
Abstract
Background
Low HbF expression in HbE-β+-thalassemia may lead to misdiagnosis of HbE heterozygosity. We aimed to characterize the β- and α-globin genes and the modifying factors related to HbF expression in patients with an Hb phenotype similar to that of HbE heterozygotes. Furthermore, screening tools for differentiating HbE-β+-thalassemia from HbE heterozygotes have been investigated.
Participants and Methods
A total of 2133 participants with HbE and HbA with varying HbF levels were recruited. Polymerase chain reaction-based DNA analysis and sequencing were performed to characterize β- and α-globin genes. DNA polymorphism at position −158 nt 5′ to Gγ-globin was performed by XmnI restriction digestion. Receiver operating characteristic (ROC) curves were constructed using the area under the curve (AUC). Cutoff values of HbA2, HbE, and HbF levels for the differentiation of HbE-β+-thalassemia from HbE heterozygotes were determined.
Results
Five β+-thalassemia mutations trans to βE-gene (β−87(C>A), β−31(A>G), β−28(A>G), β19(A>G), and β126(T>G)) were identified in 79 patients. Among these, 54 presented with low HbF levels, and 25 presented with high HbF levels. ROC curve analysis revealed an excellent AUC of 1.000 (95% confidence interval:1.000–1.000) for HbE levels, and a cut-off point of ≥35.0% had 100.0% sensitivity, specificity, and Youden’s index for differentiating HbE-β+-thalassemia from HbE heterozygotes. The proportion of α-thalassemia mutations was 46.3 and 8.0% among HbE-β+-thalassemia patients with low and high HbF levels, respectively. Two rare α-thalassemia mutations (Cap +14(C>G) and initiation codon (ATG>-TG)) of α2-globin genes were identified. The genotype and allele of the polymorphism at −158 nt 5′ to Gγ-globin was found to be negatively associated with HbF expression.
Conclusions
HbE-β+-thalassemia cannot be disregarded until appropriate DNA analysis is performed, and the detection of α-thalassemia mutations should always be performed under these conditions. An HbE level ≥35.0% may indicate screening of samples for DNA analysis for HbE-β+-thalassemia diagnosis.
KEY MESSAGES
HbE-β+-thalassemia displays a wide range of HbF expression, which may lead to the misdiagnosis of HbE heterozygosity in patients whose Hb analysis shows HbE and HbA. α-Thalassemia may be a major factor associated with decreased secondary activation of HbF expression in the disease.
HbE may be a potential indicator for effectively differentiating HbE-β+-thalassemia from HbE heterozygotes.
The high proportion and heterogeneity of α-thalassemia mutations found in patients with HbE-β+-thalassemia evoke a complex thalassemia syndrome, requiring complete DNA analysis.
Introduction
β-Thalassemia is a group of genetic disorders characterized by a quantitative deficiency of β-globin chain production and is further classified as either β0- or β+-thalassemia, based on the absence or reduction of β-globin chain production, respectively [Citation1,Citation2]. Hemoglobin E (Hb) E is the most common hemoglobinopathy in Southeast Asia and is caused by a point mutation in codon 26 (GAG to AAG) of the β-globin gene, which results in the substitution of lysine for glutamic acid. This mutation creates abnormal splicing within exon 1 of the β-globin gene, resulting in reduced production of β-globin, which manifests in the clinical phenotype of a mild form of β-thalassemia, β+-thalassemia [Citation3]. The clinical manifestation of HbE is dependent on co-inheritance of the β-globin gene defect. Heterozygous HbE is asymptomatic with normal or slight microcytosis, while homozygous HbE is clinically normal with remarkable microcytosis of red blood cells (RBCs), similar to that observed in β-thalassemia carriers. The interaction of HbE with β-thalassemia can cause HbE-β-thalassemia disease, the most common β-thalassemic disease worldwide, with the highest frequencies observed in Asia and throughout Southeast Asia, particularly in Thailand, where it is common for individuals to inherit alleles for both hemoglobin E (HbE) and beta-thalassemia [Citation4,Citation5]. HbE-β-thalassemia produces a clinical phenotype ranging from mild to severe anemia, with Hb levels ranging from 2.5 to 13.3 g/dL [Citation6]. HbE-β-thalassemia results from the co-inheritance of a β-thalassemia allele from one parent and the structural variant HbE from the other. The clinical phenotype of HbE-β0-thalassemia is usually more severe than HbE-β+-thalassemia. Hb levels of 9.4–11.1 g/dL have been observed in a cohort of Thai patients with pure HbE-β+-thalassemia [Citation7]. A presumptive diagnosis of the two diseases can be made by Hb analysis: in HbE-β0 thalassemia, βA-globin chains are not synthesized, and the condition is characterized by the production of HbE and HbF without detectable HbA; HbE constitutes 30–70% of the hemoglobin with the remainder HbF. In HbE-β+-thalassemia, variable amounts of HbA are detected, in addition to HbE and HbF. Different β+ thalassemia mutations result in variable disease severity, reflecting different levels of HbA. Interestingly, in many cases of HbE-β+-thalassemia, HbF is 0.0–5.0% of the total Hb, making it undetectable or only mildly detectable [Citation2]. Thus, HbE-β+-thalassemia with such phenotypes is difficult to differentiate from HbE heterozygotes by routine laboratory investigations based on hematological screening and Hb analysis, leading to misdiagnosis of the phenotype as HbE heterozygous, especially in areas where HbE and β-thalassemia are prevalent, such as Thailand. Misdiagnosis can lead to inaccurate genetic counseling and affect the prognosis of the disease. In such cases, appropriate DNA analysis is necessary for accurate diagnosis.
The molecular basis of HbE-β+-thalassemia with such a phenotype and modifying factors associated with decreased or abolished reactivation of the γ-globin gene leading to low HbF levels are not well understood. The genetic factors strongly associated with the regulation of HbF expression in β-thalassemia, sickle cell diseases, HbE-related disorders, and healthy individuals in various populations include three major loci: rs7482144 (Gγ-XmnI) polymorphism in the promoter region of Gγ-globin, rs4895441, rs9399137 (HBS1L-MYB), and rs4671393 (BCL11A) [Citation8–10]. Understanding the molecular basis and genetic factors that modulate phenotypic expression is important for providing appropriate counseling and planning for prenatal diagnosis and patient management. Hence, a simple initial screening tool is warranted to identify appropriate samples from patients with presumed HbE heterozygosity by Hb analysis for further DNA testing for HbE-β+-thalassemia.
