
Abstract
Aim: Non-alcoholic fatty liver disease is characterized by diffuse hepatic steatosis and has quickly risen to become the most prevalent chronic liver disease. Its incidence is increasing yearly, but the pathogenesis is still not fully understood. Porphyromonas gingivalis (P. gingivalis) is a major pathogen widely prevalent in periodontitis patients. Its infection has been reported to be a risk factor for developing insulin resistance, non-alcoholic fatty liver disease (NAFLD), non-alcoholic steatohepatitis (NASH), and metabolic syndrome. The aim of this review is to evaluate the association between P. gingivalis infection and NAFLD, identify the possible etiopathogenetic mechanisms, and raise public awareness of oral health to prevent and improve NAFLD.Methods: After searching in PubMed and Web of Science databases using ‘Porphyromonas gingivalis’, ‘non-alcoholic fatty liver disease’, and ‘hepatic steatosis’ as keywords, studies related were compiled and examined.Results: P. gingivalis infection is a direct risk factor for NAFLD based on clinical and basic research. Moreover, it induces systematic changes and systemic abnormalities by disrupting metabolic, inflammatory, and immunologic homeostasis.Conclusion: P. gingivalis-odontogenic infection promotes the occurrence and development of NAFLD. Further concerns are needed to emphasize oral health and maintain good oral hygiene for the prevention and treatment of NAFLD.
KEY MESSAGES
Porphyromonas gingivalis exacerbates the development of non-alcoholic fatty liver disease.
Porphyromonas gingivalis aggravates a homeostasis imbalance in hepatic lipid metabolism and an insulin resistance phenotype.
Introduction
In recent years, with the rapid development of the economy and the substantial improvement of people’s quality of life, the prevalence of NAFLD continues to rise and presents a trend of younger onset. NAFLD has now replaced hepatitis B as the most common chronic liver disease, and is also the leading cause of end-stage liver disease worldwide [Citation1–4]. The prevalence of NAFLD is about 25% in the general population in China, 57.5–74.0% in the obese population, and 70–75% in the diabetic population [Citation1,Citation5–8]. It is currently believed that NAFLD is closely related to obesity, type 2 diabetes, hyperlipidemia, and metabolic syndrome. The incidence rates of diabetes, arteriosclerotic cardiovascular disease, metabolic syndrome, and tumors increase significantly within 5-10 years after NAFLD diagnosis [Citation9,Citation10]. The pathogenesis of NAFLD is still incompletely understood. While there are many probably candidate contributors, one proposed underlying mechanism is the infection of Porphyromonas gingivalis (P. gingivalis).
P. gingivalis, a Gram-negative oral anaerobic bacterium, is the most important pathogenic bacterium that causes periodontal disease. The infection of P. gingivalis has been reported to be associated with an increased risk of a variety of systemic diseases including, but not limited to, cardiovascular diseases, rheumatoid arthritis, type 2 diabetes, and Alzheimer’s disease [Citation11–14]. Emerging evidence shows that P. gingivalis infection exacerbates the progression of NAFLD. P. gingivalis cell surface components such as fimbriae, outer membrane protein A, lipopolysaccharide, etc. are the main pathogenic factors [Citation15–18]. They are thought to contribute to the development of NAFLD in many ways that range from promoting endotoxemia, inflammatory response, oxidative stress, metabolic remodeling, macrophage polarization and gut microbiome [Citation11,Citation13,Citation19–26]. Therefore, studying the specific mechanism and shedding light on the direct role of P. gingivalis in the progress of NAFLD will provide a reference and basis for prevention and treatment of NAFLD.
Definition and multiple parallel hits hypothesis of nonalcoholic fatty liver disease
Non-alcoholic fatty liver disease (NAFLD), also known as metabolic dysfunction-associated fatty liver disease (MAFLD), refers to the exclusion of alcohol and other definite liver damage factors (such as drugs), is a clinicopathological syndrome characterized by diffuse lipid droplet deposition in hepatocytes [Citation1,Citation5,Citation8,Citation27]. According to the progression of the disease, NAFLD comprises a wide spectrum of pathological conditions ranging from simple steatosis to more severe forms of non-alcoholic steatohepatitis (NASH) and its related liver cirrhosis and hepatocellular carcinoma [Citation27–29].
The pathogenesis of NAFLD is still unclear, and the explanation of its pathogenesis by the ‘two-hit’ hypothesis in previous studies has been widely recognized by the academic community. The first hit is mainly caused by the excessive deposition of lipids in the liver cells induced by insulin resistance; the second hit is on the basis of the first hit, lipid peroxidation and oxidative stress occur, inducing inflammatory response and necrosis in liver parenchyma cells and fibrosis, further accelerating the progression of the disease. However, with the gradual deepening of research, some scholars believe that the ‘multiple parallel hits’ hypothesis can better explain the pathogenesis of NAFLD. These hit factors include insulin resistance, lipid metabolism disorders, and oxidative stress, mitochondrial dysfunction, pro-inflammatory cytokines, immune response, intestinal flora disturbance, etc. The vicious circle formed by the mutual influence and interaction of the above factors promotes the progression of NAFLD [Citation27,Citation30–34]. Despite a comprehensive understanding of cellular biological changes that occur during the development and progression of NAFLD, the molecular mechanisms underlying these changes remain poorly understood. In recent years, studies have found that P. gingivalis has a certain influence on the occurrence and development of NAFLD [Citation12–14,Citation20,Citation22,Citation24,Citation26,Citation35,Citation36]. However, there is still a lack of sufficient experimental data and the accumulation of clinical practice.
Porphyromonas gingivalis and systemic diseases
P. gingivalis is a non-saccharide gram-negative anaerobic coccus that is widely present in the human oral cavity and is the key pathogen that is closely related to the occurrence and development of periodontitis [Citation18]. The action of P. gingivalis is mainly achieved through numerous factors of virulence and pathogenicity such as gingivain, fimbriae, hemolysin, hemagglutinin and lipopolysaccharides [Citation18,Citation37]. Gingivain is essential for the survival and pathogenicity of P. gingivalis, and it plays key roles in oral colonization, nutrient acquisition, and tissue destruction [Citation17,Citation19]. Gingivesin is a cysteine protease composed of lysine-gingivain, arginine-gingivain A, and arginine-gingivain B, which can be secreted on the surface of biofilms or released to the extracellular environment in the form of exosomes [Citation38,Citation39].
