
Abstract
Background
The association between pulmonary involvement and microscopic polyangiitis (MPA) has been increasingly recognized in recent years. Whether interstitial lung disease (ILD) and bronchiectasis (BE) are disease manifestations of MPA, preexisting comorbidities or important complications remains unclear. The purpose of this study was to determine the clinical characteristics and prognosis of MPA with pulmonary involvement to further guide clinical management.
Methods
The data for 97 patients with a definitive diagnosis of MPA were retrospectively reviewed. The MPA diagnosis was based on the 2012 revised Chapel Hill Consensus Conference (CHCC) criteria. The baseline clinical information and laboratory parameters were collected and analysed at each patient’s initial diagnosis.
Results
Forty-seven out of the 97 (48.5%) patients who were diagnosed with MPA presented with pulmonary involvement, including 37 patients with ILD, 12 patients with BE and two patients with diffuse alveolar haemorrhage (DAH). ILD and BE antedated MPA in 56.76% and 75.00% of the patients, respectively. Compared with that in the MPA-BE group, the serum LDH level (222.86 ± 68.19 vs. 171.58 ± 31.43, p = .016) in the MPA-ILD group was significantly higher. In the multivariate Cox analysis, elevated serum creatinine (HR 4.08, confidence interval (CI) 1.38–12.05, p = .011) was an independent risk factor for shorter survival in MPA patients with pulmonary involvement, and treatment with glucocorticoid pulse cyclophosphamide therapy (HR 0.095, 95% CI 0.019–0.47, p = .004) was independently associated with prolonged survival. Among the patients in the MPA-ILD group, acute exacerbations of ILD (HR 4.55 CI 1.16–17.86, p = .029) and elevated serum creatinine (HR 4.95, CI 1.39–17.54, p = .014) were independently associated with a poor prognosis, and treatment with glucocorticoids (HR 0.057, 95% CI 0.012–0.28, p < .001) was independently associated with significant prolongation of survival.
Conclusions
Patients with MPA have a high prevalence of pulmonary involvement, and ILD is the most common subtype of MPA. ILD and BE can be considered preexisting comorbidities of MPA. Elevated serum creatinine was associated with shorter survival. However, remission induction regimens with glucocorticoids and/or immunosuppressants may improve this outcome.
Introduction
Antineutrophil cytoplasmic antibody-associated vasculitis (AAV) is a group of systemic vasculitides affecting small blood vessels and is characterized by the presence of circulating antineutrophil cytoplasmic antibodies (ANCAs). Phenotypes of the clinical disease include microscopic polyangiitis (MPA), granulomatosis with polyangiitis (GPA) and eosinophilic granulomatosis with polyangiitis (EGPA) [Citation1,Citation2]. MPA is a systemic necrotizing vasculitis that mainly involves small vessels and is mostly associated with myeloperoxidase (MPO)-ANCA [Citation1,Citation3]. The most commonly involved organs are the kidneys and lungs, and the proportion of patients with pulmonary involvement is as high as 40–69% [Citation4–6]. Recent studies have shown that interstitial lung disease (ILD) is the most common subtype of MPA with pulmonary involvement, detected in 25–55% of patients with MPA and associated with a poor prognosis [Citation4,Citation5,Citation7–10]. On chest CT, MPA-ILD mainly has either a usual interstitial pneumonia (UIP) or UIP-like radiologic pattern, followed by nonspecific interstitial pneumonia (NSIP) [Citation8,Citation11–13], and the UIP pattern is related to shortened survival compared with the NSIP pattern [Citation8]. Bronchiectasis (BE) is also a major subtype of MPA with pulmonary involvement, representing 16–42% of MPA cases [Citation5,Citation14].
The association between pulmonary involvement and MPA or AAV has been increasingly recognized in recent years [Citation4,Citation9,Citation15–17]. However, the association between pulmonary involvement, especially in ILD and BE, and MPA has only been reported in case reports and small case series. Furthermore, it remains unknown whether ILD and BE are disease manifestations of MPA, preexisting comorbidities or important complications. In clinical practice, ILD or BE patients with ANCA positivity and extrapulmonary involvement meet the AAV diagnostic criteria; however, whether glucocorticoids and/or immunosuppressants are needed remains unclear, especially for BE patients. Therefore, we retrospectively analysed the baseline clinical data of patients diagnosed with MPA with pulmonary involvement in the past eight years. The aim of our study was to assess the clinical characteristics and prognosis of MPA with pulmonary involvement to further guide clinical management.
