin signalling and type 2 diabetes risk: too little, too much or just right? Diabetologia. 2017;60(5):826–829.
- Florez JC. Newly identified loci highlight beta cell dysfunction as a key cause of type 2 diabetes: where are the insulin resistance genes? Diabetologia. 2008;51(7):1100–1110.
- Dietrich K, Birkmeier S, Schleinitz D, et al. Association and evolutionary studies of the melatonin receptor 1B gene (MTNR1B) in the self-contained population of sorbs from Germany. Diabet Med. 2011;28(11):1373–1380.
- Ji LD, Xu J, Wu DD, et al. Association of disease-predisposition polymorphisms of the melatonin receptors and sunshine duration in the global human populations. J Pineal Res. 2010;48(2):133–141.
- Woelfle MA, Ouyang Y, Phanvijhitsiri K, et al. The adaptive value of circadian clocks: an experimental assessment in cyanobacteria. Curr Biol. 2004;14(16):1481–1486.
- Ouyang Y, Andersson CR, Kondo T, et al. Resonating circadian clocks enhance fitness in cyanobacteria. Proc Natl Acad Sci U S A. 1998;95(15):8660–8664.
- Fagiani F, Di ino D, Romagnoli A, et al. Molecular regulations of circadian rhythm and implications for physiology and diseases. Signal Transduct Target Ther. 2022;7(1):41.
- Li MD, Xin H, Yuan Y, et al. Circadian Clock-Controlled checkpoints in the pathogenesis of complex disease. Front Genet. 2021;12:721231.
- Gooley JJ, Chamberlain K, Smith KA, et al. Exposure to room light before bedtime suppresses melatonin onset and shortens melatonin duration in humans. J Clin Endocrinol Metab. 2011;96(3):E463–72.
- Lockley SW, Brainard GC, Czeisler CA. High sensitivity of the human circadian melatonin rhythm to resetting by short wavelength light. J Clin Endocrinol Metab. 2003;88(9):4502–4505.
- Cipolla-Neto J, Aal FGD. Melatonin as a hormone: new physiological and clinical insights. Endocr Rev. 2018;39(6):990–1028.
- Riggs W, Jr., Seibert J. Pseudoectopic ureter on prone urogram. Radiology. 1973;106(2):391–392.
- Huang H, Wang Z, Weng SJ, et al. Neuromodulatory role of melatonin in retinal information processing. Prog Retin Eye Res. 2013;32:64–87.
- Weaver DR, Stehle JH, Stopa EG, et al. Melatonin receptors in human hypothalamus and pituitary: implications for circadian and reproductive responses to melatonin. J Clin Endocrinol Metab. 1993;76(2):295–301.
- Reppert SM, Weaver DR, Rivkees SA, et al. Putative melatonin receptors in a human biological clock. Science. 1988;242(4875):78–81.
- Vriend J, Reiter RJ. Melatonin feedback on clock genes: a theory involving the proteasome. J Pineal Res. 2015;58(1):1–11.
- Liu J, Clough SJ, Hutchinson AJ, et al. MT1 and MT2 melatonin receptors: a therapeutic perspective. Annu Rev Pharmacol Toxicol. 2016;56:361–383.
- Gobbi G, Comai S. Sleep well. Untangling the role of melatonin MT1 and MT2 receptors in sleep. J Pineal Res. 2019;66(3):e12544.
- Zlotos DP, Jockers R, Cecon E, et al. MT1 and MT2 melatonin receptors: ligands, models, oligomers, and therapeutic potential. J Med Chem. 24 2014;57(8):3161–3185.
- Cipolla-Neto J, Aal FG, Afeche SC, et al. Melatonin, energy metabolism, and obesity: a review. J Pineal Res. 2014;56(4):371–381.
- Genario R, Cipolla-Neto J, Bueno AA, et al. Melatonin supplementation in the management of obesity and obesity-associated disorders: a review of physiological mechanisms and clinical applications. Pharmacol Res. 2021;163:105254.
- Garaulet M, Qian J, Florez JC, et al. Melatonin effects on glucose metabolism: time to unlock the controversy. Trends Endocrinol Metab. 2020;31(3):192–204.
- Reiter RJ, o JC, Tan DX, et al. Melatonin as an antioxidant: under promises but over delivers. J Pineal Res. 2016;61(3):253–278.
- Claustrat B, Brun J, Chazot G. The basic physiology and pathophysiology of melatonin. Sleep Med Rev. 2005;9(1):11–24.
- Lok R, van Koningsveld MJ, Gordijn MCM, et al. Daytime melatonin and light independently affect human alertness and body temperature. J Pineal Res. 2019;67(1):e12583.
- Hardeland R. Melatonin and inflammation-story of a double-edged blade. J Pineal Res. 2018;65(4):e12525.
- Pham L, Baiocchi L, Kennedy L, et al. The interplay between mast cells, pineal gland, and circadian rhythm: links between histamine, melatonin, and inflammatory mediators. J Pineal Res. 2021;70(2):e12699.
- Ma N, Zhang J, Reiter RJ, et al. Melatonin mediates mucosal immune cells, microbial metabolism, and rhythm crosstalk: a therapeutic target to reduce intestinal inflammation. Med Res Rev. 2020;40(2):606–632.
