Abstract
There is abundant evidence that the endothelium plays a crucial role in the maintenance of vascular tone and structure. One of the major endothelium‐derived vasoactive mediators is nitric oxide (NO), an endogenous messenger molecule formed in healthy vascular endothelium from the amino acid precursor L‐arginine. Endothelial dysfunction is caused by various cardiovascular risk factors, metabolic diseases, and systemic or local inflammation. One mechanism that explains the occurrence of endothelial dysfunction is the presence of elevated blood levels of asymmetric dimethylarginine (ADMA) – an L‐arginine analogue that inhibits NO formation and thereby can impair vascular function. Supplementation with L‐arginine has been shown to restore vascular function and to improve the clinical symptoms of various diseases associated with vascular dysfunction.
| Abbreviations | ||
| ACS | = | acute coronary syndrome |
| ADMA | = | asymmetric dimethylarginine |
| DDAH | = | dimethylarginine dimethylaminohydrolase |
| ICU | = | intensive care unit |
| L‐NMMA | = | NG‐monomethyl‐L‐arginine |
| NO | = | nitric oxide |
| PRMT | = | protein‐arginine methyltransferase |
| SDMA | = | symmetric dimethylarginine |
Introduction
Methylation of the amino acid L‐arginine was discovered decades ago as a mechanism of post‐translational modification of proteins. It has been shown that within proteins, only L‐arginine residues but not free amino acid can be methylated, and that a family of enzymes named protein‐arginine‐methyltransferases catalyze this reaction Citation1,2. Methylation has been shown to affect diverse proteins such as histone proteins, myelin proteins, and many others Citation3, resulting in modified physicochemical properties and consequently, in modified functions of these proteins Citation3. Methylation of L‐arginine was shown to result in three different derivatives of this amino acid: the asymmetric isomers monomethyl‐L‐arginine (L‐NMMA) and asymmetric dimethylarginine (ADMA), as well as the symmetric dimethylarginine (SDMA). All of these L‐arginine analogues are endogenously present in the human body; however, due to its very low concentration, L‐NMMA probably plays no role in regulating NO production in vivo. Enzymes producing asymmetrically methylated arginines are called type 1 protein‐arginine methyltransferases (PRMTs), whereas enzymes that produce symmetrically methylated arginine are called type 2 PRMTs (for an overview of methyltransferases see Citation4,5). gives a schematic overview of ADMA biosynthesis and metabolism.
Figure 1. Schematic overview of biochemical pathways related to ADMA. Methylation of arginine residues within proteins or polypeptides occurs through N‐methyltransferases, which utilize S‐adenosylmethionine as a methyl group donor. After proteolytic breakdown of proteins, free ADMA is present in cytoplasm. It can also be detected circulating in human blood plasma. ADMA acts as an inhibitor of NO synthase by competing with the substrate of this enzyme, L‐arginine, and causes endothelial dysfunction and – subsequently – atherosclerosis. ADMA is eliminated from the body via urinary excretion and, alternatively, via metabolism by the enzyme dimethylarginine dimethylaminohydrolase (DDAH) to citrulline and dimethylamine. Reproduced from Citation17.
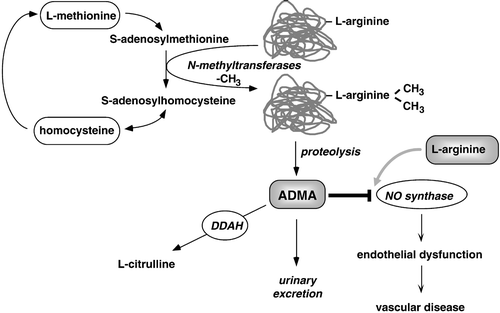
At the time when these biochemical pathways were discovered, it was not yet known that L‐arginine is the substrate of another important enzymatic pathway, namely the NO synthase pathway. L‐arginine acts as the natural precursor for the formation of nitric oxide (NO) in the endothelium and in other tissues Citation6. This reaction can be competitively inhibited by synthetic L‐NMMA, as first shown by Rees and co‐workers in 1989 Citation7.
Only in 1992 did it become clear, by the groundbreaking discovery by Patrick Vallance and colleagues Citation8, that endogenous asymmetric dimethylarginine acts as a competitive inhibitor of NO synthase. These investigators detected ADMA (and, in much lower concentrations, L‐NMMA) in human plasma and urine, and they were able to show that it inhibits endothelium‐dependent vasodilation in vitro. Moreover, they also showed that intra‐arterial infusion of ADMA results in an increased arterial tone, supposedly by blocking vasodilation mediated by endogenous NO. Their most intriguing finding, however, was that ADMA accumulates in patients with renal failure by several‐fold, suggesting that ADMA may be responsible in part for the high prevalence of hypertension and atherosclerosis associated with chronic renal failure. We and others later confirmed this finding in larger clinical studies of patients with different types of renal function impairment (reviewed in Citation9).
Since 1992, much progress has been made by an increasing number of researchers in the understanding of the functional roles of ADMA in the cardiovascular system and its role as a prognostic marker of morbidity and mortality. This review will give an updated overview of the current data on ADMA with a major focus on clinical data from prospective and cross‐sectional clinical studies.
Key messages
ADMA is asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase.
By reversibly blocking NO production from L‐arginine, ADMA induces endothelial dysfunction, which may result in the initiation and progression of atherosclerotic vascular disease.
Prospective clinical studies in various patient populations have suggested a role for ADMA as a novel cardiovascular risk factor. In these studies, elevated ADMA was associated with a mean 3‐ to 5‐fold increase in risk of experiencing a serious cardiovascular event.
ADMA is a regulator of endothelial nitric oxide (NO) formation
NO is involved in a wide variety of regulatory mechanisms of the cardiovascular system, including vascular tone. In fact, it is regarded by many as the major mediator of endothelium‐dependent vasodilation. It also regulates vascular structure (inhibition of smooth muscle cell proliferation) and cell‐cell interactions in blood vessels (inhibition of platelet adhesion and aggregation; inhibition of monocyte adhesion). Thereby, NO plays a crucial role in the endothelium‐mediated regulation of vascular homeostasis Citation10. Dysfunction of the endothelial L‐arginine/nitric oxide pathway is a common mechanism by which several cardiovascular risk factors mediate certain deleterious effects on the vascular wall. Among these are hypercholesterolemia, hypertension, smoking, diabetes mellitus, homocysteine, and vascular inflammation Citation11–16.