Methods
Study design
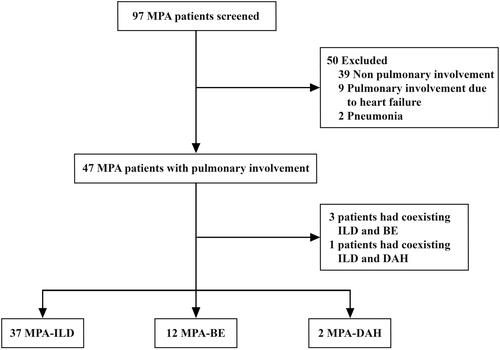
We retrospectively reviewed the medical records of patients with a definitive diagnosis of MPA at the First Affiliated Hospital of Ningbo University between March 2014 and October 2022. Ninety-seven patients diagnosed with MPA who had available chest CT or HRCT data at the time of diagnosis were screened, and 47 (48.45%) patients with MPA had pulmonary involvement. MPA diagnosis was based on the 2012 revised International Chapel Hill Consensus Conference (CHCC) Nomenclature of Vasculitides [Citation1]. Patients with pneumonia, heart failure leading to pulmonary imaging changes, and simple pleural effusion were excluded, as were patients who were ANCA-negative without histological proof of necrotizing small-vessel vasculitis and ANCA-positive without any clinical sign of AAV. A flowchart of the patient screening and classification for our study is presented in .
Figure 1. Flowchart of the study. MPA: microscopic polyangiitis; ILD: interstitial lung disease; BE: bronchiectasis; DAH: diffuse alveolar haemorrhage.
Data collection
The clinical data, including detailed patient histories, clinical manifestations, laboratory results, pulmonary function tests (PFTs) and chest HRCT, were obtained from the medical records at the time of MPA diagnosis. The data included basic patient information (age, sex, medical history, diagnosis, smoking status, occupational history) and laboratory results (blood cell count, arterial blood gas analysis, lactate dehydrogenase, erythrocyte sedimentation rate (ESR), alanine aminotransferase, aspartate aminotransferase, creatine kinase, C-reactive protein (CRP), rheumatoid factor, serologic autoantibodies, ANCA, etc.). ANCAs were assessed by enzyme-linked immunosorbent assay (ELISA) or indirect immunofluorescence assay determination of their specific antigen (anti-MPO or PR-3), according to the practices of each immunology laboratory. The results of the PFT data (FVC% predicted, FEV1% predicted, FEV1/FVC, FEV1/FVC ratio and DLCO% predicted) and the echocardiographic estimate of the systolic pulmonary artery pressure (SPAP) were also recorded.
Chest CT and high-resolution CT (HRCT) scans were evaluated by two pulmonologists, and the dominant pulmonary involvement was classified as either ILD, BE or diffuse alveolar haemorrhage (DAH). The criteria of the 2013 ATS/ESR classification of idiopathic interstitial pneumonia (IIP) were used to classify the ILD patterns [Citation18], and the criteria of the 2018 ATS/ERS/JRS/ALAT guidelines were used to classify UIP patterns [Citation19]. A combined pulmonary fibrosis and emphysema (CPFE) diagnosis was based on the 2022 ATS/ERS/JRS/ALAT guidelines [Citation20]. Pulmonary fibrosis (PF) with traction BE was not included as a BE type. A UIP-like pattern was defined as findings coinciding with a UIP or probable UIP pattern according to the modified diagnostic guidelines for the HRCT classification of IPF proposed by the Fleischner Society [Citation21]. Acute exacerbation of ILD (AE-ILD) was defined as an acute worsening or development of dyspnoea with a duration of <1 month, chest CT with new bilateral ground-glass opacity and/or consolidation superimposed on PF, and deterioration not fully explained by heart failure or fluid overload [Citation22,Citation23]. The remission induction treatment regimens were recorded. The last follow-up was in October 2022, and the outcomes were defined as death from all causes. The study was approved by the Ethics Committee of the First Affiliated Hospital of Ningbo University (IRB no.: KS202211015).