- Sato K, Meng F, Francis H, et al. Melatonin and circadian rhythms in liver diseases: functional roles and potential therapies. J Pineal Res. 2020;68(3):e12639.
- Munmun F, Witt-Enderby PA. Melatonin effects on bone: implications for use as a therapy for managing bone loss. J Pineal Res. 2021;71(1):e12749.
- Smolensky MH, Hermida RC, Portaluppi F. Circadian mechanisms of 24-hour blood pressure regulation and patterning. Sleep Med Rev. 2017;33:4–16.
- Ma Q, Reiter RJ, Chen Y. Role of melatonin in controlling angiogenesis under physiological and pathological conditions. Angiogenesis. 2020;23(2):91–104.
- Opie LH, Lecour S. Melatonin has multiorgan effects. Eur Heart J Cardiovasc Pharmacother. 2016;2(4):258–265.
- Chen HY, Chen TY, Lee MY, et al. Melatonin decreases neurovascular oxidative/nitrosative damage and protects against early increases in the blood-brain barrier permeability after transient focal cerebral ischemia in mice. J Pineal Res. 2006;41(2):175–182.
- Kong X, Gao R, Wang Z, et al. Melatonin: a potential therapeutic option for breast cancer. Trends Endocrinol Metab. 2020;31(11):859–871.
- Su SC, Hsieh MJ, Yang WE, et al. Cancer metastasis: mechanisms of inhibition by melatonin. J Pineal Res. 2017;62(1):1–40.
- Olcese JM. Melatonin and female reproduction: an expanding universe. Front Endocrinol (Lausanne). 2020;11:85.
- Vasey C, McBride J, Penta K. Circadian rhythm dysregulation and restoration: the role of melatonin. Nutrients. 2021;13(10):3480.
- Meyer N, Harvey AG, Lockley SW, et al. Circadian rhythms and disorders of the timing of sleep. Lancet. 2022;400(10357):1061–1078.
- Nishiyama K, Hirai K. The melatonin agonist ramelteon induces duration-dependent clock gene expression through cAMP signaling in pancreatic INS-1 beta-cells. PLoS One. 2014;9(7):e102073.
- Kemp DM, Ubeda M, Habener JF. Identification and functional characterization of melatonin mel 1a receptors in pancreatic beta cells: potential role in incretin-mediated cell function by sensitization of cAMP signaling. Mol Cell Endocrinol. 2002;191(2):157–166.
- Costes S, Boss M, Thomas AP, et al. Activation of melatonin signaling promotes beta-Cell survival and function. Mol Endocrinol. 2015;29(5):682–692.
- Gil-Lozano M, Wu WK, tchenko A, et al. High-Fat diet and palmitate alter the rhythmic secretion of glucagon-like peptide-1 by the rodent L-cell. Endocrinology. 2016;157(2):586–599.
- cheva B, Ramsey KM, Buhr ED, et al. Disruption of the clock components CLOCK and BMAL1 leads to hypoinsulinaemia and diabetes. Nature. 2010;466(7306):627–631.
- Picinato MC, Haber EP, Carpinelli AR, et al. Daily rhythm of glucose-induced insulin secretion by isolated islets from intact and pinealectomized rat. J Pineal Res. 2002;33(3):172–177.
- Sandu C, Liu T, Malan A, et al. Circadian clocks in rat skin and dermal fibroblasts: differential effects of aging, temperature and melatonin. Cell Mol Life Sci. 2015;72(11):2237–2248.
- Reinke H, Asher G. Crosstalk between metabolism and circadian clocks. Nat Rev Mol Cell Biol. 2019;20(4):227–241.
- Stephan FK, Zucker I. Circadian rhythms in drinking behavior and locomotor activity of rats are eliminated by hypothalamic lesions. Proc Natl Acad Sci U S A. 1972;69(6):1583–1586.
- Czeisler CA, Weitzman E, Moore-Ede MC, et al. Human sleep: its duration and organization depend on its circadian phase. Science. 1980;210(4475):1264–1267.
- Saper CB, Scammell TE, Lu J. Hypothalamic regulation of sleep and circadian rhythms. Nature. 2005;437(7063):1257–1263.
- Herrera-Moro Chao D, Leon-Mercado L, Foppen E, et al. The suprachiasmatic nucleus modulates the sensitivity of arcuate nucleus to hypoglycemia in the male rat. Endocrinology. 2016;157(9):3439–3451.
- g CM, Melanson EL, Frydendall EJ, et al. Energy expenditure during sleep, sleep deprivation and sleep following sleep deprivation in adult humans. J Physiol. 1 2011;589(Pt 1):235–244.
- Anothaisintawee T, Reutrakul S, Van Cauter E, et al. Sleep disturbances compared to traditional risk factors for diabetes development: systematic review and meta-analysis. Sleep Med Rev. 2016;30:11–24.
- Kim TW, Jeong JH, Hong SC. The impact of sleep and circadian disturbance on hormones and metabolism. Int J Endocrinol. 2015;2015:591729. .
- Moller N, Jorgensen JO. Effects of growth hormone on glucose, lipid, and protein metabolism in human subjects. Endocr Rev. 2009;30(2):152–177.
- Buckley TM, Schatzberg AF. On the interactions of the hypothalamic-pituitary-adrenal (HPA) axis and sleep: normal HPA axis activity and circadian rhythm, exemplary sleep disorders. J Clin Endocrinol Metab. 2005;90(5):3106–3114.