Currently available experimental and clinical evidence suggests that even small modifications of circulating ADMA levels significantly change vascular NO production, vascular tone, and systemic vascular resistance (for review, see Citation17). This has recently been evidenced in a mouse model in which the gene for dimethylarginine dimethylaminohydrolase (DDAH‐1), the enzyme that inactivates ADMA Citation18, was genetically overexpressed Citation19. DDAH‐1 transgenic mice displayed decreased ADMA levels, enhanced vascular NO production and lowered arterial blood pressure and systemic vascular resistance. The opposite animal model, i.e. mice in which one of the DDAH genes is lacking, is currently being generated.
Interestingly, there are a few small experimental studies in humans in which ADMA was administered in vivo in order to investigate the effects on haemodynamic parameters. Even in their first report on ADMA as an endogenous inhibitor of NO synthesis, Vallance and co‐workers reported that intra‐arterial infusion of ADMA into the brachial artery led to significantly reduced forearm blood flow Citation8. This group of investigators later confirmed their findings in a more detailed study with a larger group of subjects Citation20. However, concentrations of ADMA reached in the local circulation after intra‐arterial infusion outreach by far those that can be measured in patients. For that reason, two studies in which ADMA was administered intravenously added data to the dose‐response relationship of ADMA in humans. In one study, intravenous infusion led to circulating plasma ADMA concentrations of about 2.6 µmol/L Citation21. At this concentration level, ADMA caused a significant elevation of arterial blood pressure and systemic vascular resistance and reduced cardiac output, heart rate and the vascular response to exercise. In another study by Kielstein and co‐workers Citation22, ADMA was administered intravenously to healthy human subjects at different concentrations. These investigators found that ADMA decreased plasma cyclic GMP concentrations at pathophysiologically relevant concentrations, whereas renal plasma flow and glomerular filtration rate were only reduced at very high concentrations.
In yet another study, Chan and co‐workers studied the adhesion of monocytes isolated from peripheral blood to cultured human endothelial cells. They found that the adhesiveness index was related to the circulating ADMA level of the patient from whom monocytes were isolated. Moreover, when they influenced the ratio of L‐arginine over ADMA by administering L‐arginine, they found that monocyte adhesion normalized during 12 weeks of treatment Citation23.
ADMA is a novel cardiovascular risk factor
After the initial characterization of ADMA as endogenous inhibitor of NO synthase by Vallance et al. Citation8, we have been among the first to demonstrate that circulating ADMA levels are elevated in patients with systemic atherosclerosis Citation24, in clinically healthy human subjects with isolated hypercholesterolaemia Citation15, in patients with essential hypertension Citation25, and in patients with renal failure Citation26. The list of clinical conditions in which elevated ADMA levels were found has kept growing longer (). Most recent data suggest a pathophysiological role for ADMA in limiting NO availability in sickle cell disease Citation27, and in pulmonary hypertension. Gorenflo et al. Citation28 were the first who reported elevated ADMA levels in patients with congenital heart disease and pulmonary hypertension. These findings were extended by showing that DDAH activity is dysregulated in chronic hypoxia‐induced pulmonary hypertension Citation29 and increased levels and reduced catabolism of ADMA in patients with idiopathic pulmonary hypertension. Kielstein et al. Citation30 demonstrated that elevated ADMA levels in idiopathic pulmonary hypertension might be of prognostic relevance.
Table I. Pathophysiological conditions in which ADMA was reported to be elevated.
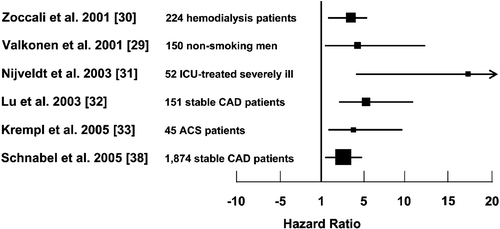
In addition, several prospective studies in which surrogate endpoints for cardiovascular disease were used have supplied evidence of a pathophysiological role of ADMA in the pathogenesis of vascular dysfunction and cardiovascular disease in humans. High ADMA levels were found to be associated with carotid artery intima‐media thickness in a study with 116 clinically healthy human subjects Citation31. Taking this observation further in a prospective design, another study performed in haemodialysis patients reported that ADMA predicts the progression of intimal thickening during one year of follow‐up Citation32. In a nested, case‐control study involving 150 middle‐aged, non‐smoking men, high ADMA levels were associated with a 3.9‐fold elevated risk for acute coronary events Citation33.
In 2001, we published the first prospective clinical trial on ADMA as a predictor of cardiovascular outcome and total mortality in 225 haemodialysis patients during three years of follow‐up Citation34. In this study, patients whose ADMA levels were within the highest quartile at the beginning of the study had a three‐fold higher risk of death from any cause than patients with ADMA levels below the median.
Another study investigated factors related to outcome of patients undergoing intensive care unit treatment for multiple causes. Patients whose ADMA levels were in the highest quartile had a 17‐fold increase in mortality compared to patients with ADMA levels in the lowest quartile Citation35. In a third prospective study, the outcome of patients with stable angina pectoris after coronary balloon angioplasty was addressed Citation36. One hundred and fifty three patients with stable angina pectoris who underwent elective coronary angioplasty were included in the study. Patients were stratified into tertiles of pre‐procedural ADMA and followed for a median of 16 months. A total of 51 major cardiovascular events occurred during follow‐up. Interestingly, in a multifactorial Cox`s regression analysis (including age, smoking, hypercholesterolemia, use of stent and others), the risk of a cardiovascular event increased with ADMA levels and was independent of other risk factors.
We have recently added to available data by showing that ADMA is higher in patients with acute coronary syndrome than in patients with stable angina pectoris. In a prospective follow‐up, patients with unstable coronary syndrome whose ADMA declined within six weeks after the acute episode to levels similar to those of patients with stable coronary artery disease experienced fewer major cardiovascular events during one year of follow‐up than those whose ADMA had remained elevated Citation37.
In an analysis of 1,874 patients from the AtheroGene study population elevated ADMA concentration was highly significantly and independently associated with increased risk of major cardiovascular events during a mean 2.6 years of follow‐up Citation38.
summarizes available data on ADMA as a cardiovascular risk factor from prospective clinical studies. In each of these studies, other cardiovascular risk factors and confounding variables were included in the analyses, and ADMA was found to be an independent predictor of cardiovascular events. Thus, it has recently been concluded that ADMA can be considered to be a novel cardiovascular risk factor Citation17,Citation39,40.