Studies in recent years have found that P.gingivalis is not only one of the main important pathogenic bacteria in the development of periodontitis, but also closely related to systemic diseases including rheumatoid arthritis (RA) [Citation40–45], cardiovascular disease [Citation46–51], diabetes [Citation36,Citation42,Citation52] and Alzheimer’s disease (AD) [Citation53–57]. The imbalance of oral microecology caused by the abnormal increase in the relative abundance of P. gingivalis can not only cause the destruction of local tissues in the oral cavity, but also damage distant tissues and organs far away from the primary lesions [Citation16,Citation47,Citation58–61]. P.gingivalis produces gingivain that can cause local tissue damage, disrupt the host’s antibacterial defense, and lead to excessive growth and dissemination of symbiotic bacteria in the oral and intestinal tract. This can lead to excessive production of toxic metabolites, promoting cardiovascular disease and autoimmune related metabolic disorders [Citation48,Citation49,Citation62,Citation63]. The gingivain produced by P.gingivalis in the brain is of great significance for the occurrence and development of AD. Gingivain promotes the migration of microglia to the infection site by activating PI3K/Akt and MEK/ERK pathways, thus accelerating the progression of AD [Citation64]. Peptide based arginine deiminase (PAD) produced by P.gingivalis is also an important pathogenic factor, which can break the immune tolerance of citrulline proteins and exacerbate the symptoms of individuals susceptible to rheumatoid arthritis (RA) [Citation41,Citation65]. Furthermore, studies have found that LPS on the surface of P.gingivalis is recognized and mediated by TLR. Through TLR-NF-kB signal axis, it promotes the secretion of inflammatory factors IL-1β, IL-6, TNF-α, IFN-γ, induces severe endothelial cell oxidative stress, and aggravates atherosclerosis [Citation66,Citation67].
Association between P. gingivalis infection and NAFLD
Periodontitis and NAFLD are both chronic inflammatory diseases with multiple risk factors, which have been confirmed to be closely related by many studies. The main pathogen of periodontitis has been confirmed to be P. gingivalis, and P. gingivalis infection is an important risk factor for the pathological progression of NAFLD. In 2012, Yoneda et al. in Japan compared 150 NAFLD patients and 60 healthy controls and found that the infection rate of P. gingivalis in NAFLD patients was significantly higher than that in healthy controls. Injection of P. gingivalis via the jugular vein into high-fat fed mice accelerated the progression of NAFLD. In contrast, non-surgical periodontal therapy in patients with NAFLD can significantly improve liver function parameters [Citation26]. In 2018, another Japanese team, Nakahara et al. found that after high-fat feeding and periodontal infection with P. gingivalis, the degree of liver steatosis and fibrosis in mice was more significant. Among 200 patients with biopsy-proven NAFLD, P. gingivalis antibody titers were also positively correlated with NAFLD progression [Citation14]. In 2017, in a study based on the US National Health and Nutrition Examination Survey (NHANES), Bhandari et al. conducted a statistical analysis of the relevant data of 1807 NAFLD patients and 2608 healthy controls and found that with serum porphyrin gingivalis monocytogenes P. gingivalis antibody level increased, the incidence of NAFLD showed an upward trend, and the higher the serum P. gingivalis antibody level, the more severe liver steatosis [Citation68]. Therefore, whether elimination of P. gingivalis infection alleviate the progress of NAFLD? Takata et al. revealed that the application of macrolide antibiotic [azithromycin (AZM)] locally and/or systemically to a high-fat-diet-induced NASH mouse model with P. gingivalis odontogenic infection inhibited NASH progression significantly [Citation24]. Taken together, these studies suggest that P. gingivalis infection may be an independent risk factor for NAFLD and may promote disease progression.
Proposed mechanisms of pathogenetic role of P. gingivalis infection on NAFLD
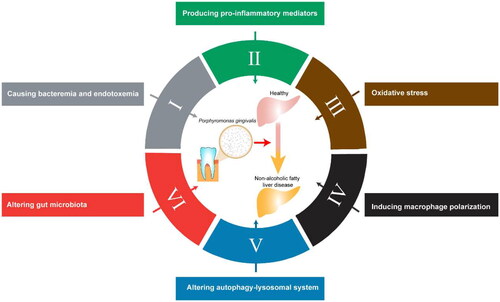
The exact link between P. gingivalis infection and the development of Non-Alcoholic Fatty Liver Disease (NAFLD) is not yet fully understood. However, there are several mechanisms through which P. gingivalis may promote the progression of NAFLD: (1) causing bacteremia and endotoxemia; (2) producing pro-inflammatory mediators; (3) oxidative stress; (4) inducing macrophage polarization; (5) altering autophagy-lysosomal system; (6) altering gut microbiota ().
Figure 1. Mechanism of Porphyromonas gingivalis mediated non-alcoholic fatty liver disease.
Causing bacteremia and endotoxemia
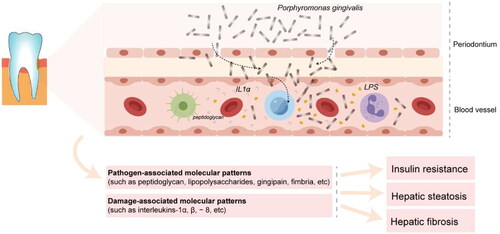
The oral mucosa acts as the first line of defense against environmental and microbial irritants through interconnected epithelial cells. Grainyhead-like transcription factor 2 (GRHL2) is an epithelial-specific transcription factor that regulates the expression of connexins and plays a critical role in the formation of the mucosal epithelial barrier. P. gingivalis has been shown to down-regulate the expression of GRHL2, leading to disruption of the mucosal epithelial barrier [Citation69]. P. gingivalis and its components, such as fimbriae, can enter the body through periodontal ulcers. The Rgp and Kgp proteases produced by P. gingivalis can degrade and inactivate various immunoglobulins and cytokines, protecting the bacterium from phagocyte attack and evading the host’s immune defense mechanism. This allows P. gingivalis to safely enter the bloodstream and produce bacteremia, which can lead to colonization of liver cells through the circulation [Citation70,Citation71]. Ishikawa et al. infected the oral cavity of mice with P. gingivalis, and then found the presence of P. gingivalis in mouse liver cells, and P. gingivalis in the liver was alive bacteria that could be cultured [Citation12] (). Hisako et al. found that P. gingivalis could be detected in the liver tissue of 52.5% of patients with nonalcoholic steatohepatitis (NASH), and the detection of P. gingivalis was positively correlated with liver fibrosis scores [Citation26,Citation35]. Endotoxemia is known to promote the development of NAFLD. The study found that the level of endotoxin in the serum of the rats in the infected group was 2.4 times higher than that of the rats not infected with P. gingivalis [Citation26]. Pathogen-associated molecular patterns (such as peptidoglycan, lipopolysaccharides, gingipain, fimbria, etc.), and damage-associated molecular patterns (such as interleukins-1α, β, −8, etc.) released by pathogens and infected periodontium in the periodontal microenvironments, enter the bloodstream, disseminate into the whole body, and induce a variety of systemic pathological effects, including hepatic steatosis and fibrosis [Citation23]. Sasaki et al. found that endotoxemia induced by intravenous injection of sonicated P. gingivalis caused impaired glucose tolerance, insulin resistance, and liver steatosis [Citation72]. Taken together, these findings suggest that P. gingivalis infection can lead to the destruction of the oral mucosal barrier, followed by bacteremia and endotoxemia, which may be an important factor in accelerating the progression of NAFLD.