Materials and methods
Study participants and hematological analysis
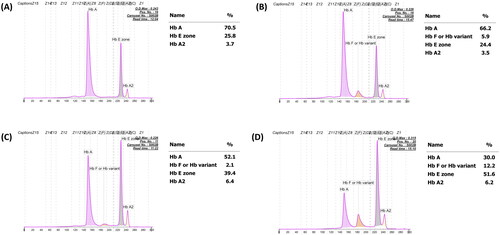
A total of 2133 unused ethylenediaminetetraacetic acid anticoagulated blood samples, whose Hb analysis showed HbE and HbA with varying HbF levels ranging from 0.0% to 40.0%, were selected from among the samples submitted for routine Hb analysis from October 2020 to September 2021 at the Division of Hematology, Lampang Hospital, Thailand (). These included patients being evaluated for anemia or antenatal screening and those who had not received a transfusion for at least 3 months before blood collection. Hb analysis was performed using automated capillary electrophoresis (CE; Capillarys 2 Flex Piercing; Sebia SA, Lisses, France). Additionally, automated cation exchange high-performance liquid chromatography (HPLC) (VARIANT II system, Bio-Rad Laboratories, Hercules, CA, USA, using the β-Thalassemia Short Program) and HPLC (Premier Resolution system from Trinity Biotech, Bray, Country Wicklow, Ireland, using the high-resolution mode) were used to separate Hb from seven samples with β-chain variants. Hematological parameters of all samples were measured using an automated blood cell counter (Coulter T series; Beckman-Coulter Co., Fullerton, CA, USA) and were originally recorded at the hospital. A total of 1946 patients were assigned to be heterozygous HbE because Hb analysis revealed both HbE and HbA (), with an HbE level <30% being considered an acceptable range for heterozygous HbE [Citation7,Citation11–13]. The remaining 187 patients, comprising those whose Hb analysis showed the following outcomes:48 had HbF >5.0% and HbE < 30.0% (); 95 had HbF <5.0% and HbE > 30.0% (); and 44 had HbF >5.0% and HbE >30.0% (). These patients were selected for further analysis of β- and α-globin gene mutations.
Figure 1. Representative capillary electropherograms of patients with classical heterozygous HbE with undetectable HbF (A), those with increasing HbF levels (B), HbE-β+-thalassemia with decreasing HbF expression (C), and classical HbE-β+-thalassemia with increasing HbF expression (D).
The research protocol was approved by the Institutional Review Board (IRB) of the University of Phayao, Thailand (approval number:1.3/004/63), and all subjects were selectively recruited after obtaining informed consent. The study was performed in accordance with the relevant guidelines and regulations.
Molecular characterization of the globin genes and genotyping of modifying factors related to HbF expression
Genomic DNA was prepared from peripheral blood leukocytes using the proteinase K method with a standard protocol [Citation14]. β-thalassemia mutations were identified by direct DNA sequencing of the entire amplified β-globin gene, and the sequence was analyzed on an ABI PRISM™ 3130 XL analyzer (Applied Biosystems, Foster City, CA, USA), as described previously [Citation15]. In addition, seven common α-thalassemia comprising α0-thalassemia (SEA, Thai, and Chiang Rai deletions), deletional α+-thalassemia (3.7 and 4.2 base kilobase [kb] deletions), Hb Constant Spring (HbCS; HBA2: c.427T>C), and Hb Paksé (HBA2: c.429A>T) mutations were also identified using appropriate polymerase chain reaction (PCR) methodology [Citation16–19]. Samples with negative results for common α-thalassemia and the α1- and α2-globin gene mutations were characterized using PCR and direct DNA sequencing methods according to the manufacturer’s protocols, as described previously [Citation20]. The inherent factors that influence HbF expression were determined by studying the −158 (C-T) Gγ XmnI polymorphism (SNP rs7482144), a modifying factor strongly associated with increased HbF expression, using PCR-restriction fragment length polymorphism (PCR-RFLP) with XmnI restriction enzyme [Citation21].
Identification of non-deletional α+-thalassemia mutations by the PCR-RFLP technique
The C to G substitution at the cap site nucleotide (nt) 14 [HBA2:c.-24C>G (or HBA1)] and the deletion of A in the translation initiation codon (ATG→-TG) [HBA2:c.1delA] in the α2-globin gene abolished the BsmFI and FatI restriction sites, respectively. These two mutations were subsequently confirmed using polymerase chain reaction-restriction fragment length polymorphism. Amplification of the 1085-base pair (bp) fragment specific for the α2-globin gene was performed using the specific primers C1 (5′-TGGAGGGTGGAGACGTCCTG-3′) and C3 (5′-CCATTGTTGGCACATTCCGG-3′) [Citation22]. The amplified fragment was then digested to completion with BsmFI (5′-GGGAC(N)10▼-3′) and FatI (5′-▼CATG-3′) to identify the respective mutations. The restriction endonucleases BsmFI and FatI were purchased from New England Biolabs Inc. (Beverly, MA, USA). The digested fragments were analyzed using 2.0% agarose gel electrophoresis and visualized under ultraviolet light after ethidium bromide staining.
Statistical analysis
Descriptive statistics including mean, median, and standard deviation were used to describe the overall characteristics of the participants. The proportion of modifying factors related to HbF expression was compared between the two groups using Fisher’s exact test. Receiver operating characteristic (ROC) curve analysis was performed to determine the optimal cut-off values of HbA2, HbE, and HbF. The diagnostic accuracy of each parameter was assessed using the area under the curve (AUC), sensitivity, specificity, positive and negative predictive values, and Youden’s index (YI) for each cut-off value. Statistical and ROC curve analyses were performed using SPSS software (version 22.0; Armonk, NY, IBM Corp). A P-value of less significance was set at p < 0.05.