Statistical analysis
Statistical analysis was performed using IBM SPSS 26 (Armonk, NY) and GraphPad Prism 9 software (La Jolla, CA). Continuous variables are presented as the mean ± standard deviation (SD), and Student’s t-test was used for comparisons. Continuous, nonnormally distributed data are presented as the median with interquartile range (IQR), and the Mann–Whitney U-test was used for comparisons between two groups. For the two-group comparisons of binary data, the chi-square test or Fisher’s exact test was used. Kaplan–Meier’s survival analysis was performed to estimate the survival rates and plot survival curves, and the log-rank test was used to detect intergroup differences. A multivariate Cox regression model was used to identify variables independently associated with death and variables associated with a given outcome at p < .1 in the Kaplan–Meier analysis. A p value < .05 was considered to indicate statistical significance.
Results
Demographic and clinical characteristics
Among the 97 patients diagnosed with MPA, 47 (48.5%) presented with pulmonary involvement, including 37 patients with ILD, 12 patients with BE and two patients with DAH. Three patients had coexisting ILD and BE, and one patient had coexisting ILD and DAH. None of the patients with BE had coexisting DAH ().
The participant demographics and main characteristics of MPA with pulmonary involvement are summarized in . The mean age at diagnosis was 70.34 ± 12.61 years (IQR, 62.50–82.00), and the ratio of males to females was 1.24:1. A total of 17 (36.17%) patients had a history of smoking. There were no significant differences in age, sex or smoking status between the MPA-ILD group and the MPA-BE group. The main clinical manifestations in our study included cough (61.70%), dyspnoea (46.81%), fever (46.81%) and arthralgia (29.79%). There was a higher proportion of patients with haemoptysis in the MPA-BE group than in the MPA-ILD group (41.67% vs. 10.81%, p = .049). Twenty-eight patients had renal involvement, 82.14% (23/28) of whom showed elevated serum creatinine, whereas 17.86% (5/28) of the patients only had haematuria and proteinuria. Most patients had systemic involvement at the time of diagnosis. However, there were no significant differences in the systemic involvement manifestations between the MPA-ILD group and the MPA-BE group. ILD and BE antedated MPA in 56.76% (21/37) and 75.00% (9/12) of the patients, respectively. However, ILD and BE were found simultaneously with MPA diagnosis in 40.54% (15/37) and 25.00% (3/12) of the patients, respectively. These patients were diagnosed with MPA at an average of 40.6 ± 24.5 months and 60.4 ± 33.4 months after the confirmation of ILD and BE, respectively. However, only one patient experienced ILD after MPA. In terms of treatment at remission induction, glucocorticoids were the most commonly used treatment and were given to 31 (65.96%) patients, and 20 (42.55%) patients received glucocorticoid pulse cyclophosphamide treatment. Six (16.22%) and four (33.33%) patients received treatment for ILD and BE alone at the time of MPA diagnosis, respectively. Twenty-five (67.57%) MPA-ILD patients and eight MPA-BE patients were treated with glucocorticoids or glucocorticoids plus cyclophosphamide.
Table 1. Comparison between the MPA-ILD group and MPA-BE group in terms of demographic and clinical characteristics.
Laboratory findings, PFTs and HRCT patterns
The laboratory tests, PFTs and HRCT patterns of MPA with pulmonary involvement are summarized in . Compared with those in the MPA-BE group, the serum LDH levels (222.86 ± 68.19 vs. 171.58 ± 31.43, p = .016) of the patients in the MPA-ILD group were significantly higher. However, the oxygenation index (331.53 ± 93.10 vs. 403.37 ± 74.06, p = .044) was significantly lower in the MPA-ILD group. Of the ANCA antibodies, the positivity rates of P-ANCA and MPO-ANCA were 89.36% and 89.36% in the MPA with pulmonary involvement patients, respectively. Only one patient was C-ANCA positive, and one patient was PR3-ANCA positive. The positivity rates of MPO-ANCA in the MPA-ILD group and in the MPA-BE group were 86.49% and 100%, respectively.