Figure 2. Porphyromonas gingivalis infection induces bacteremia and endotoxemia accelerating the progression of NAFLD.
Producing pro-inflammatory mediators
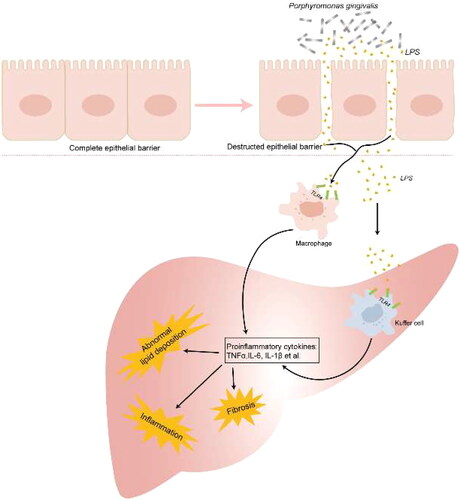
The cell wall of P. gingivalis is mainly composed of peptidoglycan, lipoproteins, lipids, and lipopolysaccharides (LPS). These substances are known to be closely associated with the release of pro-inflammatory mediators. Furusho et al. first demonstrated that P. gingivalis infection exacerbates hepatic steatosis and fibrosis through a mechanism involving the synergistic effect between NLRP3 inflammasome activation and the LPS-TLR pathway. Mice infected with P. gingivalis after a 12-week high-fat diet show significantly higher expression of hepatic cytokines (such as IL-1β, IL6, IL10, IL12, IL17, TNFα and INF-γ), chemokines (such as IL8, macrophage inflammatory protein 1α (MIP-1α), monocyte chemoattractant protein 1 (MCP-1), prostaglandin E2, and nitric oxide (NO)) and Toll-like receptor 2 levels. In vitro, the mRNA levels of Nod-like receptor 3 (NLRP3) and Caspase-1 (Casp-1) were significantly increased in hepatocytes after stimulation of hepatocytes with P. gingivalis-derived lipopolysaccharide [Citation35]. Ding et al. also found that P. gingivalis-derived lipopolysaccharide may lead to intracellular lipid accumulation and inflammatory response in hepatocytes via the activation of NF-κB and JNK signaling pathways [Citation22]. Atsuhiro et al. demonstrate that P. gingivalis odontogenic infection significantly increased the expression of pro-inflammatory factors (such as TNFα, IL1β, galectin-3), whereas eradication of their infection significantly inhibited hepatic steatosis and inflammatory progression [Citation24]. Ahn et al. also showed that P. gingivalis infection exacerbated lobular inflammation and hepatocyte ballooning [Citation20] (). Taken together, these all suggest a functional involvement of P. gingivalis infection in the pathological progression of NAFLD.
Figure 3. Porphyromonas gingivalis Derivates promote pro-inflammatory mediators leading to intracellular lipid accumulation and inflammatory reaction.
Oxidative stress
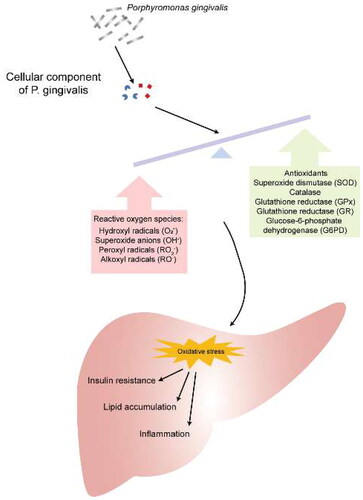
Oxidative stress is a state of imbalance between oxidative and antioxidant effects in the body, producing a large number of oxidative intermediates, such as reactive oxygen species (ROS), reactive nitrogen species (RNS) and free radicals, resulting in host DNA fragmentation, lipid peroxidation and protein oxidation, leading to loss of membrane integrity, changes in protein structure and function, and genetic mutations. ROS and RNS are considered to be the major toxicity mechanisms causing hepatocyte damage [Citation73,Citation74]. In chronic periodontitis, the cellular component of P. gingivalis, present in dental plaque, recruits and activates hyperreactive polymorphonuclear cells, thereby accelerating the process of ROS production. Excessive ROS severely damage the antioxidant system in the liver, stimulating the activation of hepatic intracellular pathways (such as c-Jun, N-terminal kinase, ERK1/2, NF- κB, etc.) and promoting chronic low-grade inflammatory responses, ultimately leading to insulin resistance and metabolic disorders [Citation75] (). These studies suggest that ROS from P. gingivalis-mediated periodontitis may be involved in liver damage.
Figure 4. Porphyromonas gingivalis infection Accelerates the process of ROS production promoting insulin resistance and metabolic disorders.
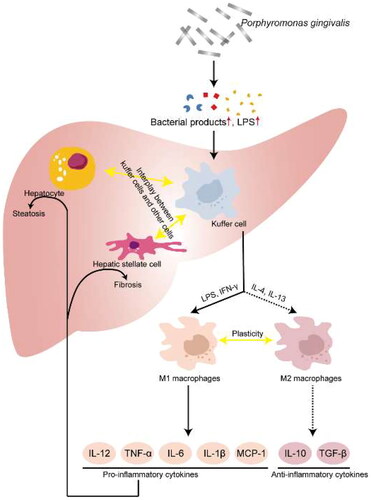
Inducing macrophage polarization
Macrophages are differentiated from bone marrow monocytes and play an important role in the innate immune response. Macrophages are a remarkable plastic cell type that can undergo morphological, functional and phenotypic differentiation after specific microenvironmental stimulation, that is, macrophage polarization [Citation76,Citation77]. Canonical and alternative pathway activated macrophages serve as two extremes of plasticity regulation of their function, termed classically activated macrophages (M1 type) and alternatively activated macrophages (M2 type), respectively.