Figure 2. Results from prospective clinical studies that have assessed ADMA as a risk marker. Data represent hazard ratios(boxes) and 95% confidence intervals (horizontal lines) for the comparison of the highest ADMA stratum with the reference group.
Mechanisms that link ADMA with cardiovascular disease
The main mechanism of action by which ADMA exerts its effects is thought to be competitive inhibition of NO synthases. Thereby, ADMA concentration‐dependently reduces NO formation and impairs the biological effects of NO in the vasculature, all of which are thought to be protective under healthy conditions. Besides direct interference with eNOŚs catalytic mechanism, experimental evidence suggests that in the presence of high ADMA levels, eNOS may be uncoupled. This means that the usual flow of electrons among the two domains of eNOS is disturbed, and eNOS becomes a generator of superoxide radicals instead of NO Citation41. A similar phenomenon has been described for eNOS under insufficient concentrations of its essential co‐factor, tetrahydrobiopterin, insufficient availability of its substrate, L‐arginine, or in the presence of the NOS inhibitor, L‐NMMA Citation42. This mechanism suggests elevated levels of oxidative stress in the presence of high ADMA concentrations, and may also explain ADMÀs detrimental effects on endothelial function. L‐arginine, the natural substrate of NO synthase, is taken up into cells by the cationic amino acid transporter system y+Citation43. This amino acid transporter has been shown to be inhibited by high concentrations of ADMA Citation44. Although the concentrations that were found to be effective in vitro are extremely high in comparison to ADMA levels circulating in vivo, the in situ concentrations in the endothelial lining are not precisely known, and levels within the endothelial cells have been estimated to be 8‐ to 10‐fold higher than circulating levels Citation45,46. Thus, it may well be that cellular L‐arginine uptake is impaired in patients with certain cardiovascular diseases in which ADMA is elevated. In support of this hypothesis, cellular L‐arginine uptake has been shown to be reduced in hypertensive patients Citation47, who are characterized by high ADMA levels Citation25,Citation48.
Inhibition of NO production or action, whichever mediates ADMA's effects, results in a shift of the homeostatic vascular balance between vasodilation and vasoconstriction, in activation of platelets and depression of cell‐cell interactions, in pro‐ and antiproliferative impacts, in pro‐ and antioxidant stimuli, and may contribute to a pro‐inflammatory state of the vascular wall, thereby promoting atherosclerosis.
Recently ADMA has been found to mediate the anti‐angiogenic effects of hypercholesterolaemia in a mouse model Citation49. In mice overexpressing human dimethylarginine, dimethylaminohydrolase reduced circulating levels of ADMA were associated with enhanced angiogenesis Citation50. These studies suggest a role for ADMA as a regulator of angiogenesis.
However, a recent study challenged the conception that all of ADMÀs effects are mediated via inhibition of NO action by showing that chronic administration of ADMA via osmotic minipumps induces similar vascular lesions in wild‐type and eNOS knockout mice Citation51. These authors reported upregulation of angiotensin converting enzyme activity during ADMA administration, and inhibition of ADMA‐induced lesion formation by angiotensin receptor blockade Citation51.
Association of ADMA with clinical conditions beyond cardiovascular medicine
Although the major focus in the research on the pathophysiological effects of ADMA has been in cardiovascular medicine, ADMA has been found to be associated with other clinical conditions as well.
ADMA has been shown to be elevated in severe liver failure. Tsikas and co‐workers demonstrated an about 10‐fold increase in circulating ADMA in eight patients with end‐stage liver disease compared with healthy controls Citation52. Siroen and colleagues Citation53 found elevated ADMA concentration in 42 patients with acute or chronic hepatic failure compared with healthy volunteers. After liver transplantation, ADMA levels immediately decreased; however, in patients who experienced a period of acute graft rejection, post‐transplantation ADMA levels were higher than in non‐rejectors. The latter group of investigators hypothesized elevation of ADMA due to impaired elimination by the kidney and the liver as one possible causative factor of multi‐organ failure Citation54.
There is one report from a clinical study in patients with hyperthyroidism Citation55. These patients had elevated ADMA concentration and diminished L‐arginine/ADMA ratio compared with euthyroid controls, which reflected a decreased NO production. Hyperthyroidism may, by inducing a catabolic state, accelerate systemic protein turnover and thereby lead to increased release of methylated arginine residues from methylated proteins. However, this hypothesis has not yet been proven.
Insulin resistance and metabolic syndrome are two important clinical entities which will have a tremendous impact on population health in the Western world in the near future. Both pre‐diabetic subjects with impaired glucose tolerance Citation56 and patients with metabolic syndrome Citation57 have been demonstrated to have elevated ADMA levels. Thus, ADMA is suggested to promote the cardiovascular complications of these metabolic diseases.
Preeclampsia is another clinical condition that is associated with impaired endothelial function, poor prognosis and ill‐explained pathophysiology. Several studies in recent years have shown elevated ADMA levels in patients with preeclampsia compared with healthy pregnant women Citation58,59. However, this finding is again restricted to women from the Western countries, as in one study with Colombian women, ADMA was found not to be different between preeclamptic and healthy pregnancies Citation60.
Finally, early studies on the biochemistry of methylated arginines have shown that neuronal proteins are methylated to a large extent Citation4. However, there appears to be more SDMA than ADMA in the central nervous system, due to the higher expression of protein methyltransferase type II enzymes Citation4. Accordingly, data from clinical studies with patients suffering from neurodegenerative diseases have been inconclusive so far: in one study, ADMA levels in the cerebrospinal fluid were significantly higher in patients with Alzheimer`s disease than in neurologically normal controls Citation61. By contrast, Mulder and colleagues Citation62 found no difference in cerebrospinal fluid ADMA levels between 20 early stage Alzheimer`s disease patients and 20 age‐matched controls. Selley Citation63 reported a highly significant elevation of ADMA plasma levels in 25 Alzheimer`s disease patients compared with 25 control subjects. This was associated with reduced plasma nitrate and nitrite levels, suggesting reduced NO production in these patients. However, according to these studies, the relationship between neurodegenerative diseases and ADMA appears uncertain.