- Liu PY, Lawrence-Sidebottom D, Piotrowska K, et al. Clamping cortisol and testosterone mitigates the development of insulin resistance during sleep restriction in men. J Clin Endocrinol Metab. 2021;106(9):e3436–e3448.
- von Gall C, Weaver DR, Kock M, et al. Melatonin limits transcriptional impact of phosphoCREB in the mouse SCN via the Mel1a receptor. Neuroreport. 000;11(9):1803–1807.
- Nelson CS, ino JL, Allen CN. Melatonin receptors activate heteromeric G-protein coupled Kir3 channels. Neuroreport. 1996;7(3):717–720.
- McArthur AJ, Hunt AE, Gillette MU. Melatonin action and signal transduction in the rat suprachiasmatic circadian clock: activation of protein kinase C at dusk and dawn. Endocrinology. 1997;138(2):627–634.
- Kandalepas PC, Mitchell JW, Gillette MU. Melatonin signal transduction pathways require E-box-mediated transcription of Per1 and Per2 to reset the SCN clock at dusk. PLoS One. 2016;11(6):e0157824.
- Pfeffer M, Rauch A, Korf HW, et al. The endogenous melatonin (MT) signal facilitates reentrainment of the circadian system to light-induced phase advances by acting upon MT2 receptors. Chronobiol Int. 2012;29(4):415–429.
- Nagy AD, Iwamoto A, Kawai M, et al. Melatonin adjusts the expression pattern of clock genes in the suprachiasmatic nucleus and induces antidepressant-like effect in a mouse model of seasonal affective disorder. Chronobiol Int. 2015;32(4):447–457.
- Stumpf I, Muhlbauer E, Peschke E. Involvement of the cGMP pathway in mediating the insulin-inhibitory effect of melatonin in pancreatic beta-cells. J Pineal Res. 2008;45(3):318–327.
- Peschke E, Muhlbauer E, Musshoff U, et al. Receptor (MT(1)) mediated influence of melatonin on cAMP concentration and insulin secretion of rat insulinoma cells INS-1. J Pineal Res. 2002;33(2):63–71.
- Peschke E, Bahr I, Muhlbauer E. Experimental and clinical aspects of melatonin and clock genes in diabetes. J Pineal Res. 2015;59(1):1–23. .
- Ha E, Yim SV, Chung JH, et al. Melatonin stimulates glucose transport via insulin receptor substrate-1/phosphatidylinositol 3-kinase pathway in C2C12 murine skeletal muscle cells. J Pineal Res. 2006;41(1):67–72.
- Shieh JM, Wu HT, Cheng KC, et al. Melatonin ameliorates high fat diet-induced diabetes and stimulates glycogen synthesis via a PKCzeta-Akt-GSK3beta pathway in hepatic cells. J Pineal Res. 2009;47(4):339–344.
- Altman J, Garay P, Papadimitriou A, et al. Alterations in 22Na fluxes of arterial smooth muscles of spontaneously hypertensive rats. Br J Pharmacol. 1977;59(3):496P.
- Nogueira TC, Lellis-Santos C, Jesus DS, et al. Absence of melatonin induces night-time hepatic insulin resistance and increased gluconeogenesis due to stimulation of nocturnal unfolded protein response. Endocrinology. 2011;152(4):1253–1263.
- Peschke E, Schucht H, Muhlbauer E. Long-term enteral administration of melatonin reduces plasma insulin and increases expression of pineal insulin receptors in both wistar and type 2-diabetic Goto-Kakizaki rats. J Pineal Res. 2010;49(4):373–381.
- Peschke E, Frese T, Chankiewitz E, et al. Diabetic Goto Kakizaki rats as well as type 2 diabetic patients show a decreased diurnal serum melatonin level and an increased pancreatic melatonin-receptor status. J Pineal Res. 2006;40(2):135–143.
- Oliveira AC, Andreotti S, Sertie RAL, et al. Combined treatment with melatonin and insulin improves glycemic control, white adipose tissue metabolism and reproductive axis of diabetic male rats. Life Sci. 2018;199:158–166.
- Sartori C, Dessen P, Mathieu C, et al. Melatonin improves glucose homeostasis and endothelial vascular function in high-fat diet-fed insulin-resistant mice. Endocrinology. 2009;150(12):5311–5317.
- Hikichi T, Tateda N, Miura T. Alteration of melatonin secretion in patients with type 2 diabetes and proliferative diabetic retinopathy. Clin Ophthalmol. 2011;5:655–660.
- Gonciarz M, Bielański W, Partyka R, et al. Plasma insulin, leptin, adiponectin, resistin, ghrelin, and melatonin in nonalcoholic steatohepatitis patients treated with melatonin. J Pineal Res. 2013;54(2):154–161.
- Kadhim HM, Ismail SH, Hussein KI, et al. Effects of melatonin and zinc on lipid profile and renal function in type 2 diabetic patients poorly controlled with metformin. J Pineal Res. 2006;41(2):189–193.
- Sparso T, Bonnefond A, Andersson E, et al. G-allele of intronic rs10830963 in MTNR1B confers increased risk of impaired fasting glycemia and type 2 diabetes through an impaired glucose-stimulated insulin release: studies involving 19,605 Europeans. Diabetes. 2009;58(6):1450–1456.