Table 2. Comparison between the MPA-ILD group and MPA-BE group in terms of laboratory tests, PFTs and HRCT patterns.
Twenty-two of the 47 patients underwent PFTs at the initial presentation, and the most common manifestation was restrictive ventilation dysfunction with diffusion dysfunction. A restrictive pattern was mainly observed in MPA-ILD patients, and an obstructive pattern was mainly observed in MPA-BE patients. The HRCT pattern was mainly UIP or UIP-like (54.05%) in the MPA-ILD group, followed by NSIP (29.73%) (Supplement 1).
Survival
The univariate and multivariate analyses of the risk of all-cause mortality in patients with MPA with pulmonary involvement are summarized in . The median follow-up time of the 47 patients with MPA with pulmonary involvement was 25 (IQR 10.0–44.0) months, and all-cause mortality was observed in 18 (38.3%) patients during follow-up. Ten patients died of pulmonary-related causes, five patients died of non-pulmonary-related causes, and the cause of death in three patients was unknown. The 1-, 3- and 5-year overall survival rates were 86.89%, 59.99% and 54.54% for patients with MPA with pulmonary involvement, respectively. The 1-, 3- and 5-year overall survival rates were 88.87%, 61.76% and 55.59% for the MPA-ILD patients and 83.33%, 55.56% and 55.56% for the MPA-BE patients, respectively. The Kaplan–Meier survival analysis showed no significant differences between the two groups (hazard ratio (HR) 0.77, 95% confidence interval (CI) 0.23–2.54, p = .617). In the univariate analysis, survival was significantly shorter for the patients who had elevated serum creatinine (), elevated IgG and PaCO2 > 45 mmHg at the time of MPA with a pulmonary involvement diagnosis. However, treatment with glucocorticoid or glucocorticoid pulse cyclophosphamide therapy () was found to prolong survival among the patients.
Figure 2. The Kaplan–Meier survival curves of the MPA with pulmonary involvement patients with different variables. MPA: microscopic polyangiitis; Cr: creatinine; GCs: glucocorticoids; CYC: cyclophosphamide.
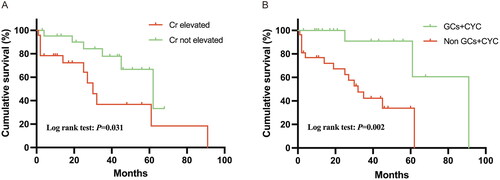
Table 3. Univariate and multivariate analyses of the risk of all-cause mortality in MPA patients with pulmonary involvement.
In the multivariable Cox analysis, elevated serum creatinine (HR 4.08, CI 1.38–12.05, p = .011) remained a significant factor that was independently associated with shorter survival, and treatment with glucocorticoid pulse cyclophosphamide (HR 0.095, 95% CI 0.019–0.47, p = .004) was independently associated with prolonged survival.
The univariate and multivariate analyses of the MPA-ILD patients are summarized according to subgroups in . In the univariate analysis, the factors significantly associated with shorter survival included acute exacerbations of ILD, elevated IgG, PaCO2 > 45 mmHg and oxygenation index < 300 mmHg. However, it was still found that treatment with glucocorticoid or glucocorticoid pulse cyclophosphamide therapy prolonged survival among MPA-ILD patients. According to the multivariate analysis, acute exacerbations of ILD (HR 4.55 CI 1.16–17.86, p = .029) and elevated serum creatinine (HR 4.95, CI 1.39–17.54, p = .014) were independently associated with a poor prognosis. However, treatment with glucocorticoids (HR 0.057, 95% CI 0.012–0.28, p < .001) was independently associated with significant prolongation of survival.
Table 4. Univariate and multivariate analyses of the risk of all-cause mortality in MPA-ILD patients.