Measurement of ADMA in human subjects: methodological considerations
Research into ADMA has been hampered in the past by the insufficient comparability of values generated with different analytical methods, which were mainly based upon HPLC technology. ADMA levels determined with different HPLC methods sometimes yielded grossly different results Citation9. The main reason for this was the protein precipitation steps, which were performed by most but not all laboratories. Protein precipitation results in lower ADMA levels, probably because only the non‐protein‐bound portion of circulating ADMA is assessed. Indeed, circulating ADMA is bound to a considerable degree to plasma proteins Citation64. Since 2002, an international research initiative has undertaken a comparison of analytical methods, in order to allow the comparison of data measured in different laboratories around the world Citation65. Moreover, novel methods have been developed which allow quantitation of ADMA with higher precision and accuracy Citation65. Among these, mass spectrometric methods like LC‐MS/MS and GC‐MS may be the analytical ‘gold standard’ if they use a true (stable isotope‐labelled) internal standard Citation66; however, all of these methods are laborious, not available in many laboratories, and therefore not readily applicable for routine diagnostic use. More available, less time consuming and therefore more affordable methods to measure ADMA are clearly needed to make ADMA determination a routine clinical tool. With the development and validation of a novel competitive enzyme immunoassay that allows to sensitively and specifically assess ADMA levels, such technology has recently been developed and validated by us Citation67,68. This ADMA ELISA kit is based on the principles of a competitive immunoassay. In contrast to previously available methods for measuring ADMA, the new ELISA is easy to use and constitutes a high throughput technique. The combination of an acylation step and the competitive design of the ELISA resulted in a specific, highly sensitive and non‐isotopic immunoassay. The selected antiserum is specific for ADMA and results in negligible cross reactivities for L‐arginine (<0.02%) and other endogenous derivatives of L‐arginine. The precision of our ELISA has been demonstrated by low intra‐ and interassay coefficients of variation (interassay, 8.3%–10.3%; intraassay, 4.5%–7.5%). ADMA concentrations can be accurately measured across the full range of physiologically relevant concentrations (i.e. 0.05 µmol/L to 2 µmol/L). The values derived from the ELISA correlate well with expected values in recovery tests (mean recovery from all serum samples was 94.6%), and they display excellent linearity in dilution studies. The correlation with LC‐MS/MS is excellent (r = 0.984; P<0.0001). The ELISA has been validated for human serum (which is the preferential matrix) and plasma, as well as for rat and mouse plasma and cell culture supernatants.
Therefore, a wide array of reliable methods to quantify ADMA is now available which all have advantages and disadvantages: Modern HPLC techniques have been shown to produce reliable results Citation69; HPLC and LC‐MS/MS techniques allow to simultaneously measure ADMA, SDMA, and L‐arginine in one sample; however, these techniques are not available in every laboratory. ELISA is rapid, easy to perform and widely available; it produces data comparable to other methods, but allows to only measure ADMA.
Assessing ADMA in humans: in whom, when?
Considering the steadily accumulating data from prospective clinical trials that suggest a role for ADMA as a marker of cardiovascular disease, one may speculate about a potential role of ADMA measurement in clinical routine. One important question to ask therefore is: which patient should be tested, and when?
ADMA is elevated in association with almost all traditional cardiovascular risk factors, and in patients with established cardiovascular or metabolic diseases. However, its concentration shows significant variability even within these patient populations. Moreover, multivariate regression analyses that have taken into consideration all of the traditional and some additional novel risk factors have shown that ADMA remains independently associated with cardiovascular risk or total mortality even after such multivariate adjustment has been carried out.
A steadily increasing number of prospective clinical trials show that ADMA does more than merely discriminating between health and cardiovascular disease (for which, by the way, there are other and established diagnostic criteria). These data point out that even within a group of patients that have previously been regarded as homogeneous for their similar risk factor profile according to traditional risk factors, ADMA is useful to identify individuals with a comparatively high probability of experiencing a major event.
Cardiovascular risk assessment is currently re‐oriented towards assessment of total risk from an array of different risk markers, as opposed to the traditional approach to assess risk factors individually. This is effected by risk factor scores such as the Framingham risk score Citation70 (http://www.nhlbi.nih.gov/about/framingham/riskabs.htm), the PROCAM risk score Citation71 (http://chdrisk.uni‐muenster.de/calculator), or the risk charts developed by the European Society of Cardiology Citation72 (http://www.escardio.org/initiatives/prevention/SCORE+Risk+Charts.htm).
Where in this spectrum is there a place for novel risk factors such as ADMA? Novel risk markers may be of little help in assessing the total cardiovascular risk of patients who clearly fall into the high‐risk (i.e. greater than 20% risk for a major event during the subsequent 10 years) or low‐risk (i.e. below 10% risk) categories. However, what about the many patients who fall within the ‘grey zone’ of intermediate risk? For example, in a patient with a 17% risk of experiencing a major adverse cardiovascular event within the next 10 years, novel risk markers like ADMA levels may be helpful in estimating the real risk. Similarly, with an ADMA level well above the normal range (which has been determined as 0.36 – 1.17 µmol/L (73)), an elevated high‐sensitive CRP level, or an elevated level of another novel risk marker may still identify this patient as a candidate for pharmacotherapeutic intervention. This conception has led to the development of the ‘multimarker’ approach for cardiovascular risk assessment, which is currently put forward by many experts in the field Citation74. Indeed, a recent study based upon data from the first prospective study on ADMA as a cardiovascular risk factor in haemodialysis patients Citation34 showed that the combined use of ADMA, BNP and CRP explained 9%–10% of the mortality variance of standard Cox regression models, as opposed to 3%–5% when these factors were added individually Citation75.
When ADMA is elevated: Therapeutic consequences?
Clinical and experimental evidence cited above suggests that elevation of ADMA causes NO deficiency, even in the presence of ‘normal’ L‐arginine plasma concentrations. As ADMA is a competitive inhibitor of NO synthase, its inhibitory action can be overcome by increasing the concentration of this enzymès natural substrate. Therefore, dietary L‐arginine supplementation may be one means of specifically antagonizing the deleterious effects of ADMA on endothelial function in primary and secondary prevention. Nutraceuticals containing high levels of L‐arginine are widely available around the globe, and dietary intervention with L‐arginine has been shown to improve the symptoms of cardiovascular disease in several studies (for a review of L‐arginine studies in the context of elevated ADMA, see Citation76).
In a more general sense, elevated ADMA indicates an elevated risk of experiencing a major adverse cardiovascular event and represents one element of a multimarker risk profile. Thus, any pharmacotherapeutic intervention that has been proven to be effective in lowering cardiovascular morbidity and mortality such as β blockers, ACE inhibitors, statins, and platelet function inhibitors, can be expected to contribute to risk reduction in patients with elevated AMDA as well.