Results
The overall median age of the 2133 participants was 39 years (range: 1–98 years). Of these, 1946 patients were heterozygous for HbE, whereas the remaining 187 were completely diagnosed by DNA analysis. Molecular analysis of the β-globin mutations revealed that 79 patients (42.3%) carried β-thalassemia mutations identified trans to the βE allele. These included three mutations in the promoter region of the β-globin gene, nt-87(HBB:c.-137C > A), nt-31(HBB:c.-81A > G), and nt-28 (HBB:c.-78A > G), and two missense mutations, Hb Malay (HBB:c.59A > G) and Hb Dhonburi (HBB:c.380T > G) (). These mutations are responsible for β+-thalassemia [Citation23]. The proportion of HbE-β+-thalassemia patients identified according to our selection criteria (using HbF and HbE levels) is shown in . Among the 79 patients with HbE-β+-thalassemia, 35 (44.3%) had HbF levels less than 5.0% and HbE levels > 30.0%. The remaining 44 (55.7%) patients were identified in the group with HbF levels > 5.0% and HbE levels > 30.0% (). These results indicate that the patients in our study with HbE-β+-thalassemia had a wide-range expression of HbF and HbE of 0.0–40.7% and 38.7–69.4%, respectively. Based on HbF levels, these patients were categorized into two groups: low HbF levels (<10.0%, n = 54) and high HbF levels (≥10.0%, n = 25), as shown in . Statistical analysis of hematological parameters and Hb analysis demonstrated that Hb, hematocrit (Hct), mean corpuscular volume (MCV), and mean corpuscular Hb (MCH) of patients with HbE-β+-thalassemia were significantly lower than those of patients with heterozygous HbE. Furthermore, significantly increased red cell distribution width (RDW) was observed in patients with HbE-β+-thalassemia. These hematological parameters were similar to those observed in patients with low and high HbF levels. Interestingly, we found one patient carrying a β-genotype with βE/β−31(A>G) among the group with low HbF levels and secondary erythrocytosis (RBC count: 11.1 × 1012/L; Hb: 22.4 g/dL; Hct: 75.3%). This indicated the possibility of polycythemia vera (PV), as the values for the parameters were within the accepted range specified by the World Health Organization [Citation24]. Extensive testing of JAK2V167F and JAK2 exon 12 mutations, commonly found in PV, and the CALR gene mutation, rarely found in PV, was performed; however, these analyses were negative. Presumably, this patient had JAK2 unmutated erythrocytosis. Therefore, comprehensive molecular analysis to identify probable specific gene mutations of JAK2 unmutated erythrocytosis may provide a key for better diagnosis and explain the pathogenesis of the disease [Citation25]. However, further analysis in the search for these specific gene mutations could not be performed in this study.
Figure 2. The molecular characterization of the β-globin gene performed by DNA sequencing. Mutations in the promotor region of β-globin gene; (A) nt-87(HBB:c.-137C>a), (B) nt-31(HBB:c.-81A>G), (C) nt-28(HBB:c.-78A>G), (D) Hb Malay (beta 19(B1) Asn>ser; HBB:c.59A>G), and (E) Hb Dhonburi (beta 126(H4) Val>gly;HBB:c.380T>G).
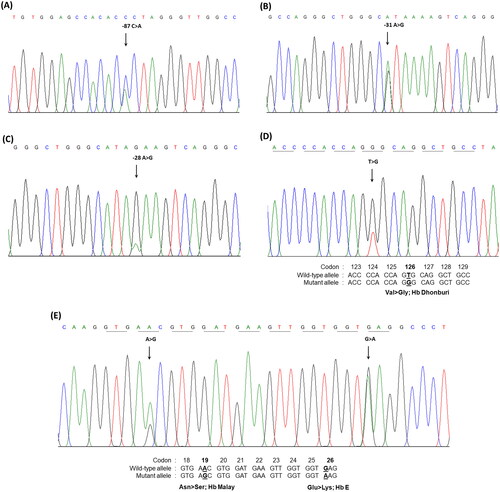
Figure 3. Distribution of HbF and HbE levels among the study population. The patients are Subdivided into four groups using the HbF and HbE levels obtained by capillary electrophoresis. Hb: hemoglobin.
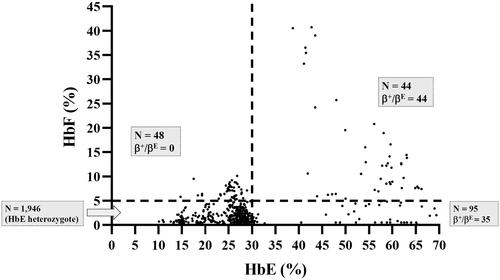
Table 1. Hematological parameters, HbE, HbA2, and HbF levels in HbE heterzygosity, HbE-β+-thalassemia with low HbF level, and HbE-β+-thalassemia with high HbF level. Data are expressed as means ± standard deviations (min-max).
The distributions of HbA2, HbE, and HbF levels in patients with HbE heterozygosity and HbE-β+-thalassemia (with low- and high-HbF levels) are shown in . The percentage of HbA2 observed among patients with HbE heterozygosity, low-HbF HbE-β+-thalassemia, and high-HbF HbE-β+-thalassemia was 3.4 ± 0.5, 7.0 ± 1.5, and 6.7 ± 1.3, respectively. The percentage of HbE in patients with HbE heterozygosity, low-HbF HbE-β+-thalassemia, and high-HbF HbE-β+-thalassemia was 25.2 ± 4.4, 57.6 ± 7.1, and 52.5 ± 8.3, respectively. The percentage of HbA2 and HbE in patients with HbE-β+-thalassemia, with both low and high HbF levels, was significantly higher than that in patients with HbE heterozygotes (p < 0.0001). HbA2 levels exhibited no statistically significant differences between HbE-β+-thalassemia patients with low and high HbF levels (p = 0.6057) (). However, the HbE levels of patients with HbE-β+-thalassemia with low HbF levels were significantly higher than those of patients with high HbF levels (p = 0.0169) (). Additionally, the percentage of HbF in patients with HbE-β+-thalassemia with low and high HbF levels was significantly higher than that in patients with HbE heterozygotes (). We observed that, although the HbF level was significantly different between patients with HbE heterozygosity and HbE-β+-thalassemia with low HbF levels, there was some overlap in HbF levels among these patients. The proportion of HbE-β+-thalassemia patients disguised with low HbF levels was 2.6% (54/2108). ROC curves were analyzed to investigate the cut-off values of HbA2, HbE, and HbF for ascertaining HbE-β+-thalassemia ( and ). The AUC for HbF level was 0.937 (95% confidence interval (CI):0.911–0.963), and the cutoff value was found to be 0.05, with a sensitivity of 97.5%, specificity of 20.2%, and Youden’s index of 17.7%. For the HbA2 level, the AUC was 0.998 (95% CI:0.997–1.000) and the cut-off value was found to be 4.25, with a sensitivity of 100.0%, specificity of 3.5%, and Youden’s index of 5.1%. Furthermore, the HbE level was found to be an excellent parameter with an AUC of 1.000 (95% CI:1.000–1.000), and the cut-off point was between 33.05 and 36.05, with 100.0% sensitivity, specificity, and Youden’s index. To accommodate the variation in Hb analysis results among different analyzers, we proposed a cut-off HbE level of 35.0%. Therefore, participants with an HbE level < 35.0% were likely to be HbE heterozygotes, whereas those with HbE levels ≥35.0% were suspected of having HbE-β+-thalassemia, requiring further mutation analysis.