Discussion
In the present study, 47 out of 97 (48.5%) patients who were diagnosed with MPA presented with pulmonary involvement, including 37 patients with ILD, 12 patients with BE and two patients with DAH. ILD and BE antedated MPA in 56.76% and 75.00% of the patients, respectively. Compared with that in the MPA-BE group, the serum LDH levels in the MPA-ILD group were significantly higher. Elevated serum creatinine was an independent risk factor associated with shorter survival in MPA patients with pulmonary involvement, and treatment with glucocorticoid pulse cyclophosphamide was independently associated with prolonged survival. Among the patients in the MPA-ILD group, acute exacerbations of ILD and elevated serum creatinine were independently associated with a poor prognosis, and treatment with glucocorticoids was independently associated with significant prolongation of survival.
As reported previously [Citation4,Citation5,Citation16], the prevalence of pulmonary involvement in MPA is high, and ILD is the most common pulmonary manifestation. However, whether there is a pathogenic relationship between MPA and pulmonary involvement remains unclear. It has been clarified that DAH is an important manifestation of MPA, but whether ILD and BE are disease manifestations of MPA or are independent important complications remains unknown. Our present study showed that ILD and BE usually occur before MPA; patients were diagnosed with MPA at an average of 3.4 years and 5.0 years after the confirmation of ILD and BE, respectively, and these findings were consistent with those of previous studies [Citation5,Citation7,Citation8,Citation10,Citation15,Citation24,Citation25]. An increased percentage of neutrophils are usually observed in the bronchoalveolar lavage (BAL) fluid of PF and BE patients [Citation26]. Once neutrophils are activated, which could be due to pulmonary tissue injury and fibrosis or chronic airway inflammation, MPO may become exposed at the membrane [Citation8,Citation27]. In addition, previous studies evaluating surgical lung biopsy findings in patients positive for MPA-ILD and ANCA with ILD mainly showed a UIP histologic pattern, with accompanying areas of NSIP, organizing pneumonia (OP), bronchiolitis, lymphoid hyperplasia and lymphoid follicles. However, none of the patients had findings of vasculitis [Citation28–30]. This is essentially different from DAH patients with pathological manifestations of alveolar capillaritis. It is believed that ILD and BE could induce MPO-ANCA production as a result of neutrophil destruction during the chronic inflammation process, which may explain the appearance of ANCA after the onset of ILD and BE. In addition, MPA with ILD and BE have similar clinical and laboratory manifestations of vasculitis, and the rate of extrapulmonary system involvement was not significantly different in our study, which also supports this hypothesis. Thus, we believe that ILD and BE were not manifestations of MPA; instead, they should be classified as preexisting comorbidities or important complications.
In our present study, the frequency of smoking was obviously higher in MPA-ILD patients than in MPA-BE patients, although there was no statistically significant difference between the two groups. Cigarette smoking is considered a principal pathogenetic factor for the development of certain diffuse interstitial and bronchiolar lung diseases, such as respiratory bronchiolitis-interstitial lung disease (RB-ILD), desquamative interstitial pneumonia (DIP) and adult pulmonary Langerhans cell histiocytosis (PLCH) [Citation31]. In addition, previous studies have shown that cigarette smoking has an independent detrimental effect on the risk of IPF and works synergistically [Citation31,Citation32]. In our opinion, multiple factors, such as the environment, smoking, infection and genetic susceptibility, lead to the occurrence and development of ILD, with a small number of ILD patients being ANCA positive and diagnosed with AAV-ILD. In addition, a recent study showed that patients with AAV were more likely to be current or former smokers and that this association was largely driven by MPO-ANCA-positive AAV [Citation33]. This finding indicated that cigarette smoking may also induce ANCA production, especially MPO-ANCA.