- Voight BF, Scott LJ, Steinthorsdottir V, et al. Twelve type 2 diabetes susceptibility loci identified through large-scale association analysis. Nat Genet. 2010;42(7):579–589.
- Sabatti C, Service SK, Hartikainen AL, et al. Genome-wide association analysis of metabolic traits in a birth cohort from a founder population. Nat Genet. 2009;41(1):35–46.
- Ronn T, Wen J, Yang Z, et al. A common variant in MTNR1B, encoding melatonin receptor 1B, is associated with type 2 diabetes and fasting plasma glucose in Han Chinese individuals. Diabetologia. 2009;52(5):830–833.
- Liu C, Wu Y, Li H, et al. MTNR1B rs10830963 is associated with fasting plasma glucose, HbA1C and impaired beta-cell function in chinese hans from shanghai. BMC Med Genet. 2010;11:59.
- Takeuchi F, Katsuya T, Chakrewarthy S, et al. Common variants at the GCK, GCKR, G6PC2-ABCB11 and MTNR1B loci are associated with fasting glucose in two Asian populations. Diabetologia. 2010;53(2):299–308.
- Fujita H, Hara K, Shojima N, et al. Variations with modest effects have an important role in the genetic background of type 2 diabetes and diabetes-related traits. J Hum Genet. 2012;57(12):776–779.
- Palmer ND, Goodarzi MO, Langefeld CD, et al. Genetic variants associated with quantitative glucose homeostasis traits translate to type 2 diabetes in Mexican Americans: the GUARDIAN (genetics underlying diabetes in hispanics) consortium. Diabetes. 2015;64(5):1853–1866.
- Ramos E, Chen G, Shriner D, et al. Replication of genome-wide association studies (GWAS) loci for fasting plasma glucose in African-Americans. Diabetologia. 2011;54(4):783–788.
- Liu CT, Ng MC, Rybin D, et al. Transferability and fine-mapping of glucose and insulin quantitative trait loci across populations: CARe, the candidate gene association resource. Diabetologia. 2012;55(11):2970–2984.
- Chambers JC, Zhang W, Zabaneh D, et al. Common genetic variation near melatonin receptor MTNR1B contributes to raised plasma glucose and increased risk of type 2 diabetes among Indian Asians and European Caucasians. Diabetes. 2009;58(11):2703–2708.
- Soranzo N, Sanna S, Wheeler E, et al. Common variants at 10 genomic loci influence hemoglobin A(1)(C) levels via glycemic and nonglycemic pathways. Diabetes. 2010;59(12):3229–3239.
- Kraja AT, Vaidya D, Pankow JS, et al. A bivariate genome-wide approach to metabolic syndrome: STAMPEED consortium. Diabetes. 2011;60(4):1329–1339.
- Kim YJ, Go MJ, Hu C, MAGIC consortium, et al. Large-scale genome-wide association studies in east Asians identify new genetic loci influencing metabolic traits. Nat Genet. 2011;43(10):990–995.
- Kettunen J, Tukiainen T, Sarin AP, et al. Genome-wide association study identifies multiple loci influencing human serum metabolite levels. Nat Genet. 2012;44(3):269–276.
- Rasmussen-Torvik LJ, Guo X, Bowden DW, et al. Fasting glucose GWAS candidate region analysis across ethnic groups in the multiethnic study of atherosclerosis (MESA). Genet Epidemiol. 2012;36(4):384–391.
- Manning AK, Hivert MF, Scott RA, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012;44(6):659–669.
- Comuzzie AG, Cole SA, Laston SL, et al. Novel genetic loci identified for the pathophysiology of childhood obesity in the Hispanic population. PLoS One. 2012;7(12):e51954.
- Kwak SH, Kim SH, Cho YM, et al. A genome-wide association study of gestational diabetes mellitus in Korean women. Diabetes. . 2012;61(2):531–541.
- Kristiansson K, Perola M, Tikkanen E, et al. Genome-wide screen for metabolic syndrome susceptibility loci reveals strong lipid gene contribution but no evidence for common genetic basis for clustering of metabolic syndrome traits. Circ Cardiovasc Genet. 2012;5(2):242–249.
- Morris CJ, Yang JN, Scheer F. The impact of the circadian timing system on cardiovascular and metabolic function. Prog Brain Res. 2012;199:337–358.
- Go MJ, Hwang JY, Kim YJ, et al. New susceptibility loci in MYL2, C12orf51 and OAS1 associated with 1-h plasma glucose as predisposing risk factors for type 2 diabetes in the korean population. J Hum Genet. 2013;58(6):362–365.
- Hayes MG, Urbanek M, Hivert MF, HAPO Study Cooperative Research Group, et al. Identification of HKDC1 and BACE2 as genes influencing glycemic traits during pregnancy through genome-wide association studies. Diabetes. 2013;62(9):3282–3291.
- Prokopenko I, Poon W, Magi R, et al. A Central role for GRB10 in regulation of islet function in man. PLoS Genet. 2014;10(4):e1004235.
- Maha A, Go MJ, Zhang W. Genome-wide trans-ancestry meta-analysis provides insight into the genetic architecture of type 2 diabetes susceptibility. Nat Genet. 2014;46(3):234–244.
- Wessel J, Chu AY, Willems SM, et al. Low-frequency and rare exome chip variants associate with fasting glucose and type 2 diabetes susceptibility. Nat Commun. 2015;6:5897.