Figure 4. Comparison of HbA2 levels (A), HbE levels (B), and HbF levels (C) quantified by the capillary electrophoresis system among patients with heterozygous HbE (1), HbE-β+-thalassemia with low HbF expression (2), and HbE-β+-thalassemia with high HbF expression (3). Hb: hemoglobin.
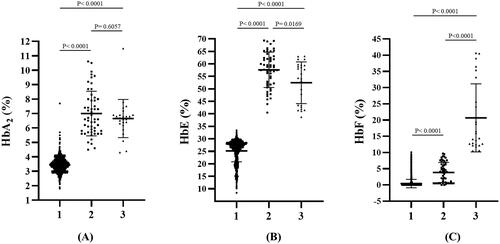
Figure 5. ROC analyses of the HbE, HbF, and HbA2 levels for differentiating patients with HbE-β+-thalassemia from HbE heterozygotes.
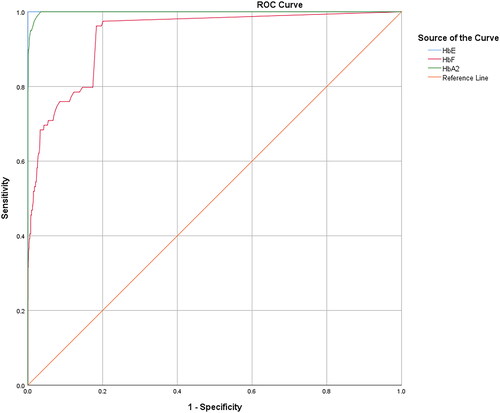
Table 2. Sensitivity, specificity, false positive rate (FP), false negative rate (FN), and youden’s index based on varying cutoffs of HbE, HbF, and HbA2 values.
Among the 79 patients with HbE-β+-thalassemia, various β-thalassemia mutations were identified in patients with low HbF levels compared with those with high HbF levels. The most common β+-thalassemia mutation was the nt-28(HBB:c.-78A > G) mutation, which was observed in 59.3 and 92.0% of the patients with low and high HbF levels, respectively. Other mutations with different frequencies and hematological parameters are presented in . The G to A transition in codon 19 and the T to G transition in codon 126 of the β-globin gene, corresponding to Hb Malay (HBB:c.59A > G) and Hb Dhonburi (HBB:c.380T > G), respectively, were identified in only seven patients with low HbF levels. These mutations produced a β-globin variant with the same migration time as HbA in the electropherogram (). The electropherogram was likely to be an HbE heterozygote owing to a fraction of HbE, and another Hb that seemed to be assigned as HbA was distinctly observed. Similarly, Hb analysis using HPLC methods (VARIANT II, Premier resolution) revealed a high peak of HbE (retention time (RT) 3.70–3.71 min for VARIANT II, and 5.45–5.47 min for Premier resolution) and a high-value peak of another Hb was eluted at RT 2.51–2.54 min (VARIANT II) and 4.66–4.69 min (Premier resolution), which was the position of HbA. This Hb pattern was observed in both variants ().
Figure 6. Hb analysis of Hb Malay (A-C) and Hb Dhonburi (D-F) using capillary electrophoresis (A&D), HPLC-VARIANT II (B&E), and HPLC-Premier resolution (C&F).
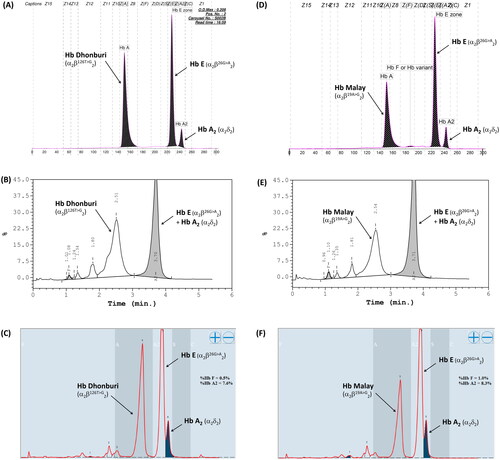
Table 3. β-globin mutations, frequency, hematological data, HbA2, HbE, and HbF levels in participants having HbE-β+-thalassemia with low- and high-HbF expression. Values are presented as means ± standard deviations or as raw data where appropriate.
DNA analysis of the α-globin gene showed α-thalassemia mutations in 46.3% (25/54) of patients with HbE-β+-thalassemia with low HbF levels and 8.0% (2/25) of those with high HbF levels. HbCS had the highest proportion of common α-thalassemia mutations found in the former groups. In addition, we also identified the compound heterozygous HbCS with 3.7 kb deletional α+-thalassemia and α-thalassemia 1 (SEA), the latter expressed the HbH-CS phenotype. Remarkably, no HbCS peaks were observed in the CE electropherogram of all patients with HbCS. An unexpected mutation, located in the promoter region of the α2-globin gene, Cap +14(C>G) [HBA2:c.-24C>G], was identified in two patients with HbE-β+-thalassemia with low HbF levels, whose β-globin genotype was βE/βMalay and βE/βDhonburi. This α-globin mutation has not previously been described in the Thai population. Additionally, a deletional A at the translation initiation codon of the α2-globin gene (ATG>-TG) [HBA2:c.1delA] was identified in one patient with HbE-β+-thalassemia with a low HbF level, whose β-globin genotype was βE/β−28. These mutations were confirmed by PCR-RFLP using BsmFI and FatI. As shownin , digestion of the 1085-bp fragment with BsmFI gave rise to three different fragments and the 983-bp fragment in the mutated allele, indicating the presence of the [HBA2:c.-24C>G (or HBA1)] mutation. Digestion with FatI resulted in four fragments; the 582-bp fragment indicating the presence of a mutation in the translation codon. The hematological parameters, HbE, HbA2, and HbF, of each corresponding HbE and α-globin genotype are summarized in . Remarkably, patients with HbE-β+-thalassemia with low and high HbF levels who had normal α-globin genes showed no differences in hematological parameters, HbE, and HbA2 levels.