Studies concerning MPA with pulmonary involvement, MPA-ILD and MPA-BE are limited. Our study showed that patients diagnosed with MPA with pulmonary involvement who had haemoptysis symptoms accounted for a minority of the overall patient population. These symptoms mainly occurred in patients with BE, suggesting that haemoptysis may be related to the presence of BE rather than vasculitis. In the present study, LDH elevation was more prevalent in patients with MPA-ILD than in patients with MPA-BE, which may be related to the presence of elevated LDH in idiopathic pulmonary fibrosis (IPF) or PF patients [Citation12,Citation13,Citation30]. In previous studies, investigators have reported that the kidneys are involved in more than 90% of MPA cases [Citation34,Citation35]. However, studies have shown that 57–86.9% of MPA-ILD patients have renal involvement [Citation9,Citation36]. In the present study, the rates of renal involvement were 59.6%, 56.8% and 66.7% in the patients with MPA with pulmonary involvement, MPA-ILD and MPA-BE, respectively. The multivariate Cox regression analysis showed that elevated serum creatinine was an independent risk factor associated with shorter survival for the patients in both the MPA with pulmonary involvement group and the MPA-ILD group. A recent study also showed that poor renal function was a risk factor for long-term death [Citation17]. These findings indicate that more attention should be given to patients with poor renal function, and glucocorticoids and/or cyclophosphamide or rituximab should be used in a timely manner if possible.
Glucocorticoids plus immunosuppressants (cyclophosphamide, methotrexate and azathioprine) or glucocorticoids plus rituximab were administered to patients with MPA at remission induction, in accordance with the International Consensus Statement for treatment of AAV [Citation37,Citation38]. According to our survival analyses, treatment with glucocorticoids or glucocorticoids plus cyclophosphamide was associated with significant prolongation of survival in MPA patients with pulmonary involvement. However, there are conflicting views regarding the treatment of MPA with pulmonary involvement. Comarmond et al. [Citation36] showed that glucocorticoids in combination with cyclophosphamide or rituximab could improve the outcome at induction compared with treatment with only glucocorticoids. Maillet et al. [Citation8] came to the opposite conclusion that immunosuppressants in the induction regimen were associated with shorter survival. These studies suggest that individualized treatment dependent on disease severity may be necessary for patients with MPA-ILD or MPA-BE. Because ILD and BE are preexisting comorbidities of MPA, early antifibrotic treatment for fibrotic ILD patients and appropriate phlegm treatment for BE patients are necessary. Standardized treatment regimens for patients with MPA-ILD or MPA-BE should be retrospectively clarified by studies with larger samples and randomized controlled studies.
Our retrospective cohort study had several limitations. First, this was a single-centre study with a small sample size; therefore, it is possible that our findings would not be consistent with findings in larger patient populations. Second, this study enrolled only patients who underwent chest CT retrospectively. There may be a selection bias, which would cause MPA with pulmonary involvement to be overestimated. Third, due to the historically inadequate understanding of MPA with pulmonary involvement, 34% of the patients received no treatment, which may have led to bias in the univariate and multivariate survival analysis results. Finally, PFTs and arterial blood gas analysis were performed for some patients, and related variables were not analysed by univariate and/or multivariate survival analysis. The results of the multivariate analysis of risk factors for prognosis should be carefully interpreted. Thus, larger-scale prospective studies and clinical randomized controlled trials are needed for further evaluation.
Conclusions
In conclusion, patients with MPA have a high prevalence of pulmonary involvement, and ILD is the most common subtype of MPA. ILD and BE can be considered preexisting comorbidities or important complications of MPA. Elevated serum creatinine was associated with shorter survival for the patients in both the MPA with pulmonary involvement group and the MPA-ILD group. However, remission induction regimens with glucocorticoids and/or immunosuppressants may improve this outcome. Based on the hypothesis that ILD and BE are preexisting comorbidities of MPA, early antifibrotic treatment for patients with MPA and fibrotic ILD and appropriate phlegm treatment for patients with MPA-BE are necessary.
Ethical approval
All study was conducted in accordance with the Declaration of Helsinki. The cohort study was approved by the institutional review board of the First Affiliated Hospital of Ningbo University, Ningbo, China (IRB no.: KS202211015), and all methods were carried out in accordance with relevant guidelines and regulations.
Consent form
Written informed consent from study subjects was waived because of the retrospective design.
Author contributions
Tingting Wu and Yun Zhang were involved in the conception and design of the study. Yun Zhang, Qunli Ding, Yanan Ying and Zekai Cen were responsible for the data collection. Tingting Wu wrote the first draft. Yun Zhang, Haijun Zhou and Chengna Lv were in charge of the analysis. Tingting Wu accessed and verified the data. All authors were involved in the interpretation, critically reviewed the first draft. All authors read and approved the final manuscript.