- Kettunen J, Demirkan A, Wurtz P, et al. Genome-wide study for circulating metabolites identifies 62 loci and reveals el systemic effects of LPA. Nat Commun. 2016;7:11122.
- Horikoshi M, Beaumont RN, Day FR, et al. Genome-wide associations for birth weight and correlations with adult disease. Nature. 2016;538(7624):248–252.
- Wood AR, Jonsson A, Jackson AU, et al. A genome-wide association study of IVGTT-based measures of first-phase insulin secretion refines the underlying physiology of type 2 diabetes variants. Diabetes. 2017;66(8):2296–2309.
- Wheeler E, Leong A, Liu CT, et al. Impact of common genetic determinants of hemoglobin A1c on type 2 diabetes risk and diagnosis in ancestrally diverse populations: a transethnic genome-wide meta-analysis. PLoS Med. 2017;14(9):e1002383.
- Zhao W, Rasheed A, Tikkanen E, Michigan Biobank, et al. Identification of new susceptibility loci for type 2 diabetes and shared etiological pathways with coronary heart disease. Nat Genet. 2017;49(10):1450–1457.
- Qi Q, Stilp AM, Sofer T, et al. Genetics of type 2 diabetes in U.S. Hispanic/latino individuals: results from the hispanic community health study/study of latinos (HCHS/SOL). Diabetes. . 2017;66(5):1419–1425.
- Beaumont RN, Warrington NM, Cavadino A, et al. Genome-wide association study of offspring birth weight in 86 577 women identifies five el loci and highlights maternal genetic effects that are independent of fetal genetics. Hum Mol Genet. 2018;27(4):742–756.
- Kanai M, Akiyama M, Takahashi A, et al. Genetic analysis of quantitative traits in the Japanese population links cell types to complex human diseases. Nat Genet. 2018;50(3):390–400.
- Wu B, Pankow JS. Fast and accurate genome-wide association test of multiple quantitative traits. Comput Math Methods Med. 2018;2018:2564531.
- Loomis SJ, Li M, uthur NM, et al. Genome-wide association study of serum fructosamine and glycated albumin in adults without diagnosed diabetes: results from the atherosclerosis risk in communities study. Diabetes. 2018;67(8):1684–1696.
- Kichaev G, Bhatia G, Loh PR, et al. Leveraging polygenic functional enrichment to improve GWAS power. Am J Hum Genet. 2019;104(1):65–75.
- Keaton JM, Gao C, Guan M, et al. Genome-wide interaction with the insulin secretion locus MTNR1B reveals CMIP as a el type 2 diabetes susceptibility gene in african americans. Genet Epidemiol. 2018;42(6):559–570.
- Bonas-Guarch S, Guindo-tinez M, Miguel-Escalada I, et al. Re-analysis of public genetic data reveals a rare X-chromosomal variant associated with type 2 diabetes. Nat Commun. 22 2018;9(1):321.
- van Zuydam NR, Ahlqvist E, Sandholm N, et al. A genome-wide association study of diabetic kidney disease in subjects with type 2 diabetes. Diabetes. . 2018;67(7):1414–1427.
- Xue A, Wu Y, Zhu Z, et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat Commun. 2018;9(1):2941.
- Maha A, Taliun D, Thurner M, et al. Fine-mapping type 2 diabetes loci to single-variant resolution using high-density imputation and islet-specific epigenome maps. Nat Genet. 2018;50(11):1505–1513.
- Masotti M, Guo B, Wu B. Pleiotropy informed adaptive association test of multiple traits using genome-wide association study sumy data. Biometrics. 2019;75(4):1076–1085.
- Warrington NM, Beaumont RN, Horikoshi M, et al. Maternal and fetal genetic effects on birth weight and their relevance to cardio-metabolic risk factors. Nat Genet. 2019;51(5):804–814.
- Wojcik GL, Graff M, Nishimura KK, et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019;570(7762):514–518.
- Gallois A, Mefford J, Ko A, et al. A comprehensive study of metabolite genetics reveals strong pleiotropy and heterogeneity across time and context. Nat Commun. 2019;10(1):4788.
- Zhu Z, Guo Y, Shi H, et al. Shared genetic and experimental links between obesity-related traits and asthma subtypes in UK biobank. J Allergy Clin Immunol. 2020;145(2):537–549.
- ienne H, Lechat P, Guillemot V, et al. JASS: command line and web interface for the joint analysis of GWAS results. NAR Genom Bioinform. 2020;2(1):lqaa003.
- Oh SW, Lee JE, Shin E, et al. Genome-wide association study of metabolic syndrome in korean populations. PLoS One. 2020;15(1):e0227357.
- Vujkovic M, Keaton JM, Lynch JA, et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet. 2020;52(7):680–691.
- Spracklen CN, Horikoshi M, Kim YJ, et al. Identification of type 2 diabetes loci in 433,540 east asian individuals. Nature. 2020;582(7811):240–245.
- Lagou V, Magi R, Hottenga JJ, et al. Sex-dimorphic genetic effects and el loci for fasting glucose and insulin variability. Nat Commun. J2021;12(1):24.
- Sinnott-Armstrong N, Tanigawa Y, A D, et al. Genetics of 35 blood and urine biokers in the UK biobank. Nat Genet. 2021;53(2):185–194.