Figure 7. Identification and Confirmation of the Cap +14(C>G) mutation and initiation codon (ATG>-TG) of the α2-globin gene by nucleotide sequencing and PCR-RFLP.
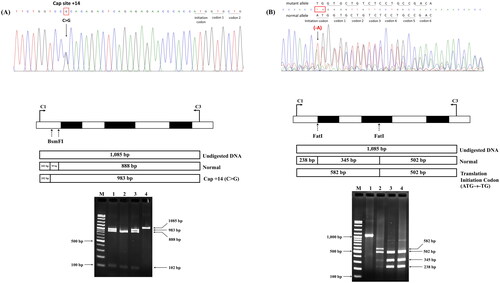
Table 4. α-globin genotypes, hematological parameters, HbE, HbA2, and HbF levels among HbE-β+-thalassemia with low- and high-HbF levels subjects. Values are presented as means ± standard deviations or as raw data where applicable.
The distribution of the genotypes and alleles of the −158 (C-T) Gγ XmnI polymorphism (SNP rs7482144) among the 79 patients with HbE-β+-thalassemia with low and high HbF levels is summarized in . The 54 patients with low HbF levels showed a relatively high proportion of the CT/TT genotype (66.7%), whereas the allelic C frequency was relatively high (63.9%). Among the 24 patients with high HbF levels, the CT/TT genotype was identified as relatively high (approximately 80.0%), and the C allele was identified in approximately 60% of patients. Comparison of the proportion of genotypes and alleles of the examined Gγ-XmnI polymorphism between the low and high HbF level groups revealed no statistically significant difference (p > 0.05).
Table 5. The genotypes and allele proportion of -158 (C-T) Gγ XmnI polymorphism among patients with HbE-β+-thalassemia with low- and high-HbF expression.
Discussion
HbE (beta26(B8)Glu > Lys;HBB:c.79G > A) is a β-thalassemic Hb variant whose frequency throughout Thailand is relatively high, together with a high prevalence of β-thalassemia [Citation4]. Thus, the coexistence of HbE with β-thalassemia leading to HbE-β-thalassemia disease is common in this country. The clinical and hematological severity of the disease are variable. The type of β-thalassemia mutation and DNA polymorphisms within the β-globin gene cluster determine the clinical severity [Citation8,Citation26,Citation27]. The characteristic phenotype of HbE-β+-thalassemia usually presents as mild-to-moderate anemia (Hb levels ranging from 7.0–9.0 g/dL), markedly low MCV and MCH, and elevated HbF expression (>10% of total Hb) with Hb analysis exhibiting an EFA pattern [Citation2,Citation26]. However, patients presented with major peaks of HbE and HbA on Hb analysis, quite similar to that of the HbE heterozygote, but the level of HbE was relatively high compared to subjects who were pure HbE heterozygotes. Furthermore, non-elevated HbF levels are regularly encountered in routine laboratories for thalassemia diagnosis. Based on the Hb analysis results, these patients were misdiagnosed as HbE heterozygotes or those with abnormal Hb, which had motility behavior similar to that of HbA. Therefore, the patient did not receive appropriate genetic counseling, treatment, or disease management.
In this study, 79 (3.7%) out of 2133 patients had varying HbF levels and β+-thalassemia mutations in trans to the βE-globin allele. Of these, 54 had relatively low HbF levels, whose Hb type was read as HbE and HbA (), while the remaining 25 patients had relatively high HbF levels, with HbE, HbA, and HbF (). This demonstrated that approximately 2.6% (54/2108) of the patients in our cohort could be misdiagnosed as HbE heterozygotes unless DNA analysis was performed. The hematological parameters of HbE-β+-thalassemia with both low and high HbF levels showed a more severe thalassemia syndrome phenotype, with average Hb concentrations <10 g/dL, markedly decreased MCV and MCH, and distinctly increased RDW-CV compared to HbE heterozygotes (). Nevertheless, differentiation of HbE-β+-thalassemia from HbE heterozygosity using hematologic parameters alone is challenging because we found that some patients with HbE-β+-thalassemia were not anemic or slightly anemic and had slightly abnormal red cell parameters, similar to those observed in HbE heterozygotes [Citation7,Citation28]. Remarkably, a markedly elevated HbE level of 38.7–69.4% was observed in patients who had HbE-β+-thalassemia, while HbE levels of approximately 8.4–33.4% were observed in HbE heterozygotes (). This is similar to the results of previous studies [Citation2,Citation7,Citation11]. Additionally, relatively high HbA2 levels, separated and calculated by CE, were observed in patients with HbE-β+-thalassemia (). Thus, Hb levels may be useful markers for differentiating HbE-β+-thalassemia from HbE heterozygotes. The ROC curves constructed to calculate the AUC for HbA2, HbE, and HbF revealed an excellent AUC (1.000) for HbE (, ). An accurate cut-off of ≥ 35.0% for HbE levels was proposed to differentiate patients with HbE-β+-thalassemia from HbE heterozygotes, which exhibited 100.0% sensitivity, specificity, and Youden’s index. Although this differentiation was not evaluated in independent prospective cases, our results showed that HbE level may be a robust indicator for differentiating patients with HbE-β+-thalassemia from HbE heterozygotes, which is simple, less time-consuming, and inexpensive to evaluate. This HbE level could be applied in clinical settings to screen cases for the possibility of HbE-β+-thalassemia requiring definite diagnosis by DNA analysis, leading to a significant reduction in the misdiagnosis of HbE heterozygosity and resulting in DNA analysis oversight.
The types of β-thalassemia mutations identified in the 79 patients with β+-thalassemia were similar to the β-thalassemia mutations most commonly identified previously in Thai β-thalassemia carriers and patients with HbE-β+-thalassemia [Citation15,Citation29]. Remarkably, two β-globin chain variants (Hb Malay and Hb Dhonburi) were identified in only seven patients with low HbF levels presenting the thalassemia intermedia phenotype (). This finding is concordant with previous reports [Citation2,Citation30–32]. Hb Malay is caused by a mutation in codon 19 (AAC > AGC) () and usually presents with a thalassemia intermedia phenotype when co-inherited with HbE and has more severe anemia when associated with β0-thalassemia [Citation2,Citation30,Citation31].