M1-type macrophages express specific markers such as MHC-II molecules, CD80 and CD86 on the surface. Studies have shown that Toll-like receptor ligands, interferon-γ (IFN-γ), pathogen-associated molecular complex (PAMC), bacterial lipopolysaccharide (LPS), and lipoproteins can induce macrophage polarization to the M1 type. Activated M1-type macrophages produce and secrete a series of pro-inflammatory cytokines such as interleukin (IL)-1α, IL-1β, IL-6, IL-12, IL-23, TNF-α and cycloheximide enzyme-2 (COX-2), which exert pro-inflammatory effects. M2-type macrophages express CD163, mannose receptor 1 (MR1), resistin-like molecule-β and arginase-1 (Arg-1) on the surface. Studies have shown that IL-4, IL-10, IL-13, TLR agonists or IL-1 receptor ligands can induce macrophage polarization to the M2 type. Polarized M2 macrophages can inhibit the production of pro-inflammatory cytokines and induce the secretion of anti-inflammatory cytokines such as IL-10 and IL-12 [Citation78]. Yu et al. found that after infection of bone marrow-derived macrophages with P. gingivalis, macrophage could be polarized to the M1 type, but not the M2 type [Citation79]. Polarized M1 macrophages can produce a series of pro-inflammatory cytokines (including IL-1β, IL-6, inducible nitric oxide synthase, etc.), which further promote the activation of inflammatory signals and increase lipid deposition in hepatocytes. Animal experiments have also shown that M1-type polarization of macrophages can increase hepatic lipid deposition, and inhibition of its polarization can ameliorate hepatic lipid accumulation [Citation80] (). These suggest that P. gingivalis infection may be involved in the pathogenesis of NAFLD by inducing the polarization of macrophages.
Figure 5. Porphyromonas gingivalis infection Involves in the pathogenesis of NAFLD by inducing the polarization of macrophages.
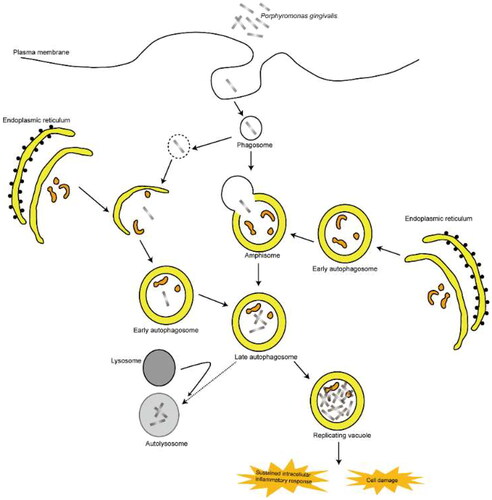
Altering autophagy-lysosomal system
Autophagy is a highly conserved intracellular self-protection mechanism. It mainly degrades damaged organelles and macromolecules through autophagy lysosomes, and recycles small molecules such as amino acids and nucleic acids, and plays an important role in maintaining cellular homeostasis. Autophagy efficiently degrades normal cellular metabolites that, when accumulated, can cause cytotoxicity, such as redox-active protein aggregates and damaged mitochondria [Citation81–84]. Many studies show that autophagy can reduce the content of lipid droplets in hepatocytes and alleviate hepatic steatosis by degrading intracellular lipid droplets (lipophagy). When autophagy is impaired, the ability to degrade intrahepatic lipid droplets is reduced, which would cause a sustained increase in lipid droplets and the development of steatosis. Studies have shown that autophagy is inhibited in vivo in NAFLD patients. In vitro and in vivo experiments have shown that inhibition of autophagy can exacerbate hepatic steatosis. Therefore, modulation of hepatocyte autophagy may be an effective and protective way to treat NAFLD [Citation85,Citation86]. P. gingivalis infection can inhibit the formation of autophagolysosomes while activating autophagy and colonizing late autophagosomes, resulting in persistent infection. Zaitsu et al. found that accumulation of intracellular lipid droplets in HepG2 cells can delay the clearance of P. gingivalis by inhibiting the formation of lysosomes and autophagolysosomes. These lead to sustained intracellular inflammatory response and cell damage, which is a risk factor for the development of NAFLD [Citation87,Citation88] (). Therefore, the inhibition of autophagy-lysosome formation may play an important role in the development of NAFLD caused by P. gingivalis infection.
Figure 6. Porphyromonas gingivalis infection Alters autophagy-lysosomal system accelerating the development of NAFLD.
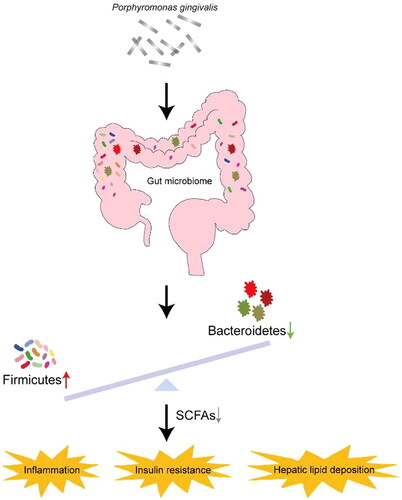
Altering gut microbiota
The gut microbiota participates in and regulates many life activities throughout the life process of the human body, and is closely related to host health [Citation89]. The maintenance of gut microbiota homeostasis is influenced by a combination of factors. A large number of clinical studies have shown that the composition or dysfunction of the gut microbiota plays a key role in the development and progression of NAFLD. Bacteroidetes and Firmicutes are the two dominant bacterial groups in the human gut, and the relative abundances of Firmicutes and Bacteroidetes differ in NAFLD patients and healthy controls. Compared to healthy controls, the relative abundance of Bacteroidetes was decreased in the gut of NAFLD patients, while the relative abundance of Firmicutes was increased. When NAFLD patients consumed a low-calorie diet or exercised to lose weight, the abundance of Bacteroidetes increased and the abundance of Firmicutes decreased [Citation90]. Nakajima et al. found that a single feeding of P.gingivalis could significantly induce changes in the gut flora, with an increase in the abundance of Bacteroidetes and a decrease in the abundance of Firmicutes [Citation25,Citation91–93]. Alexandra et al. found that both acute and chronic P. gingivalis infection disrupted the gut microbiota composition of mice and reduced the abundance of short-chain fatty acid-producing microbiota, resulting in functional changes associated with exacerbated NAFLD [Citation94]. Arimatsu et al. found that oral administration of P. gingivalis could not only induced similar changes in gut microbiota of mice, but also weakened the barrier function of intestinal epithelial cells, increased intestinal wall permeability, increased serum endotoxin levels, and induced systemic inflammation, insulin resistance, and hepatic lipid deposition [Citation95]. Serum endotoxin levels began to increase 1 h after a single oral administration of P. gingivalis, reached a peak after 3 h, and remained at the peak for 12 h. Sasaki et al. found that after intravenous injection of ultrasonically inactivated P. gingivalis into mice fed a high-fat diet, the intestinal flora of the mice changed significantly compared with the control group. Specifically, the numbers of Firmicutes and Proteobacteria decreased, the numbers of Alcaligenaceae and Erysipelotrichaceae increased significantly; and the numbers of Dehalobacteriaceae and Ruminococcaceae decreased, and the numbers of Bilophila and Dehalobacterium decreased significantly, while the numbers of Sutterella and Allobaculum increased significantly. In addition, treatment with P. gingivalis resulted in increased body weight, increased accumulation of subcutaneous fat and visceral fat, impaired glucose and insulin tolerance, and significant hepatic steatosis in mice (). These all suggest that P. gingivalis infection may promote the occurrence and development of NAFLD by altering the gut microbiome.