Nonetheless, a few small sample sized clinical studies suggested that certain drugs may decrease ADMA levels. This has been shown for ACE inhibitors and angiotensin receptor blockers in two studies Citation77,78. However, patient numbers were 20 and 19, respectively, and we were recently unable to reproduce these data in a study involving 24 hypertensive patients treated with the angiotensin receptor blocker telmisartan (R. Benndorf and R.H. Böger, unpublished data). The data from the large multinational VIVALDI trial that are expected for 2006 will more clearly show whether angiotensin receptor blockers exert an effect on ADMA levels Citation79.
Certain antidiabetic drugs like rosiglitazone Citation56 and metformin Citation80 have also been suggested to lower ADMA. The former is believed to exert this action via activation of PPARγ receptors, while the latter has been shown to have some structural similarities with ADMA, and may thus interfere with ADMA metabolism via currently unknown mechanisms.
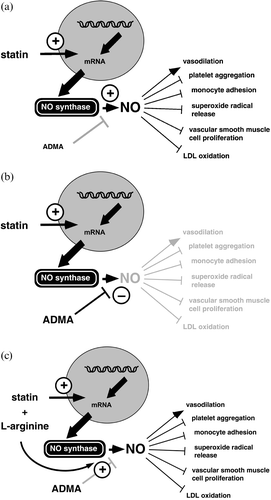
Although a close relationship has been established between ADMA and LDL cholesterol levels in some studies Citation15, the only statin that has been shown to reduce ADMA levels in one study is rosuvastatin Citation81. Simvastatin, pravastatin, and atorvastatin had no effect on ADMA levels Citation82,83. Notwithstanding this, ADMA levels may be important for statin treatment. The beneficial vascular effects of statins depend in part on their ability to upregulate endothelial NO synthase gene expression () Citation84. However, there are about as many clinical studies in which statins failed to improve endothelium‐dependent vasodilation as there are in which they failed. ADMA may help to resolve this discrepancy: upregulation of eNOS gene expression by statins may be functionally useless when the enzymès catalytic activity is blocked by ADMA () Citation85. Indeed, Janatuinen et al. Citation82 recently found that six months of treatment with pravastatin resulted in enhanced myocardial blood flow in patients with low ADMA levels, whereas the drug was ineffective in patients with elevated ADMA. We took up the idea that ADMA may block the NO synthase despite its upregulated gene expression after statin treatment, and that this blockade may be overcome by L‐arginine supplementation () Citation85. Indeed, we found, in a randomized controlled trial, that in patients with elevated ADMA concentration, simvastatin only enhanced endothelium‐dependent vasodilation when it was combined with supplemental nutritional L‐arginine Citation86.
Figure 3. ADMA may modify the statins' therapeutic effects. Statins are suggested to exert at least part of their vascular protective effects via upregulation of NO synthase gene expression (a). In the presence of high circulating ADMA levels, upregulation of the eNOS's gene by statins may still result in functionally inactive enzyme, since it is inhibited by ADMA (b). Excess exogenous L‐arginine may overcome inhibition by ADMA and thus restore the statins' effects on endothelial function (c). Reproduced from Citation75.
In conclusion, the role of ADMA as a novel cardiovascular risk factor is becoming increasingly clear with the publication of additional prospective clinical studies in various patient populations. However, data are still too sparse to draw definite conclusions as to how to incorporate ADMA into a multimarker concept of cardiovascular risk assessment. More and yet larger clinical trials are necessary to make it possible. As a tool for experimental studies in which endothelial regulation of vascular function is addressed, as a means to understand the pathophysiological regulation of the vascular regulator NO, and as a diagnostic approach in patients with unexplained cardiovascular disease, ADMA can be helpful at the moment. As far as therapeutic interventions to prevent the deleterious cardiovascular effects of ADMA are concerned, the long road from small, experimental clinical studies to large prospective clinical trials needs to be taken before we ultimately understand how to specifically deal with elevated ADMA levels.
References
- Kakimoto Y., Akazawa S. Isolation and identification of NG,NG‐ and NG,N ´G‐dimethyl‐arginine, N‐epsilon‐mono‐, di‐, and trimethyllysine, and glucosylgalactosyl‐ and galactosyl‐delta‐hydroxylysine from human urine. J Biol Chem 1970; 245: 5751–8
- McDermott J. R. Studies on the catabolism of NG‐methylarginine, NG, NG‐dimethylarginine and NG, NG ´‐dimethylarginine in the rabbit. Biochem J 1976; 154: 179–84
- Rawal N., Rajpurohit R., Lischwe M. A., Williams K. R., Paik W. K., Kim S. Structural specifity of substrate for S‐adenosylmethionine:protein arginine N‐methyltransferases. Biochim Biophys Acta 1995; 1248: 11–18
- Gary J. D., Clarke S. RNA and protein interactions modulated by protein arginine methylation. Prog Nucleic Acid Res Mol Biol 1998; 61: 65–131
- Pawlak M. R., Banik‐Maiti S., Pietenpol J. A., Ruley H. E. Protein arginine methyltransferase I: substrate specificity and role in hnRNP assembly. J Cell Biochem 2002; 87: 394–407
- Palmer R. M., Rees D. D., Ashton D. S., Moncada S. L‐arginine is the physiological precursor for the formation of nitric oxide in endothelium‐dependent relaxation. Biochem Biophys Res Commun 1988; 153: 1251–6
- Rees D. D., Palmer R. M., Hodson H. F., Moncada S. A specific inhibitor of nitric oxide formation from L‐arginine attenuates endothelium‐dependent relaxation. Br J Pharmacol 1989; 96: 418–24