Hb Dhonburi or Hb Neapolis are caused by a GTG to GGG mutation at codon 126 associated with a mild β-thalassemia phenotype [Citation32], which is rarely reported in the Thai population. A previous study on the α/β globin mRNA ratio revealed that the average ratio observed in Hb Dhonburi carriers was relatively high (1.7) compared with that of the normal group (1.1), confirming that this variant is a β+-thalassemia [Citation2]. In our study, the three patients with coinheritance of HbE and Hb Dhonburi were presented with an intermediate phenotype with an RBC count of 4.7 ± 0.6 × 1012/L, Hb level of 8.0 ± 0.2 g/dL, and Hct of 26.0 ± 1.5. This result was similar to that observed in another Thai patient who coinherited HbE and Hb Dhonburi [Citation33]. Hb separation in Hb Malay and Hb Dhonburi using CE and HPLC (VARIANT II) was also studied. The results showed that they could not identify aberrant Hb representing Hb Malay and Hb Dhonburi in either HbE heterozygotes or HbE compound heterozygotes [Citation33]. However, we could separate these variants using the new HPLC (Premier resolution with high-resolution mode), which, to the best of our knowledge, has not been previously used and revealed that Hb Dhonburi eluted at the same RT as HbA (). Our results emphasize that these two variants are difficult to distinguish from HbA using Hb electrophoresis and HPLC because βMalay and βDhonburi globin chains have biochemical properties similar to those of HbA [Citation32].
In our study, variable amounts of HbF were detected in addition to HbE in patients with HbE-β+-thalassemia. However, similar β+-thal mutations were identified in 54 and 25 patients with markedly different HbF expression levels, respectively. Therefore, the β-thal mutation alone does not account for the observed wide expression of HbF, and other modifying genetic factors may be responsible. Previous studies have demonstrated that many genetic factors are associated with HbF expression in HbE-related disorders [Citation8–10,Citation26,Citation34]. The αβ-thalassemia mutation has also been explained as one of the factors contributing to HbF expression. As expected, we found a relatively high proportion of αβ-thalassemia mutations (approximately 46.3%) among the 54 patients with low HbF levels, whereas it was only 8.0% among the 24 patients with high HbF levels. It is noteworthy that the HbE level among patients co-inheriting the α-thalassemia mutation was consistently elevated, albeit with more pronounced defects in α-globin gene chain synthesis (). HbCS was the highest proportion with missing HbCS peak on the CE electropherogram. Interestingly, we found that two patients had complex thalassemia syndrome: one had co-inheritance of compound heterozygous HbCS with -α3.7, having genotype -α3.7/αCSα, βE/β−28, and the other had co-inheritance of compound heterozygous HbCS with α-thalassemia 1 (–SEA/αCSα, βE/β−28), diagnosed hematologically as CSEFABart’s disease, an uncommon form of thalassemia intermedia rarely observed in the Thai population. Defects in the three α-globin genes in this patient did not appear to affect HbE expression because of the consistently elevated HbE observed (). Interestingly, HbCS was not seen on the CE electropherogram from either patient, and additional HbBart’s (0.4%) were observed in the latter. Therefore, there is a possibility of misdiagnosis in these patients unless a complete DNA analysis is performed. Unexpectedly, we found that the Cap +14(C>G) mutation located in the promoter region of the α2-globin gene might be responsible for α+-thalassemia because of the partially abolished translation identified in two patients, one with the βE/βMalay genotype and the other with the βE/βDhonburi genotype. The Cap +14(C>G) mutation has been found in patients with Hb J-Meerut and Hb Winnipeg [α75(EF4)Asp→Tyr (α2); HBA2: c.226G > T (or HBA1)] [Citation35,Citation36], but this mutation has not yet been observed in the Thai population. To the best of our knowledge, this is the first report of doubly coinherited HbE with Hb Malay and Hb Dhonburi with the Cap +14(C>G) mutation causing the β-thalassemia intermedia phenotype in Thai patients. Additionally, we found that the ATG to -TG mutation in the initiation codon of the α2-globin gene completely abolished translation in the βE/β−28 patient without an apparent effect on HbE expression. This mutation causes α+-thalassemia, which has been reported in Thai patients with HbH [Citation37]. The interactions between β-thalassemia and different α-thalassemia mutations identified in patients with HbE-β+-thalassemia in our study led to more than nine genotypes (), which cause complex thalassemia syndromes, making accurate diagnosis with routine laboratory testing, using hematological parameters alone, difficult.
The markedly different proportion of α-thalassemia observed between patients with low and high HbF levels supports the hypothesis that α-thalassemia mutations have a favorable influence on HbF expression in patients with HbE-β+-thalassemia in our study. The coinheritance of α-thalassemia has been reported to be strongly associated with amelioration of clinical severity in HbE-β-thalassemia syndrome [Citation8,Citation26,Citation38–40]. Therefore, the patients in this study with low HbF levels, presenting with hematological features similar to those with high HbF levels, may have co-inherited α-thalassemia ( and ). Concomitant inheritance of α-thalassemia should lead to a more balanced β-globin and α-globin chain synthesis and should be responsible for decreased reactivation of the γ-globin gene in the disease. However, co-inheritance of α the-thalassemia mutation was not observed in 29 (53.7%) patients with low HbF levels, possibly because of other inherent factors associated with low HbF expression in those patients. Additionally, an investigation of the −158 (C-T) Gγ XmnI polymorphism revealed no significant difference in the proportion of the genotype and allele between patients with low and high HbF levels (). In our study, this polymorphism was less associated with HbF levels in patients with HbE-β+-thalassemia. However, we found a high proportion of the C allele (63.9% and 60.0%) among patients with low and high HbF levels, respectively, which was similar to that observed by Winichagoon et al. [Citation39]. The C allele is associated with β+-thalassemia. Our results emphasized that the C allele was associated with the type of βmutation rather than with HbF expression in patients with HbE-β+-thalassemia. Previous studies have demonstrated that in HbE-β-thalassemia, the T allele or T/T genotype is also associated with increased HbF and milder anemia [Citation6,Citation41]. However, in our study, the T allele was identified in a relatively low proportion of patients with both low and high HbF levels (). Overall, our results indicate that α-thalassemia mutations favorably influenced HbF levels in patients with HbE-β+-thalassemia and increased susceptibility to low HbF levels. However, additional unknown modulating factors in patients require further investigation.