Figure 7. Porphyromonas gingivalis infection may promote the occurrence and development of NAFLD by Changing gut microbiota.
Other non-alcoholic fatty liver disease related periodontal pathogens
The relationship between periodontal disease and non-alcoholic fatty liver disease has been discussed from in vitro, in vivo, and epidemiologic perspectives. In addition to P. gingivalis, studies have found that other periodontal pathogens are similarly involved in the occurrence and development of NAFLD. For example, Rina et al. found that in the NAFLD population, the antibody titer of Aggregatibacter actinomycetemcommitans (Aa) was positively correlated with visual fat, fast plasma insulin and HOMA-IR. After infected with Aggregatibacter actinomycetemcomitans, the mice showed abnormalities in glucose tolerance, insulin resistance and liver steatosis, and these abnormalities were mainly mediated by the regulation of glucose metabolism regulation, fatty acid metabolism and intestinal flora. Whereas, antibiotic treatment with norfloxacin could improve glucose tolerance and liver lipid accumulation, and also lead to changes in the gut microbiota [Citation96]. This suggest that periodontal pathogens are important factors leading to NAFLD, and maintaining oral hygiene is crucial in the prevention and treatment of NAFLD. Given the oral cavity being a complex microbiome, further insights into the alterations of the oral microbial community under the pathological state of NAFLD are hence warranted.
Concluding remarks
Although there are currently many studies on the relationship between NAFLD and gut microbiota, the relationship between oral microbial community and NAFLD has received growing attention in recent years. P. gingivalis is the main pathogen of periodontitis, which may influence the occurrence and development of NAFLD. More research is needed to confirm the role of P. gingivalis in the pathogenesis of NAFLD and clarify the underlying mechanisms. In addition, long-term chronic infection with P. gingivalis has also been reported to damage various systems of the body, such as the cardiovascular system, immune system, and metabolic system. Therefore, emphasizing oral health, maintaining good oral hygiene, seeking timely and standardized treatment for various periodontal diseases, especially P. gingivalis-odontogenic infection, can have a positive impact on overall health.
Authors contributions
All authors provided substantial intellectual contribution to the study, approved the final version and agreed to be accountable for the work. Linbo Liu and Yan Geng wrote the original draft and designed the schematic figures. Linbo Liu, Yan Geng and Chaoliang Xiong edited the figures and revised the manuscript. All authors read and approved the final manuscript.
Acknowledgments
We would like to express our gratitude to the research team of our lab for their collaboration and support on every step of this work.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
Additional information
Funding
References
- Lazarus JV, Mark HE, Anstee QM, et al. Advancing the global public health agenda for NAFLD: a consensus statement. Nat Rev Gastroenterol Hepatol. 2022;19(1):1–12. doi:10.1038/s41575-021-00523-4.
- Hernaez R, Peck-Radosavljevic M. MAFLD, HCC and the dilemma of (changing) terminology in liver diseases. Gut. 2023;72(1):9–11. doi:10.1136/gutjnl-2022-326992.
- Kanneganti M, Singal AG. Incidence of hepatocellular carcinoma in nonalcoholic fatty liver disease. Gastroenterology. 2022;162(6):1772–1774. doi:10.1053/j.gastro.2022.01.037.
- Foerster F, Gairing SJ, Muller L, et al. NAFLD-driven HCC: safety and efficacy of current and emerging treatment options. J Hepatol. 2022;76(2):446–457. doi:10.1016/j.jhep.2021.09.007.
- Younossi Z, Tacke F, Arrese M, et al. Global perspectives on nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Hepatology. 2019;69(6):2672–2682. doi:10.1002/hep.30251.
- Wong WK, Chan WK. Nonalcoholic fatty liver disease: a global perspective. Clin Ther. 2021;43(3):473–499. doi:10.1016/j.clinthera.2021.01.007.
- Zhou J, Zhou F, Wang W, et al. Epidemiological features of NAFLD from 1999 to 2018 in China. Hepatology. 2020;71(5):1851–1864. doi:10.1002/hep.31150.
- Gallage S, Avila JEB, Ramadori P, et al. A researcher’s guide to preclinical mouse NASH models. Nat Metab. 2022;4(12):1632–1649. doi:10.1038/s42255-022-00700-y.
- Mantovani A, Byrne CD, Benfari G, et al. Risk of heart failure in patients with nonalcoholic fatty liver disease: JACC review topic of the week. J Am Coll Cardiol. 2022;79(2):180–191. doi:10.1016/j.jacc.2021.11.007.
- Tsochatzis EA. Natural history of NAFLD: knowns and unknowns. Nat Rev Gastroenterol Hepatol. 2022;19(3):151–152. doi:10.1038/s41575-021-00565-8.
- Hatasa M, Yoshida S, Takahashi H, et al. Relationship between NAFLD and periodontal disease from the view of clinical and basic research, and immunological response. Int J Mol Sci. 2021;22(7):22.
- Ishikawa M, Yoshida K, Okamura H, et al. Oral Porphyromonas gingivalis translocates to the liver and regulates hepatic glycogen synthesis through the akt/GSK-3beta signaling pathway. Biochim Biophys Acta. 2013;1832(12):2035–2043. doi:10.1016/j.bbadis.2013.07.012.
- Kuraji R, Ito H, Fujita M, et al. Porphyromonas gingivalis induced periodontitis exacerbates progression of non-alcoholic steatohepatitis in rats. Clin Exp Dent Res. 2016;2(3):216–225. doi:10.1002/cre2.41.
- Nakahara T, Hyogo H, Ono A, et al. Involvement of Porphyromonas gingivalis in the progression of non-alcoholic fatty liver disease. J Gastroenterol. 2018;53(2):269–280. doi:10.1007/s00535-017-1368-4.