- Vallance P., Leone A., Calver A., Collier J., Moncada S. Accumulation of an endogenous inhibitor of NO synthesis in chronic renal failure. Lancet 1992; 339: 572–5
- Böger R. H., Zoccali C. ADMA: a novel risk factor that explains excess cardiovascular event rate in patients with end‐stage renal disease. Atherosclerosis Suppl 2003; 4: 23–8
- Böger R. H., Bode‐Böger S. M., Frölich J. C. The L‐arginine – nitric oxide pathway: Role in atherosclerosis and therapeutic implications. Atherosclerosis 1996; 127: 1–11
- Panza J. A., Quyyumi A. A., Brush J. E., Jr., Epstein S. E. Abnormal endothelium‐dependent vascular relaxation in patients with essential hypertension. N Engl J Med 1990; 323: 22–7
- Celermajer D. S., Sorensen K. E., Georgakopoulos D., et al. Cigarette smoking is associated with dose‐related and potentially reversible impairment of endothelium‐dependent dilation in healthy young adults. Circulation 1993; 88: 2149–55
- Johnstone M. T., Creager S. J., Scales K. M., Cusco J. A., Lee B. K., Creager M. A. Impaired endothelium‐dependent vasodilation in patients with insulin‐dependent diabetes mellitus. Circulation 1993; 88: 2510–6
- Tawakol A., Omland T., Gerhard M., Wu J. T., Creager M. A. Hyperhomocyst(e)inemia is associated with impaired endothelium‐dependent vasodilation in humans. Circulation 1997; 95: 1119–21
- Böger R. H., Bode‐Böger S. M., Szuba A., et al. Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation 1998; 98: 1842–7
- Hingorani A. D., Cross J., Kharbanda R. K., Mullen M. J., Bhagat K., Taylor M., Donald A. E., Palacios M., Griffin G. E., Deanfield J. E., MacAllister R. J., Vallance P. Acute systemic inflammation impairs endothelium‐dependent dilatation in humans. Circulation 2000; 102: 994–9
- Böger R. H. The emerging role of ADMA as a novel cardiovascular risk factor. Cardiovasc Res 2003; 59: 824–33
- Ogawa T., Kimoto M., Sasaoka K. Occurrence of a new enzyme catalysing the direct conversion of NG, NG‐dimethyl‐L‐arginine to L‐citrulline in rats. Biochem Biophys Res Commun 1987; 148: 671–7
- Dayoub H., Achan V., Adimoolam S., Jacobi J., Stühlinger M. C., Wang B. Y., et al. Dimethylarginine dimethylaminohydrolase regulates nitric oxide synthesis: genetic and physiological evidence. Circulation 2003; 108: 3042–7
- Calver A., Collier J., Leone A., Moncada S., Vallance P. Effect of local intra‐arterial asymmetric dimethylarginine (ADMA) on the forearm arteriolar bed of healthy volunteers. J Hum Hypertens 1993; 7: 193–4
- Achan V., Broadhead M., Malaki M., Whitley G., Leiper J., MacAllister R., et al. Asymmetric dimethylarginine causes hypertension and cardiac dysfunction in humans and is actively metabolised by dimethylarginine dimethylaminohydolase. Arterioscler Thromb Vasc Biol 2003; 23: 1455–9
- Kielstein J. T., Impraim B., Simmel S., Bode‐Boger S. M., Tsikas D., Frolich J. C., et al. Cardiovascular effects of systemic nitric oxide synthase inhibition with asymmetrical dimethylarginine in humans. Circulation 2004; 109: 172–7
- Chan J. R., Böger R. H., Bode‐Böger S. M., Tangphao O., Tsao P. S., Blaschke T. F., et al. Asymmetric dimethylarginine increases mononuclear cell adhesiveness in hypercholesterolemic humans. Arterioscler Thromb Vasc Biol 2000; 20: 1040–6
- Böger R. H., Bode‐Böger S. M., Thiele W., Junker W., Alexander K., Frölich J. C. Biochemical evidence for impaired nitric oxide synthesis in patients with peripheral arterial occlusive disease. Circulation 1997; 95: 2068–74
- Surdacki A., Nowicki M., Sandmann J., Tsikas D., Böger R. H., Bode‐Böger S. M., et al. Reduced urinary excretion of nitric oxide metabolites and increased plasma levels of asymmetrical dimethylarginine in men with essential hypertension. J Cardiovasc Pharmacol 1999; 33: 652–8
- Kielstein J. T., Böger R. H., Bode‐Böger S. M., Schäffer J., Barbey M., Koch K. M., Frölich J. C. Asymmetric dimethylarginine plasma concentrations differ in patients with end‐stage renal disease: Relationship to treatment method and atherosclerotic disease. J Am Soc Nephrol 1999; 10: 594–600
- Schnog J. B., Teerlink T., van der Dijs F. P., Duits A. J., Muskiet F. A., CURAMA Study Group. Plasma levels of asymmetric dimethylarginine (ADMA), an endogenous nitric oxide synthase inhibitor, are elevated in sickle cell disease. Ann Hematol 2005; 84: 282–6
- Gorenflo M., Zheng C., Werle E., Fiehn W., Ulmer H. E. Plasma levels of asymmetrical dimethyl‐L‐arginine in patients with congenital heart disease and pulmonary hypertension. J Cardiovasc Pharmacol 2001; 37: 489–92
- Millatt L. J., Whitley G. S., Li D., Leiper J. M., Siragy H. M., Carey R. M., et al. Evidence for dysregulation of dimethylarginine dimethylaminohydrolase I in chronic hypoxia‐induced pulmonary hypertension. Circulation 2003; 108: 1493–8
- Kielstein J. T., Bode‐Böger S. M., Hesse G., Martens‐Lobenhöffer J., Takacs A., Fliser D., et al. Asymmetrical dimethylarginine in idiopathic pulmonary arterial hypertension. Arterioscler Thromb Vasc Biol 2005; 25: 1414–8
- Miyazaki H., Matsuoka H., Cooke J. P. Endogenous nitric oxide synthase inhibitor. A novel marker of atherosclerosis. Circulation 1999; 99: 1141–6
- Zoccali C., Benedetto F. A., Maas R., Mallamaci F., Tripepi G., Malatino L., et al. Asymmetric dimethylarginine (ADMA), C‐reactive protein, and carotid intima media‐thickness in end‐stage renal disease. J Am Soc Nephrol 2002; 13: 490–6
- Valkonen V. P., Päivä H., Salonen J. T., Lakka T. A., Lehtimaki T., Laakso J., et al. Risk of acute coronary events and serum concentration of asymmetrical dimethylarginine. Lancet 2001; 358: 2127–8