Conclusion
HbE-β+-thalassemia has a widely variable expression of HbF, and many patients whose Hb type is read as HbE and/or HbA can be misdiagnosed with HbE-β+-thalassemia. Therefore, molecular analysis of the β-globin gene should be performed to improve the accuracy of diagnosis. We also found that α-thalassemia mutations are a common factor favorably decreasing HbF expression, resulting in complex thalassemia syndromes requiring complete DNA analysis; therefore, the presence of α-thalassemia mutations should always be assessed to provide complete genetic information. Furthermore, an HbE cutoff value of ≥35.0% may be a potential indicator for effectively differentiating HbE-β+-thalassemia from HbE heterozygosity. Patients whose Hb analysis showed HbE and HbA and elevated HbE, regardless of the amount of HbF, could not be concluded to have HbE heterozygosity unless DNA analysis was performed. This study provides details regarding HbE-β+-thalassemia that may help in diagnosis, especially in areas where HbE and β-thalassemia are both prevalent.
Consent to publish
Consent to publish patient details was obtained from all individuals included in the study.
Authors’ contributions
Jomoui W: acquisition of the grant, performed investigations, identified the modifying factors associated with Hb F expression, analyzed the data and statistics, Satthakarn S performed the α- and β-globin gene and statistics. Panyasai S: acquired the grant, designed the study, performed investigations, developed methods, analyzed the data, and wrote, reviewed, and edited the manuscript. All the authors have read and approved the final manuscript.
Acknowledgements
We wish to express our appreciation to Biozen Co., Ltd. (Bangkok, Thailand) for their help with Hb analysis using the Premier Resolution HPLC system.
Disclosure statement
No potential conflict of interest was reported by the author(s). The authors of this article and the planning committee members and staff have no relevant financial relationships with the commercial interests in the products or companies described in this article.
Data availability statement
Data supporting the findings of this study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Weatherall DJ, Clegg JB. The thalassemia syndromes., 4th ed. Blackwell Science, Oxford, 2001;Database]
- Yamsri S, Singha K, Prajantasen T, et al. A large cohort of β(+)-thalassemia in Thailand: molecular, hematological and diagnostic considerations. Blood Cells Mol Dis. 2015;54(2):1–14. doi: 10.1016/j.bcmd.2014.11.008.
- Vichinsky E. Hemoglobin E syndromes. Hematology Am Soc Hematol Educ Program. 2007;2007(1):79–83. doi: 10.1182/asheducation-2007.1.79.
- Fucharoen S, Winichagoon P. Hemoglobinopathies in Southeast Asia. Hemoglobin. 1987;11(1):65–88. doi: 10.3109/03630268709036587.
- Fucharoen S, Winichagoon P. Haemoglobinopathies in Southeast Asia. Indian J Med Res. 2011;134(4):498–506.
- Winichagoon P, Thonglairoam V, Fucharoen S, et al. Severity differences in beta-thalassaemia/haemoglobin E syndromes: implication of genetic factors. Br J Haematol. 1993;83(4):633–639. doi: 10.1111/j.1365-2141.1993.tb04702.x.
- Sae-Ung N, Srivorakun H, Fucharoen G, et al. Phenotypic expression of hemoglobins A2, E and F in various hemoglobin E related disorders. Blood Cells Mol Dis. 2012;48(1):11–16. doi: 10.1016/j.bcmd.2011.09.008.
- Yamsri S, Pakdee N, Fucharoen G, et al. Molecular understanding of non-transfusion-dependent thalassemia associated with hemoglobin E-β-thalassemia in Northeast Thailand. Acta Haematol. 2016;136(4):233–239. doi: 10.1159/000449120.
- Jomoui W, Tepakhan W, Yamsri S, et al. A novel SNP rs11759328 on Rho GTPase-activating protein 18 gene is associated with the expression of Hb F in hemoglobin E-related disorders. Ann Hematol. 2020;99(1):23–29. doi: 10.1007/s00277-019-03862-0.
- Tepakhan W, Yamsri S, Sanchaisuriya K, et al. Nine known and five novel mutations in the erythroid transcription factor KLF1 gene and phenotypic expression of fetal hemoglobin in hemoglobin E disorder. Blood Cells Mol Dis. 2016;59:85–91. doi: 10.1016/j.bcmd.2016.04.010.
- Fucharoen S, Winichagoon P. Clinical and hematologic aspects of hemoglobin E beta-thalassemia. Curr Opin Hematol. 2000;7(2):106–112. doi: 10.1097/00062752-200003000-00006.
- Fucharoen S, Weatherall DJ. The hemoglobin E thalassemias. Cold Spring Harb Perspect Med. 2012;2(8):a011734–a011734. doi: 10.1101/cshperspect.a011734.
- Sanchaisuriya K, Chirakul S, Srivorakun H, et al. Effective screening for double heterozygosity of Hb E/alpha0-thalassemia. Ann Hematol. 2008;87(11):911–914. doi: 10.1007/s00277-008-0520-x.
- Ahmad NN, Cu-Unjieng AB, Donoso LA. Modification of standard proteinase K/phenol method for DNA isolation to improve yield and purity from frozen blood. J Med Genet. 1995;32(2):129–130. doi: 10.1136/jmg.32.2.129.
- Sirichotiyakul S, Saetung R, Sanguansermsri T. Analysis of beta-thalassemia mutations in Northern Thailand using an automated fluorescence DNA sequencing technique. Hemoglobin. 2003;27(2):89–95. doi: 10.1081/hem-120021541.
- Sae-Ung N, Fucharoen G, Sanchaisuriya K, et al. Alpha(0)-thalassemia and related disorders in northeast Thailand: a molecular and hematological characterization. Acta Haematol. 2007;117(2):78–82. doi: 10.1159/000096857.
- Fucharoen S, Fucharoen G, Sanchaisuriya K, et al. Molecular analysis of a Thai beta-thalassaemia heterozygote with normal haemoglobin A2 level: implication for population screening. Ann Clin Biochem. 2002;39(Pt 1):44–49. doi: 10.1258/0004563021901720.
- Fucharoen S, Sanchaisuriya K, Fucharoen G, et al. Interaction of hemoglobin E and several forms of alpha-thalassemia in cambodian families. Haematologica. 2003;88(10):1092–1098.
- Ruengdit C, Khamphikham P, Jinorose N, et al. Hb bart’s hydrops fetalis syndrome and Hb H disease caused by deletional Chiang Rai (- -CR) α0-Thalassemia in two unrelated Thai families. Hemoglobin. 2021;45(2):75–79. doi: 10.1080/03630269.2021.1906269.
- Khalil MS, Timbs A, Henderson S, et al. Haemoglobin (Hb) G-Philadelphia, Hb Stanleyville-II, Hb G-Norfolk, Hb Matsue-Oki and Hb Mizushi can form a panel of α-chain variants that overlap in their phenotype: the novel use of StyI to screen for Hb G-Philadelphia. Int J Lab Hematol. 2011;33(3):318–325. doi: 10.1111/j.1751-553X.2010.01289.x.