Supplemental Material
Download JPEG Image (17.4 MB){kind=link}
Supplemental Material
Download MS Word (11.7 KB)Acknowledgements
The authors thank all patients and their families involved in this research. The authors also thank all members of Department of Respiratory, the First Affiliated Hospital of Ningbo University.
Data availability statement
The data supporting our findings are available from the corresponding author on reasonable request.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised international chapel hill consensus conference nomenclature of vasculitides. Arthritis Rheum. 2013;65(1):1–10.
- Geetha D, Jefferson JA. ANCA-associated vasculitis: core curriculum 2020. Am J Kidney Dis. 2020;75(1):124–137.
- Cornec D, Cornec-Le Gall E, Fervenza FC, et al. ANCA-associated vasculitis – clinical utility of using ANCA specificity to classify patients. Nat Rev Rheumatol. 2016;12(10):570–579.
- Makhzoum JP, Grayson PC, Ponte C, et al. Pulmonary involvement in primary systemic vasculitides. Rheumatology. 2021;61(1):319–330.
- Tashiro H, Takahashi K, Tanaka M, et al. Characteristics and prognosis of microscopic polyangiitis with bronchiectasis. J Thorac Dis. 2017;9(2):303–309.
- Mohammad AJ, Mortensen KH, Babar J, et al. Pulmonary involvement in antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitis: the influence of ANCA subtype. J Rheumatol. 2017;44(10):1458–1467.
- Alba MA, Flores-Suarez LF, Henderson AG, et al. Interstitial lung disease in ANCA vasculitis. Autoimmun Rev. 2017;16(7):722–729.
- Maillet T, Goletto T, Beltramo G, et al. Usual interstitial pneumonia in ANCA-associated vasculitis: a poor prognostic factor. J Autoimmun. 2020;106:102338.
- Hozumi H, Kono M, Hasegawa H, et al. Clinical significance of interstitial lung disease and its acute exacerbation in microscopic polyangiitis. Chest. 2021;159(6):2334–2345.
- Fernandez Casares M, Gonzalez A, Fielli M, et al. Microscopic polyangiitis associated with pulmonary fibrosis. Clin Rheumatol. 2015;34(7):1273–1277.
- Wu TT, Cen ZK, Zhou HJ, et al. Clinical features and survival analysis of microscopic polyangiitis-associated interstitial lung disease: a retrospective study of 28 patients. Zhonghua Jie He He Hu Xi Za Zhi. 2022;45(10):1022–1030.
- Komatsu M, Yamamoto H, Kitaguchi Y, et al. Clinical characteristics of non-idiopathic pulmonary fibrosis, progressive fibrosing interstitial lung diseases: a single-center retrospective study. Medicine. 2021;100(13):e25322.
- Wu T, Zhang Y, Cen Z, et al. Clinical significance of acute exacerbation in interstitial lung disease with antineutrophil cytoplasmic antibody: an indicator of poor prognosis. Ther Adv Respir Dis. 2022;16:17534666221140974.
- Suzuki A, Sakamoto S, Kurosaki A, et al. Chest high-resolution CT findings of microscopic polyangiitis: a Japanese first nationwide prospective cohort study. AJR Am J Roentgenol. 2019;213(1):104–114.
- Neel A, Espitia-Thibault A, Arrigoni PP, et al. Bronchiectasis is highly prevalent in anti-MPO ANCA-associated vasculitis and is associated with a distinct disease presentation. Semin Arthritis Rheum. 2018;48(1):70–76.
- Biedroń G, Włudarczyk A, Wawrzycka-Adamczyk K, et al. Respiratory involvement in antineutrophil cytoplasmic antibody-associated vasculitides: a retrospective study based on POLVAS registry. Clin Exp Rheumatol. 2022;40(4):720–726.
- Gu Y, Zhang T, Peng M, et al. Characteristics and prognosis of microscopic polyangiitis patients with diffuse alveolar hemorrhage and interstitial lung disease. Chin Med Sci J. 2022;37(4):293–302.