- Zoccali C., Bode‐Böger S. M., Mallamaci F., Benedetto F. A., Tripepi G., Malatino L., et al. Asymmetric dimethylarginine (ADMA): An endogenous inhibitor of nitric oxide synthase predicts mortality in end‐stage renal disease (ESRD). Lancet 2001; 358: 2113–7
- Nijveldt R. J., Teerlink T., van der Hoven B., Siroen M. P., Kuik D. J., Rauwerda J. A., et al. Asymmetrical dimethylarginine (ADMA) in critically ill patients: high plasma ADMA concentration is an independent risk factor of ICU mortality. Clin Nutr 2003; 22: 23–30
- Lu T. M., Ding Y. A., Lin S. J., Lee W. S., Tai H. C. Plasma levels of asymmetrical dimethylarginine and adverse cardiovascular events after percutaneous coronary intervention. Eur Heart J 2003; 24: 1912–9
- Krempl T. K., Maas R., Sydow K., Meinertz T., Böger R. H., Kähler J. Elevation of asymmetric dimethylarginine in patients with unstable angina and recurrent cardiovascular events. Eur Heart J 2005; 26: 1846–51
- Schnabel R., Blankenberg S., Lubos E., Lackner K. J., Rupprecht H. J., Espinola‐Klein C., et al. Asymmetric dimethylarginine and the risk of cardiovascular events and death in patients with coronary artery disease: results from the AtheroGene Study. Circ Res 2005; 97: e53–9
- Cooke J. P. Asymmetrical dimethylarginine: the Über marker?. Circulation 2004; 109: 1813–8
- Vallance P., Leiper J. Cardiovascular biology of the asymmetric dimethylarginine:dimethylarginine dimethylaminohydrolase pathway. Arterioscler Thromb Vasc Biol 2004; 24: 1023–30
- Sydow K., Münzel T. ADMA and oxidative stress. Atherosclerosis 2003; 4((Suppl. 4))41–51
- Pritchard K. A., Jr., Groszek L., Smalley D. M., Sessa W. C., Wu M., Villalon P., et al. Native low‐density lipoprotein increases endothelial cell nitric oxide synthase generation of superoxide anion. Circ Res 1995; 77: 510–8
- Closs E. I., Simon A., Vekony N., Tormann A. Plasma membrane transporters for arginine. J Nutr 2004; 134((10 Suppl.))2752S–9
- Closs E. I., Basha F. Z., Habermeier A., Förstermann U. Interference of L‐arginine analogues with L‐arginine transport mediated by the y+ carrier hCAT‐2B. Nitric Oxide 1997; 1: 65–73
- Bogle R. G., MacAllister R. J., Whitley G. S. J., Vallance P. Induction of NG‐monomethyl‐L‐arginine uptake: a mechanism for differential inhibiton of NO synthases?. Am J Physiol 1995; 269: C750–6
- Böger R. H., Sydow K., Borlak J., Thum T., Lenzen H., Schubert B., et al. LDL Cholesterol upregulates synthesis of asymmetric dimethylarginine (ADMA) in human endothelial cells. Involvement of S‐adenosylmethionine‐dependent methyltransferases. Circ Res 2000; 87: 99–105
- Moss M. B., Brunini T. M. C., Soares de Moura R., Novaes Malagris L. E., Roberts N. B., Ellory J. C., Mann G. E., Mendres Ribeiro A. C. Diminished L‐arginine bioavailability in hypertension. Clin Sci 2004; 107: 391–7
- Goonasekera C. D., Rees D. D., Woolard P., Frend A., Shah V., Dillon M. J. Nitric oxide synthase inhibitors and hypertension in children and adolescents. J Hypertens 1997; 15: 901–9
- Jang J. J., Ho H. K., Kwan H. H., Fajardo L. F., Cooke J. P. Angiogenesis is impaired by hypercholesterolemia: role of asymmetric dimethylarginine. Circulation 2000; 102: 1414–9
- Jacobi J., Sydow K., von Degenfeld G., Zhang Y., Dayoub H., Wang B., et al. Overexpression of dimethylarginine dimethylaminohydrolase reduces tissue asymmetric dimethylarginine levels and enhances angiogenesis. Circulation 2005; 111: 1431–8
- Suda O., Tsutsui M., Morishita T., Tasaki H., Ueno S., Nakata S., et al. Asymmetric dimethylarginine produces vascular lesions in endothelial nitric oxide synthase‐deficient mice: involvement of renin‐angiotensin system and oxidative stress. Arterioscler Thromb Vasc Biol 2004; 24: 1682–8
- Tsikas D., Rode I., Becker T., Nashan B., Klempnauer J., Frölich J. C. Elevated plasma and urine levels of ADMA and 15(S)‐8‐iso‐PGF2alpha in end‐stage liver disease. Hepatology 2003; 38: 1063–4
- Siroen M. P., Warle M. C., Teerlink T., Nijveldt R. J., Kuipers E. J., Metselaar H. J., et al. The transplanted liver is capable of clearing asymmetric dimethylarginine. Liver transpl 2004; 10: 1524–30
- Nijveldt R. J., Siroen M. P., Teerlink T., van Leeuwen P. A. Elimination of asymmetric dimethylarginine by the kidney and the liver: a link to the development of multiple organ failure?. J Nutr 2004; 134((10 Suppl.))2848S–52
- Hermenegildo C., Medina P., Peiro M., Segarra G., Vila J. M., Ortega J., et al. Plasma concentration of asymmetric dimethylarginine, an endogenous inhibitor of nitric oxide synthase, is elevated in hyperthyroid patients. J Clin Endocrinol Metab 2002; 87: 5636–40
- Stühlinger M. C., Abbasi F., Chu J. W., Lamendola C., McLaughlin T. L., Cooke J. P., et al. Relationship between insulin resistance and an endogenous nitric oxide synthase inhibitor. JAMA 2002; 287: 1420–6
- Abbasi F., Asagami T., Cooke J. P., Lamendola C., McLaughlin T., Reaven G. M., et al. Plasma concentrations of asymmetric dimethylarginine are increased in patients with type 2 diabetes mellitus. Am J Cardiol 2001; 88: 1201–3
- Pettersson A., Hedner T., Milsom I. Increased circulating concentrations of asymmetric dimethyl arginine (ADMA), an endogenous inhibitor of nitric oxide synthesis, in preeclampsia. Acta Obstet Gynecol Scand 1998; 77: 808–13
- Savvidou M. D., Hingorani A. D., Tsikas D., Frölich J. C., Vallance P., Nicolaides K. H. Endothelial dysfunction and raised plasma concentrations of asymmetric dimethylarginine in pregnant women who subsequently develop pre‐eclampsia. Lancet 2003; 361: 1511–7