- Fucharoen S, Shimizu K, Fukumaki Y. A novel C-T transition within the distal CCAAT motif of the G gamma-globin gene in the Japanese HPFH: implication of factor binding in elevated fetal globin expression. Nucleic Acids Res. 1990;18(17):5245–5253. doi: 10.1093/nar/18.17.5245.
- Kattamis AC, Camaschella C, Sivera P, et al. Human alpha-thalassemia syndromes: detection of molecular defects. American J Hematol. 1996;53(2):81–91. doi: 10.1002/(SICI)1096-8652(199610)53:2≤81::AID-AJH5≥.0.CO;2-#.
- Singha K, Taweenan W, Fucharoen G, et al. Erythrocyte indices in a large cohort of β-thalassemia carrier: implication for population screening in an area with high prevalence and heterogeneity of thalassemia. Int J Lab Hematol. 2019;41(4):513–518. doi: 10.1111/ijlh.13035.
- Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi: 10.1182/blood-2016-03-643544.
- Gangat N, Szuber N, Pardanani A, et al. JAK2 unmutated erythrocytosis: current diagnostic approach and therapeutic views. Leukemia. 2021;35(8):2166–2181. doi: 10.1038/s41375-021-01290-6.
- Nuntakarn L, Fucharoen S, Fucharoen G, et al. Molecular, hematological and clinical aspects of thalassemia major and thalassemia intermedia associated with Hb E-beta-thalassemia in Northeast Thailand. Blood Cells Mol Dis. 2009;42(1):32–35. doi: 10.1016/j.bcmd.2008.09.002.
- Nuinoon M, Makarasara W, Mushiroda T, et al. A genome-wide association identified the common genetic variants influence disease severity in beta0-thalassemia/hemoglobin E. Hum Genet. 2010;127(3):303–314. doi: 10.1007/s00439-009-0770-2.
- Sanchaisuriya K, Fucharoen G, Sae-Ung N, et al. Molecular and hematologic features of hemoglobin E heterozygotes with different forms of alpha-thalassemia in Thailand. Ann Hematol. 2003;82(10):612–616. doi: 10.1007/s00277-003-0689-y.
- Satthakarn S, Panyasai S, Pornprasert S. Molecular characterization of β- and α-globin gene mutations in individuals with borderline Hb A2 levels. Hemoglobin. 2020;44(5):349–353. doi: 10.1080/03630269.2020.1826327.
- Yang KG, Kutlar F, George E, et al. Molecular characterization of beta-globin gene mutations in Malay patients with Hb E-beta-thalassaemia and thalassaemia major. Br J Haematol. 1989;72(1):73–80. doi: 10.1111/j.1365-2141.1989.tb07655.x.
- Ma SK, Chow EY, Chan AY, et al. Beta-thalassemia intermedia caused by compound heterozygosity for Hb Malay (beta codon 19 AAC–>AGC; asn–>Ser) and codons 41/42 (-CTTT) beta(0)-thalassemia mutation. American J Hematol. 2000;64(3):206–209. doi: 10.1002/1096-8652(200007)64:3≤206::aid-ajh12≥3.0.co;2-#.
- Bardakdjian-Michau J, Fucharoen S, Delanoe-Garin J, et al. Hemoglobin Dhonburi alpha 2 beta 2 126 (H4) Val–Gly: a new unstable beta variant producing a beta-thalassemia intermedia phenotype in association with beta zero-thalassemia. Am J Hematol. 1990;35(2):96–99. doi: 10.1002/ajh.2830350206.
- Viprakasit V, Chinchang W. Two independent origins of Hb Dhonburi (Neapolis) [beta 126 (H4) val–>gly]: an electrophoretically silent hemoglobin variant. Clin Chim Acta. 2007;376(1-2):179–183. doi: 10.1016/j.cca.2006.08.016.
- Tepakhan W, Yamsri S, Fucharoen G, et al. Krüppel-like factor 1 mutations and expression of hemoglobins F and A2 in homozygous hemoglobin E syndrome. Ann Hematol. 2015;94(7):1093–1098. doi: 10.1007/s00277-015-2335-x.
- Khalil MSM, Timbs AT, Henderson SJ, et al. Fifteen cases of Hb J-Meerut: the rare association with Hb E and/or HBA1: c. 24C>G (or HBA2) variants. Hemoglobin. 2020;44(5):364–367. doi: 10.1080/03630269.2020.1817755.
- Khalil MSM, Timbs AT, Henderson SJ, et al. Eight cases of Hb Winnipeg [HBA2: c.226G > T (or HBA1)]: a detailed study. Hemoglobin. 2021;45(4):256–258. doi: 10.1080/03630269.2021.1976203.
- Viprakasit V, Ekwattanakit S, Chalaow N, et al. Clinical presentation and molecular identification of four uncommon alpha globin variants in Thailand. Initiation codon mutation of α2-globin gene (HBA2:c.1delA), donor splice site mutation of α1-globin gene (IVSI-1, HBA1:c.95 + 1G > a), hemoglobin Queens Park/Chao pra Ya (HBA1:c.98T > A) and hemoglobin Westmead (HBA2:c.369C>G). Acta Haematol. 2014;131(2):88–94. doi: 10.1159/000353119.
- Winichagoon P, Fucharoen S, Chen P, et al. Genetic factors affecting clinical severity in beta-thalassemia syndromes. J Pediatr Hematol Oncol. 2000;22(6):573–580. doi: 10.1097/00043426-200011000-00026.
- Winichagoon P, Fucharoen S, Weatherall D, et al. Concomitant inheritance of alpha-thalassemia in beta 0- thalassemia/Hb E disease. Am J Hematol. 1985;20(3):217–222. doi: 10.1002/ajh.2830200303.
- Olivieri NF, Pakbaz Z, Vichinsky E. HbE/β-thalassemia: basis of marked clinical diversity. Hematol Oncol Clin North Am. 2010;24(6):1055–1070. doi: 10.1016/j.hoc.2010.08.008.
- Mohammad SNNA, Iberahim S, Wan Ab Rahman WS, et al. Single nucleotide polymorphisms in XMN1-HBG2, HBS1L-MYB, and BCL11A and their relation to high fetal hemoglobin levels that alleviate anemia. Diagnostics. 2022;12(6):1374. doi: 10.3390/diagnostics12061374.