- Hasegawa Y, Nagano K. Porphyromonas gingivalis FimA and Mfa1 fimbriae: current insights on localization, function, biogenesis, and genotype. Jpn Dent Sci Rev. 2021;57:190–200. doi:10.1016/j.jdsr.2021.09.003.
- Bostanci N, Belibasakis GN. Porphyromonas gingivalis: an invasive and evasive opportunistic oral pathogen. FEMS Microbiol Lett. 2012;333(1):1–9. doi:10.1111/j.1574-6968.2012.02579.x.
- Yoshimura F, Murakami Y, Nishikawa K, et al. Surface components of Porphyromonas gingivalis. J Periodontal Res. 2009;44(1):1–12. doi:10.1111/j.1600-0765.2008.01135.x.
- Lunar Silva I, Cascales E. Molecular strategies underlying Porphyromonas gingivalis virulence. J Mol Biol. 2021;433(7):166836. doi:10.1016/j.jmb.2021.166836.
- Mei F, Xie M, Huang X, et al. Porphyromonas gingivalis and its systemic impact: current status. Pathogens. 2020;9(11):944. doi:10.3390/pathogens9110944.
- Ahn J-S, Yang JW, Oh S-J, et al. Porphyromonas gingivalis exacerbates the progression of fatty liver disease via CD36-PPARγ pathway. BMB Rep. 2021;54(6):323–328. doi:10.5483/BMBRep.2021.54.6.050.
- Alakhali MS, Al-Maweri SA, Al-Shamiri HM, et al. The potential association between periodontitis and non-alcoholic fatty liver disease: a systematic review. Clin Oral Investig. 2018;22(9):2965–2974. doi:10.1007/s00784-018-2726-1.
- Ding LY, Liang LZ, Zhao YX, et al. Porphyromonas gingivalis-derived lipopolysaccharide causes excessive hepatic lipid accumulation via activating NF-kappaB and JNK signaling pathways. Oral Dis. 2019;25(7):1789–1797. doi:10.1111/odi.13153.
- Ezhilarasan D. Deciphering the toxicological role of Porphyromonas gingivalis derived endotoxins in liver diseases. Environ Toxicol Pharmacol. 2021;88:103755. doi:10.1016/j.etap.2021.103755.
- Nagasaki A, Sakamoto S, Arai T, et al. Elimination of Porphyromonas gingivalis inhibits liver fibrosis and inflammation in NASH. J Clin Periodontol. 2021;48(10):1367–1378. doi:10.1111/jcpe.13523.
- Yamazaki K, Kato T, Tsuboi Y, et al. Oral Pathobiont-Induced changes in gut microbiota aggravate the pathology of nonalcoholic fatty liver disease in mice. Front Immunol. 2021;12:766170. doi:10.3389/fimmu.2021.766170.
- Yoneda M, Naka S, Nakano K, et al. Involvement of a periodontal pathogen, Porphyromonas gingivalis on the pathogenesis of non-alcoholic fatty liver disease. BMC Gastroenterol. 2012;12(1):16. doi:10.1186/1471-230X-12-16.
- Loomba R, Friedman SL, Shulman GI. Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell. 2021;184(10):2537–2564. doi:10.1016/j.cell.2021.04.015.
- Kang SH, Cho Y, Jeong SW, et al. From nonalcoholic fatty liver disease to metabolic-associated fatty liver disease: big wave or ripple? Clin Mol Hepatol. 2021;27(2):257–269. doi:10.3350/cmh.2021.0067.
- Gerges SH, Wahdan SA, Elsherbiny DA, et al. Non-alcoholic fatty liver disease: an overview of risk factors, pathophysiological mechanisms, diagnostic procedures, and therapeutic interventions. Life Sci. 2021;271:119220. doi:10.1016/j.lfs.2021.119220.
- Musso G, Cassader M, Gambino R. Non-alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov. 2016;15(4):249–274. doi:10.1038/nrd.2015.3.
- Longo M, Meroni M, Paolini E, et al. Mitochondrial dynamics and nonalcoholic fatty liver disease (NAFLD): new perspectives for a fairy-tale ending? Metabolism. 2021;117:154708. doi:10.1016/j.metabol.2021.154708.
- Lebeaupin C, Vallee D, Hazari Y, et al. Endoplasmic reticulum stress signalling and the pathogenesis of non-alcoholic fatty liver disease. J Hepatol. 2018;69(4):927–947. doi:10.1016/j.jhep.2018.06.008.
- Friedman SL, Neuschwander-Tetri BA, Rinella M, et al. Mechanisms of NAFLD development and therapeutic strategies. Nat Med. 2018;24(7):908–922. doi:10.1038/s41591-018-0104-9.
- Tilg H, Adolph TE, Moschen AR. Multiple parallel hits hypothesis in nonalcoholic fatty liver disease: revisited after a decade. Hepatology. 2021;73(2):833–842. doi:10.1002/hep.31518.
- Furusho H, Miyauchi M, Hyogo H, et al. Dental infection of Porphyromonas gingivalis exacerbates high fat diet-induced steatohepatitis in mice. J Gastroenterol. 2013;48(11):1259–1270. doi:10.1007/s00535-012-0738-1.
- Tian J, Liu C, Zheng X, et al. Porphyromonas gingivalis induces insulin resistance by increasing BCAA levels in mice. J Dent Res. 2020;99(7):839–846. doi:10.1177/0022034520911037.
- Aleksijevic LH, Aleksijevic M, Skrlec I, et al. Porphyromonas gingivalis virulence factors and clinical significance in periodontal disease and coronary artery diseases. Pathogens. 2022;11(10):1173. doi:10.3390/pathogens11101173.
- Tribble GD, Kerr JE, Wang BY. Genetic diversity in the oral pathogen Porphyromonas gingivalis: molecular mechanisms and biological consequences. Future Microbiol. 2013;8(5):607–620. doi:10.2217/fmb.13.30.
- Atanasova KR, Yilmaz O. Looking in the Porphyromonas gingivalis cabinet of curiosities: the microbium, the host and cancer association. Mol Oral Microbiol. 2014;29(2):55–66. doi:10.1111/omi.12047.
- Ahmadi P, Mahmoudi M, Kheder RK, et al. Impacts of Porphyromonas gingivalis periodontitis on rheumatoid arthritis autoimmunity. Int Immunopharmacol. 2023;118:109936. doi:10.1016/j.intimp.2023.109936.
- Li Y, Guo R, Oduro PK, et al. The relationship between Porphyromonas gingivalis and rheumatoid arthritis: a meta-analysis. Front Cell Infect Microbiol. 2022;12:956417. doi:10.3389/fcimb.2022.956417.