- Chung RH, Chiu YF, Wang WC, et al. Multi-omics analysis identifies CpGs near G6PC2 mediating the effects of genetic variants on fasting glucose. Diabetologia. 2021;64(7):1613–1625.
- Chen J, Spracklen CN, enne G, et al. The trans-ancestral genomic architecture of glycemic traits. Nat Genet. 2021;53(6):840–860.
- Sakaue S, Kanai M, Tanigawa Y, et al. A cross-population atlas of genetic associations for 220 human phenotypes. Nat Genet. 2021;53(10):1415–1424.
- Li-Gao R, Hughes DA, van Klinken JB, et al. Genetic studies of metabolomics change after a liquid meal illuminate el pathways for glucose and lipid metabolism. Diabetes. 2021;70(12):2932–2946.
- Downie CG, Dimos SF, Bien SA, et al. Multi-ethnic GWAS and fine-mapping of glycaemic traits identify el loci in the PAGE study. Diabetologia. 2022;65(3):477–489.
- DiCorpo D, Gaynor SM, Russell EM, et al. Whole genome sequence association analysis of fasting glucose and fasting insulin levels in diverse cohorts from the NHLBI TOPMed program. Commun Biol. 2022;5(1):756.
- Tchio C, Musani SK, Quarshie A, et al. Association between MTNR1B polymorphisms and obesity in African American: findings from the jackson heart study. BMC Med Genomics. 2021;14(1):136.
- Song JF, Zhang J, Zhang MZ, et al. Evaluation of the effect of MTNR1B rs10830963 gene variant on the therapeutic efficacy of nateglinide in treating type 2 diabetes among chinese han patients. BMC Med Genomics. 2021;14(1):156.
- Tan X, Benedict C. Does the common type 2 Diabetes-Susceptibility variant in the MTNR1B gene matter for glycemic control among patients on antidiabetic pharmacotherapy? Clin Proc. 2021;96(5):1372–1374.
- Garaulet M, Gomez-Abellan P, Rubio-Sastre P, et al. Common type 2 diabetes risk variant in MTNR1B worsens the deleterious effect of melatonin on glucose tolerance in humans. Metabolism. 2015;64(12):1650–1657.
- Kampmann U, Lauritzen ES, Grarup N, et al. Acute metabolic effects of melatonin – a randomized crossover study in healthy young men. J Pineal Res. 2021;70(2):e12706.
- Cherrington AD. Banting lecture 1997. Control of glucose uptake and release by the liver in vivo. Diabetes. 1999;48(5):1198–1214.
- Magnusson I, Rothman DL, Katz LD, et al. Increased rate of gluconeogenesis in type II diabetes mellitus. A 13C nuclear magnetic resonance study. J Clin Invest. 1992;90(4):1323–1327.
- Vangipurapu J, Stancakova A, Pihlajamaki J, et al. Association of indices of liver and adipocyte insulin resistance with 19 confirmed susceptibility loci for type 2 diabetes in 6,733 non-diabetic finnish men. Diabetologia. 2011;54(3):563–571.
- Staiger H, Machicao F, Schafer SA, et al. Polymorphisms within the el type 2 diabetes risk locus MTNR1B determine beta-cell function. PLoS One. 2008;3(12):e3962.
- Lyssenko V, Nagorny CL, Erdos MR, et al. Common variant in MTNR1B associated with increased risk of type 2 diabetes and impaired early insulin secretion. Nat Genet. 2009;41(1):82–88.
- van de Bunt M, Manning Fox JE, Dai X, et al. Transcript expression data from human islets links regulatory signals from Genome-Wide association studies for type 2 diabetes and glycemic traits to their downstream effectors. PLoS Genet. 2015;11(12):e1005694.
- Li Y, Wu H, Liu N, et al. Melatonin exerts an inhibitory effect on insulin gene transcription via MTNR1B and the downstream Raf1/ERK signaling pathway. Int J Mol Med. 2018;41(2):955–961.
- Gaulton KJ, Ferreira T, Lee Y, et al. Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat Genet. 2015;47(12):1415–1425.
- Bonnefond A, Clement N, Fawcett K, et al. Rare MTNR1B variants impairing melatonin receptor 1B function contribute to type 2 diabetes. Nat Genet. 2012;44(3):297–301.
- Zibolka J, Bazwinsky-Wutschke I, Muhlbauer E, et al. Distribution and density of melatonin receptors in human main pancreatic islet cell types. J Pineal Res. 2018;65(1):e12480.
- Bahr I, Muhlbauer E, Schucht H, et al. Melatonin stimulates glucagon secretion in vitro and in vivo. J Pineal Res. 2011;50(3):336–344.
- Zibolka J, Muhlbauer E, Peschke E. Melatonin influences somatostatin secretion from human pancreatic Delta-cells via MT1 and MT2 receptors. J Pineal Res. 2015;58(2):198–209.
- Jonsson A, Ladenvall C, Ahluwalia TS, et al. Effects of common genetic variants associated with type 2 diabetes and glycemic traits on alpha- and beta-cell function and insulin action in humans. Diabetes. . 2013;62(8):2978–2983.
- You H, Hu J, Liu Y, et al. Risk of type 2 diabetes mellitus after gestational diabetes mellitus: a systematic review & meta-analysis. Indian J Med Res. 2021;154(1):62–77.