- Maas R., Serrano N., Schwedhelm E., Diaz L. A., Casas J. P., Böger R. H., et al. Plasma concentration of asymmetrical dimethylarginine (ADMA) in women with pre‐eclampsia from a high‐risk population. JAMA 2004; 291: 823–4
- Abe T., Tohgi H., Murata T., Isobe C., Sato C. Reduction in asymmetrical dimethylarginine, an endogenous nitric oxide synthase inhibitor, in the cerebrospinal fluid during aging and in patients with Alzheimer's disease. Neurosci Lett 2001; 312: 177–9
- Mulder C., Wahlund L. O., Blomberg M., de Jong S., van Kamp G. J., Scheltens P., et al. Alzheimer's disease is not associated with altered concentrations of the nitric oxide synthase inhibitor asymmetric dimethylarginine in cerebrospinal fluid. J Neural Transm 2002; 109: 1203–8
- Selley M. L. Increased concentrations of homocysteine and asymmetric dimethylarginine and decreased concentrations of nitric oxide in the plasma of patients with Alzheimer's disease. Neurobiol Aging 2003; 24: 903–7
- Kielstein J. T., Böger R. H., Bode‐Böger S. M., Martens‐Lobbenhöfer J., Lonnemann G., Frölich J. C., et al. Low dialysance of asymmetric dimethylarginine (ADMA) – in vivo and in vitro evidence of significant protein binding. Clin Nephrol 2004; 62: 295–300
- Schwedhelm E. Quantification of ADMA: Analytical approaches. Vasc Med 2005; 10((Suppl. 1))S89–95
- Albsmeier J., Schwedhelm E., Schulze F., Kastner M., Böger R. H. Determination of NG,NG‐dimethyl‐L‐arginine, an endogenous NO synthase inhibitor, by gas chromatography‐mass spectrometry. J Chromatogr B 2004; 809: 59–65
- Schulze F., Wesemann R., Schwedhelm E., Sydow K., Albsmeier J., Cooke J. P., et al. Determination of ADMA using a novel ELISA assay. Clin Chem Lab Med 2004; 42: 1377–83
- Böger RH. All about ADMA. www.allaboutadma.com (last accessed by the author on 10 November 2005).
- Teerlink T. Measurement of asymmetric dimethylarginine in plasma: methodological considerations and clinical relevance. Clin Chem Lab Med 2005; 43: 1130–8
- NHLBI Framingham Heart Study. Estimating Coronary Heart Disease (CHD) Risk Using Framingham Heart Study Prediction Score Sheets. December 2002. http://www.nhlbi.nih.gov/about/framingham/riskabs.htm (last accessed on 13 June 2005 by the author).
- International Task Force for Prevention of Coronary Heart Disease, PROCAM Risk Calculator. 2004. http://www.chd‐taskforce.de/calculator/calculator.htm (last accessed on 13 June 2005 by the author).
- European Society of Cardiology, CVD Prevention Score Risk Charts, 2004. http://www.escardio.org/initiatives/prevention/SCORE+Risk+Charts.htm (last accessed on 13 June 2005 by the author).
- Schulze F., Maas R., Freese R., Schwedhelm E., Silberhorn L., Böger R. H. Determination of a reference value for N,N‐dimethyl‐L‐arginine in 500 subjects. Eur J Clin Invest 2005; 35: 622–6
- Lee D. S., Vasan R. S. Novel markers for heart failure diagnosis and prognosis. Curr Opin Cardiol 2005; 20: 201–10
- Mallamaci F., Tripepi G., Cutrupi S., Malatino L. S., Zoccali C. Prognostic value of combined use of biomarkers of inflammation, endothelial dysfunction, and myocardiopathy in patients with ESRD. Kidney Int 2005; 67: 2330–7
- Böger R. H., Ron E. S. L‐Arginine improves vascular function by overcoming the deleterious effects of ADMA, a novel cardiovascular risk factor. Altern Med Rev 2005; 10: 14–23
- Delles C., Schneider M. P., John S., Gekle M., Schmieder R. E. Angiotensin converting enzyme inhibition and angiotensin II AT1‐receptor blockade reduce the levels of asymmetrical N(G), N(G)‐dimethylarginine in human essential hypertension. Am J Hypertens 2002; 15: 590–3
- Ito A., Egashira K., Narishige T., Muramatsu K., Takeshita A. Angiotensin‐converting enzyme activity is involved in the mechanism of increased endogenous nitric oxide synthase inhibitor in patients with type 2 diabetes mellitus. Circ J 2002; 66: 811–5
- Böger R. H., Schwedhelm E., Maas R., Quispe‐Bravo S., Skamira C. ADMA and oxidative stress may relate to the progression of renal disease: Rationale and design of the VIVALDI study. Vasc Med 2005, In press
- Asagami T., Abbasi F., Stuelinger M., Lamendola C., McLaughlin T., Cooke J. P., et al. Metformin treatment lowers asymmetric dimethylarginine concentrations in patients with type 2 diabetes. Metabolism 2002; 51: 843–6
- Lu T. M., Ding Y. A., Leu H. B., Yin W. H., Sheu W. H., Chu K. M. Effect of rosuvastatin on plasma levels of asymmetric dimethylarginine in patients with hypercholesterolemia. Am J Cardiol 2004; 94: 157–61
- Janatuinen T., Laakso J., Laaksonen R., Vesalainen R., Nuutila P., Lehtimaki T., et al. Plasma asymmetric dimethylarginine modifies the effect of pravastatin on myocardial blood flow in young adults. Vasc Med 2003 2003; 8: 185–9
- Paiva H., Laakso J., Lehtimaki T., Isomustajarvi M., Ruokonen I., Laaksonen R. Effect of high‐dose statin treatment on plasma concentrations of endogenous nitric oxide synthase inhibitors. J Cardiovasc Pharmacol 2003; 41: 219–22
- Laufs U., La F., Plutzky J., Liao J. K. Upregulation of endothelial nitric oxide synthase by HMG CoA reductase inhibitors. Circulation 1998; 97: 1129–35
- Böger R. H. Asymmetric dimethylarginine (ADMA) modulates endothelial function – therapeutic implications. Vasc Med 2003; 8: 149–51
- Böger G. I., Maas R., Schwedhelm E., Bierend A., Benndorf R., Kastner M., et al. Improvement of endothelium‐dependent vasodilation by simvastatin is potentiated by combination with L‐arginine sustained release in patients with elevated ADMA levels. [Abstract]. J Am Coll Cardiol 2004; Suppl: 525A