- Travis W D, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society Statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med. 2013;188(6):733–748.
- Raghu G, Remy-Jardin M, Myers JL, et al. Diagnosis of idiopathic pulmonary fibrosis. An official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med. 2018;198(5):e44–e68.
- Cottin V, Selman M, Inoue Y, et al. Syndrome of combined pulmonary fibrosis and emphysema: an official ATS/ERS/JRS/ALAT research statement. Am J Respir Crit Care Med. 2022;206(4):e7–e41.
- Lynch DA, Sverzellati N, Travis WD, et al. Diagnostic criteria for idiopathic pulmonary fibrosis: a Fleischner Society white paper. Lancet Respir Med. 2018;6(2):138–153.
- Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am J Respir Crit Care Med. 2016;194(3):265–275.
- Kolb M, Bondue B, Pesci A, et al. Acute exacerbations of progressive-fibrosing interstitial lung diseases. Eur Respir Rev. 2018;27(150):180071.
- Takahashi K, Hayashi S, Ushiyama O, et al. Development of microscopic polyangiitis in patients with chronic airway disease. Lung. 2005;183(4):273–281.
- Lhote R, Chilles M, Groh M, et al. Spectrum and prognosis of antineutrophil cytoplasmic antibody-associated vasculitis-related bronchiectasis: data from 61 patients. J Rheumatol. 2020;47(10):1522–1531.
- Meyer KC, Raghu G, Baughman RP, et al. An official American Thoracic Society Clinical Practice Guideline: the clinical utility of bronchoalveolar lavage cellular analysis in interstitial lung disease. Am J Respir Crit Care Med. 2012;185(9):1004–1014.
- Borie R, Crestani B. Antineutrophil cytoplasmic antibody-associated lung fibrosis. Semin Respir Crit Care Med. 2018;39(4):465–470.
- Tanaka T, Otani K, Egashira R, et al. Interstitial pneumonia associated with MPO-ANCA: clinicopathological features of nine patients. Respir Med. 2012;106(12):1765–1770.
- Baqir M, Yi EE, Colby TV, et al. Radiologic and pathologic characteristics of myeloperoxidase–antineutrophil cytoplasmic antibody-associated interstitial lung disease: a retrospective analysis. Sarcoidosis Vasc Diffuse Lung Dis. 2019;36(3):195–201.
- Hosoda C, Baba T, Hagiwara E, et al. Clinical features of usual interstitial pneumonia with anti-neutrophil cytoplasmic antibody in comparison with idiopathic pulmonary fibrosis. Respirology. 2016;21(5):920–926.
- Margaritopoulos GA, Vasarmidi E, Jacob J, et al. Smoking and interstitial lung diseases. Eur Respir Rev. 2015;24(137):428–435.
- Bellou V, Belbasis L, Evangelou E. Tobacco smoking and risk for pulmonary fibrosis: a prospective cohort study from the UK biobank. Chest. 2021;160(3):983–993.
- Mcdermott G, Fu X, Stone JH, et al. Association of cigarette smoking with antineutrophil cytoplasmic antibody-associated vasculitis. JAMA Intern Med. 2020;180(6):870–876.
- Villiger PM, Guillevin L. Microscopic polyangiitis: clinical presentation. Autoimmun Rev. 2010;9(12):812–819.
- Karras A. Microscopic polyangiitis: new insights into pathogenesis, clinical features and therapy. Semin Respir Crit Care Med. 2018;39(4):459–464.
- Comarmond C, Crestani B, Tazi A, et al. Pulmonary fibrosis in antineutrophil cytoplasmic antibodies (ANCA)-associated vasculitis: a series of 49 patients and review of the literature. Medicine. 2014;93(24):340–349.
- Yates M, Watts RA, Bajema IM, et al. EULAR/ERA-EDTA recommendations for the management of ANCA-associated vasculitis. Ann Rheum Dis. 2016;75(9):1583–1594.
- Chung SA, Langford CA, Maz M, et al. 2021 American College of Rheumatology/Vasculitis Foundation Guideline for the management of antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheumatol. 2021;73(8):1366–1383.