- Pervjakova N, Moen GH, Borges MC, et al. Multi-ancestry genome-wide association study of gestational diabetes mellitus highlights genetic links with type 2 diabetes. Hum Mol Genet. 2022;31(19):3377–3391.
- Powe CE, Kwak SH. Genetic studies of gestational diabetes and glucose metabolism in pregnancy. Curr Diab Rep. 2020;20(12):69.
- Liao S, Liu Y, Chen X, et al. The impact of genetic variants for different physiological characterization of type 2 diabetes loci on gestational insulin signaling in nondiabetic pregnant chinese women. Reprod Sci. 2015;22(11):1421–1428.
- Vlassi M, Gazouli M, Paltoglou G, et al. The rs10830963 variant of melatonin receptor MTNR1B is associated with increased risk for gestational diabetes mellitus in a Greek population. Hormones (Athens). - 2012;11(1):70–76.
- Kim JY, Cheong HS, Park BL, et al. Melatonin receptor 1 B polymorphisms associated with the risk of gestational diabetes mellitus. BMC Med Genet. 10 2011;12:82.
- Huopio H, Cederberg H, Vangipurapu J, et al. Association of risk variants for type 2 diabetes and hyperglycemia with gestational diabetes. Eur J Endocrinol. 2013;169(3):291–297.
- Ding M, Chavarro J, Olsen S, et al. Genetic variants of gestational diabetes mellitus: a study of 112 SNPs among 8722 women in two independent populations. Diabetologia. 2018;61(8):1758–1768.
- Grotenfelt NE, Wasenius NS, Rono K, et al. Interaction between rs10830963 polymorphism in MTNR1B and lifestyle intervention on occurrence of gestational diabetes. Diabetologia. 2016;59(8):1655–1658.
- van Poppel MNM, Corcoy R, Hill D, DALI Core Investigator Group, et al. Interaction between rs10830962 polymorphism in MTNR1B and lifestyle intervention on maternal and neonatal outcomes: secondary analyses of the DALI lifestyle randomized controlled trial. Am J Clin Nutr. 9 2022;115(2):388–396.
- Liang Z, Liu H, Wang L, et al. Maternal MTNR1B genotype, maternal gestational weight gain, and childhood obesity. Am J Clin Nutr. 1 2020;111(2):360–368.
- Nisa H, Qi KHT, Leng J, et al. The circadian rhythm-related MTNR1B genotype, gestational weight gain, and postpartum glycemic changes. J Clin Endocrinol Metab. 2018;103(6):2284–2290. .
- Firneisz G, Rosta K, Rigo J, et al. Identification and potential clinical utility of the MTNR1B rs10830963 core gene variant associated to endophenotypes in gestational diabetes mellitus. Front Genet. 2020;11:332.
- Zhang C, Bao W, Rong Y, et al. Genetic variants and the risk of gestational diabetes mellitus: a systematic review. Hum Reprod Update. - 2013;19(4):376–390.
- Wu L, Cui L, Tam WH, et al. Genetic variants associated with gestational diabetes mellitus: a meta-analysis and subgroup analysis. Sci Rep. 29 2016;6:30539.
- Vejrazkova D, Lukasova P, Vankova M, et al. MTNR1B genetic variability is associated with gestational diabetes in czech women. Int J Endocrinol. 2014;2014:508923.
- Alharbi KK, Al-Sulaiman AM, Shedaid KMB, et al. MTNR1B genetic polymorphisms as risk factors for gestational diabetes mellitus: a case-control study in a single tertiary care center. Ann Saudi Med. 2019;39(5):309–318.
- Zhang Y, Sun CM, Hu XQ, et al. Relationship between melatonin receptor 1B and insulin receptor substrate 1 polymorphisms with gestational diabetes mellitus: a systematic review and meta-analysis. Sci Rep. 2014;4:6113.
- Rosta K, Al-Aissa Z, Hadarits O, et al. Association study with 77 SNPs confirms the robust role for the rs10830963/G of MTNR1B variant and identifies two el associations in gestational diabetes mellitus development. PLoS One. 2017;12(1):e0169781.
- Nakamura Y, Tamura H, Kashida S, et al. Changes of serum melatonin level and its relationship to feto-placental unit during pregnancy. J Pineal Res. 2001;30(1):29–33.
- Li C, Zhou Y, Qiao B, et al. Association between a melatonin receptor 1B genetic polymorphism and its protein expression in gestational diabetes mellitus. Reprod Sci. 2019;26(10):1382–1388.
- Holzapfel C, Siegrist M, Rank M, et al. Association of a MTNR1B gene variant with fasting glucose and HOMA-B in children and adolescents with high BMI-SDS. Eur J Endocrinol. 2011;164(2):205–212.
- de Luis Roman DA, Primo D, Aller R, et al. Association of the rs10830963 polymorphism in MTNR1B with fasting glucose, serum adipokine levels and components of metabolic syndrome in adult obese subjects. rs10830963 en MTNR1B con los niveles de glucosa en ayunas, de adipocitoquinas y componentes del sindrome metabolico en sujetos obesos adultos. Nutr Hosp. 2019;36(1):60–65. Asociacion del polimorfi smo
- Zheng C, Dalla Man C, Cobelli C, et al. A common variant in the MTNR1b gene is associated with increased risk of impaired fasting glucose (IFG) in youth with obesity. Obesity. 2015;23(5):1022–1029.