- Zhou N, Zou F, Cheng X, et al. Porphyromonas gingivalis induces periodontitis, causes immune imbalance, and promotes rheumatoid arthritis. J Leukoc Biol. 2021;110(3):461–473. doi:10.1002/JLB.3MA0121-045R.
- Bartold PM, Marino V, Cantley M, et al. Effect of Porphyromonas gingivalis-induced inflammation on the development of rheumatoid arthritis. J Clin Periodontol. 2010;37(5):405–411. doi:10.1111/j.1600-051X.2010.01552.x.
- Mikuls TR, Payne JB, Yu F, et al. Periodontitis and Porphyromonas gingivalis in patients with rheumatoid arthritis. Arthritis Rheumatol. 2014;66(5):1090–1100. doi:10.1002/art.38348.
- Yamakawa M, Ouhara K, Kajiya M, et al. Porphyromonas gingivalis infection exacerbates the onset of rheumatoid arthritis in SKG mice. Clin Exp Immunol. 2016;186(2):177–189. doi:10.1111/cei.12847.
- Nakano K, Wada K, Nomura R, et al. Characterization of aortic aneurysms in cardiovascular disease patients harboring Porphyromonas gingivalis. Oral Dis. 2011;17(4):370–378. doi:10.1111/j.1601-0825.2010.01759.x.
- Deshpande RG, Khan M, Genco CA. Invasion strategies of the oral pathogen porphyromonas gingivalis: implications for cardiovascular disease. Invasion Metastasis. 1998;18(2):57–69. doi:10.1159/000024499.
- Ruan Q, Guan P, Qi W, et al. Porphyromonas gingivalis regulates atherosclerosis through an immune pathway. Front Immunol. 2023;14:1103592. doi:10.3389/fimmu.2023.1103592.
- Zhang J, Xie M, Huang X, et al. The effects of Porphyromonas gingivalis on atherosclerosis-related cells. Front Immunol. 2021;12:766560. doi:10.3389/fimmu.2021.766560.
- Li L, Messas E, Batista EL, Jr., et al. Porphyromonas gingivalis infection accelerates the progression of atherosclerosis in a heterozygous apolipoprotein E-deficient murine model. Circulation. 2002;105(7):861–867. doi:10.1161/hc0702.104178.
- Hayashi C, Viereck J, Hua N, et al. Porphyromonas gingivalis accelerates inflammatory atherosclerosis in the innominate artery of ApoE deficient mice. Atherosclerosis. 2011;215(1):52–59. doi:10.1016/j.atherosclerosis.2010.12.009.
- Blasco-Baque V, Garidou L, Pomie C, et al. Periodontitis induced by Porphyromonas gingivalis drives periodontal microbiota dysbiosis and insulin resistance via an impaired adaptive immune response. Gut. 2017;66(5):872–885. doi:10.1136/gutjnl-2015-309897.
- Singhrao SK, Harding A, Poole S, et al. Porphyromonas gingivalis periodontal infection and its putative links with alzheimer’s disease. Mediators Inflamm. 2015;2015:137357–137310. doi:10.1155/2015/137357.
- Liu S, Butler CA, Ayton S, et al. Porphyromonas gingivalis and the pathogenesis of alzheimer’s disease. Crit Rev Microbiol. 2023. doi:10.1080/1040841X.2022.2163613.
- Elwishahy A, Antia K, Bhusari S, et al. Porphyromonas Gingivalis as a risk factor to alzheimer’s disease: a systematic review. J Alzheimers Dis Rep. 2021;5(1):721–732. doi:10.3233/ADR-200237.
- Olsen I. Porphyromonas gingivalis-Induced neuroinflammation in alzheimer’s disease. Front Neurosci. 2021;15:691016. doi:10.3389/fnins.2021.691016.
- Olsen I. Possible effects of Porphyromonas gingivalis on the blood-brain barrier in alzheimer’s disease. Expert Rev anti Infect Ther. 2021;19(11):1367–1371. doi:10.1080/14787210.2021.1925540.
- Olsen I, Yilmaz O. Possible role of Porphyromonas gingivalis in orodigestive cancers. J Oral Microbiol. 2019;11(1):1563410. doi:10.1080/20002297.2018.1563410.
- Dominy SS, Lynch C, Ermini F, et al. Porphyromonas gingivalis in alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019;5(1):eaau3333. doi:10.1126/sciadv.aau3333.
- Gao S, Li S, Ma Z, et al. Presence of Porphyromonas gingivalis in esophagus and its association with the clinicopathological characteristics and survival in patients with esophageal cancer. Infect Agent Cancer. 2016;11:3.
- Gnanasekaran J, Binder Gallimidi A, Saba E, et al. Intracellular Porphyromonas gingivalis promotes the tumorigenic behavior of pancreatic carcinoma cells. Cancers (Basel). 2020;12(8):2331. doi:10.3390/cancers12082331.
- Da Venezia C, Hussein N, Hernandez M, et al. Assessment of cardiovascular risk in women with periodontal diseases according to C-reactive protein levels. Biomolecules. 2021;11(8):1238. doi:10.3390/biom11081238.
- Stanisic D, Jeremic N, Singh M, et al. Porphyromonas gingivalis induces cardiovascular dysfunction. Can J Physiol Pharmacol. 2023;101(8):413–424. doi:10.1139/cjpp-2022-0392.
- Olsen I, Taubman MA, Singhrao SK. Porphyromonas gingivalis suppresses adaptive immunity in periodontitis, atherosclerosis, and alzheimer’s disease. J Oral Microbiol. 2016;8(1):33029. doi:10.3402/jom.v8.33029.
- Tang Z, Liang D, Cheng M, et al. Effects of Porphyromonas gingivalis and its underlying mechanisms on Alzheimer-like tau hyperphosphorylation in Sprague-Dawley rats. J Mol Neurosci. 2021;71(1):89–100. doi:10.1007/s12031-020-01629-1.
- Xie M, Tang Q, Nie J, et al. BMAL1-Downregulation aggravates Porphyromonas gingivalis-induced atherosclerosis by encouraging oxidative stress. Circ Res. 2020;126(6):e15–e29. doi:10.1161/CIRCRESAHA.119.315502.
- Wan M, Liu J, Ouyang X. Nucleotide-binding oligomerization domain 1 regulates Porphyromonas gingivalis-induced vascular cell adhesion molecule 1 and intercellular adhesion molecule 1 expression in endothelial cells through NF-kappaB pathway. J Periodontal Res. 2015;50(2):189–196. doi:10.1111/jre.12192.
- Bhandari S, Gupta E, Abdul MKM, et al. Association ofPorphyromonas gingivalisSerum antibody levels and non-alcoholic fatty liver disease (NAFLD): an NHANES study: 1004. Off J Am Coll Gastroenterol| ACG. 2017;112:S559. doi:10.14309/00000434-201710001-01005.
- Chen W, Alshaikh A, Kim S, et al. Porphyromonas gingivalis impairs oral epithelial barrier through targeting GRHL2. J Dent Res. 2019;98(10):1150–1158. doi:10.1177/0022034519865184.
- Bregaint S, Boyer E, Fong SB, et al. Porphyromonas gingivalis outside the oral cavity. Odontology. 2022;110(1):1–19. doi:10.1007/s10266-021-00647-8.
- Abe N, Baba A, Takii R, et al. Roles of arg- and lys-gingipains in coaggregation of Porphyromonas gingivalis: identification of its responsible molecules in translation products of rgpA, kgp, and hagA genes. Biol Chem. 2004;385(11):1041–1047. doi:10.1515/BC.2004.135.
- Sasaki N, Katagiri S, Komazaki R, et al. Endotoxemia by Porphyromonas gingivalis injection aggravates non-alcoholic fatty liver disease, disrupts glucose/lipid metabolism, and alters gut microbiota in mice. Front Microbiol. 2018;9:2470. doi:10.3389/fmicb.2018.02470.
- Sumida Y, Niki E, Naito Y, et al. Involvement of free radicals and oxidative stress in NAFLD/NASH. Free Radic Res. 2013;47(11):869–880. doi:10.3109/10715762.2013.837577.
- Narasimhan S, Gokulakrishnan K, Sampathkumar R, et al. Oxidative stress is independently associated with non-alcoholic fatty liver disease (NAFLD) in subjects with and without type 2 diabetes. Clin Biochem. 2010;43(10-11):815–821. doi:10.1016/j.clinbiochem.2010.04.003.
- Kumar J, Teoh SL, Das S, et al. Oxidative stress in oral diseases: understanding its relation with other systemic diseases. Front Physiol. 2017;8:693. doi:10.3389/fphys.2017.00693.
- Gordon S. Alternative activation of macrophages. Nat Rev Immunol. 2003;3(1):23–35. doi:10.1038/nri978.
- Murray PJ. Macrophage polarization. Annu Rev Physiol. 2017;79(1):541–566. doi:10.1146/annurev-physiol-022516-034339.
- Kazankov K, Jorgensen SMD, Thomsen KL, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol. 2019;16(3):145–159. doi:10.1038/s41575-018-0082-x.
- Yu S, Ding L, Liang D, et al. Porphyromonas gingivalis inhibits M2 activation of macrophages by suppressing alpha-ketoglutarate production in mice. Mol Oral Microbiol. 2018;33(5):388–395. doi:10.1111/omi.12241.
- Wan J, Benkdane M, Teixeira-Clerc F, et al. M2 kupffer cells promote M1 kupffer cell apoptosis: a protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology. 2014;59(1):130–142. doi:10.1002/hep.26607.
- Saha S, Panigrahi DP, Patil S, et al. Autophagy in health and disease: a comprehensive review. Biomed Pharmacother. 2018;104:485–495. doi:10.1016/j.biopha.2018.05.007.
- Kitada M, Koya D. Autophagy in metabolic disease and ageing. Nat Rev Endocrinol. 2021;17(11):647–661. doi:10.1038/s41574-021-00551-9.
- Mizushima N, Levine B. Autophagy in human diseases. N Engl J Med. 2020;383(16):1564–1576. doi:10.1056/NEJMra2022774.
- Hazari Y, Bravo-San Pedro JM, Hetz C, et al. Autophagy in hepatic adaptation to stress. J Hepatol. 2020;72(1):183–196. doi:10.1016/j.jhep.2019.08.026.
- Gonzalez-Rodriguez A, Mayoral R, Agra N, et al. Impaired autophagic flux is associated with increased endoplasmic reticulum stress during the development of NAFLD. Cell Death Dis. 2014;5(4):e1179–e1179. doi:10.1038/cddis.2014.162.
- Czaja MJ. Function of autophagy in nonalcoholic fatty liver disease. Dig Dis Sci. 2016;61(5):1304–1313. doi:10.1007/s10620-015-4025-x.
- Zaitsu Y, Iwatake M, Sato K, et al. Lipid droplets affect elimination of Porphyromonas gingivalis in HepG2 cells by altering the autophagy-lysosome system. Microbes Infect. 2016;18(9):565–571. doi:10.1016/j.micinf.2016.05.004.
- Rodrigues PH, Belanger M, Dunn W, Jr., et al. Porphyromonas gingivalis and the autophagic pathway: an innate immune interaction? Front Biosci. 2008;13(13):178–187. doi:10.2741/2668.
- Abu-Shanab A, Quigley EM. The role of the gut microbiota in nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2010;7(12):691–701. doi:10.1038/nrgastro.2010.172.
- Compare D, Coccoli P, Rocco A, et al. Gut–liver axis: the impact of gut microbiota on non alcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2012;22(6):471–476. doi:10.1016/j.numecd.2012.02.007.
- Nakajima M, Arimatsu K, Kato T, et al. Oral administration of P. gingivalis induces dysbiosis of gut microbiota and impaired barrier function leading to dissemination of enterobacteria to the liver. PLoS One. 2015;10(7):e0134234. doi:10.1371/journal.pone.0134234.
- Lang S, Schnabl B. Microbiota and fatty liver disease—the known, the unknown, and the future. Cell Host Microbe. 2020;28(2):233–244. doi:10.1016/j.chom.2020.07.007.
- Aron-Wisnewsky J, Vigliotti C, Witjes J, et al. Gut microbiota and human NAFLD: disentangling microbial signatures from metabolic disorders. Nat Rev Gastroenterol Hepatol. 2020;17(5):279–297. doi:10.1038/s41575-020-0269-9.
- Simas AM, Kramer CD, Genco CA. Diet-Induced non-alcoholic fatty liver disease and associated gut dysbiosis are exacerbated by oral infection. Front Oral Health. 2021;2:784448. doi:10.3389/froh.2021.784448.
- Arimatsu K, Yamada H, Miyazawa H, et al. Oral pathobiont induces systemic inflammation and metabolic changes associated with alteration of gut microbiota. Sci Rep. 2014;4(1):4828. doi:10.1038/srep04828.
- Komazaki R, Katagiri S, Takahashi H, et al. Periodontal pathogenic bacteria, Aggregatibacter actinomycetemcomitans affect non-alcoholic fatty liver disease by altering gut microbiota and glucose metabolism. Sci Rep. 2017;7(1):13950. doi:10.1038/s41598-017-14260-9.