- Reinehr T, Scherag A, Wang HJ, et al. Relationship between MTNR1B (melatonin receptor 1B gene) polymorphism rs10830963 and glucose levels in overweight children and adolescents. Pediatr Diabetes. 2011;12(4 Pt 2):435–441.
- Firneisz G, Rosta K, Al-Aissa Z, et al. The MTNR1B rs10830963 variant in interaction with Pre-Pregnancy BMI is a pharmacogenetic ker for the initiation of antenatal insulin therapy in gestational diabetes mellitus. Int J Mol Sci. 2018;19(12):3734.
- Jensen AC, Barker A, Kui M, et al. Associations of common genetic variants with age-related changes in fasting and postload glucose: evidence from 18 years of follow-up of the whitehall II cohort. Diabetes. 2011;60(5):1617–1623.
- Walford GA, Green T, Neale B, et al. Common genetic variants differentially influence the transition from clinically defined states of fasting glucose metabolism. Diabetologia. 2012;55(2):331–339.
- Vejrazkova D, Vankova M, Vcelak J, et al. The rs10830963 polymorphism of the MTNR1B gene: association with abnormal glucose, insulin and C-peptide kinetics. Front Endocrinol (Lausanne). 2022;13:868364.
- Linder K, Wagner R, Hatziagelaki E, et al. Allele summation of diabetes risk genes predicts impaired glucose tolerance in female and obese individuals. PLoS One. 2012;7(6):e38224.
- Stancakova A, Kuulasmaa T, Paananen J, et al. Association of 18 confirmed susceptibility loci for type 2 diabetes with indices of insulin release, proinsulin conversion, and insulin sensitivity in 5,327 nondiabetic Finnish men. Diabetes. 2009;58(9):2129–2136.
- Winkler C, Raab J, Grallert H, et al. Lack of association of type 2 diabetes susceptibility genotypes and body weight on the development of islet autoimmunity and type 1 diabetes. PLoS One. 2012;7(4):e35410.
- Andersen MK, Sterner M, Forsen T, et al. Type 2 diabetes susceptibility gene variants predispose to adult-onset autoimmune diabetes. Diabetologia. 2014;57(9):1859–1868.
- Ahlqvist E, Storm P, Karajamaki A, et al. el subgroups of adult-onset diabetes and their association with outcomes: a data-driven cluster analysis of six variables. Lancet Diabetes Endocrinol. 2018;6(5):361–369.
- Neel JV, JV. Diabetes mellitus: a "thrifty" genotype rendered detrimental by "progress”?. Am J Hum Genet. 1962;14(4):353–362.
- Ayub Q, Moutsianas L, Chen Y, et al. Revisiting the thrifty gene hypothesis via 65 loci associated with susceptibility to type 2 diabetes. Am J Hum Genet. 6 2014;94(2):176–185.
- Li H, Durbin R. Inference of human population history from individual whole-genome sequences. Nature. 2011;475(7357):493–496.
- Grant SF, Thorleifsson G, Reynisdottir I, et al. Variant of transcription factor 7-like 2 (TCF7L2) gene confers risk of type 2 diabetes. Nat Genet. 2006;38(3):320–323.
- Helgason A, Palsson S, Thorleifsson G, et al. Refining the impact of TCF7L2 gene variants on type 2 diabetes and adaptive evolution. Nat Genet. 2007;39(2):218–225.
- Southam L, Soranzo N, Montgomery SB, et al. Is the thrifty genotype hypothesis supported by evidence based on confirmed type 2 diabetes- and obesity-susceptibility variants? Diabetologia. 2009;52(9):1846–1851.
- Klimentidis YC, Abrams M, Wang J, et al. Natural selection at genomic regions associated with obesity and type-2 diabetes: east asians and Sub-Saharan africans exhibit high levels of differentiation at type-2 diabetes regions. Hum Genet. 2011;129(4):407–418.
- Chen R, Corona E, Sikora M, et al. Type 2 diabetes risk alleles demonstrate extreme directional differentiation among human populations, compared to other diseases. PLoS Genet. 2012;8(4):e1002621.
- Gibbons A. 12th international congress of human genetics. Diabetes genes line out of Africa. Science. 2011;334(6056):583.
- Neel JV. The "thrifty genotype" in 1998. Nutr Rev. 1999;57(5 Pt 2):S2–S9.
- Reppert SM, Godson C, Mahle CD, et al. Molecular characterization of a second melatonin receptor expressed in human retina and brain: the Mel1b melatonin receptor. Proc Natl Acad Sci U S A. 995;92(19):8734–8738.
- Mars G, Nourizadeh-Lillabadi R, Weltzien FA. New insights into the evolutionary history of melatonin receptors in vertebrates, with particular focus on teleosts. Front Endocrinol. 2020;11:538196.
- Yoshiuchi I. Analysis of evolution and ethnic diversity at glucose-associated SNPs of circadian clock-related loci with cryptochrome 1, cryptochrome 2, and melatonin receptor 1b. Biochem Genet. 2021;59(5):1173–1184.
- De Oliveira JW. [An undetermined form of chagas’ disease: medico-occupational implications]. arq bras cardiol. Forma Indeterminada da Doenca de Chagas. Implicacaoes Medico-Trabalhistas. 1990;54(2):89–91.