Abstract
Background. Inhibition of cytokine‐induced expression of adhesion molecules is one of the atheroprotective mechanisms of high‐density lipoprotein (HDL).
Aim. We investigated whether increased endothelial activation and low‐grade inflammation are present in Finnish subjects with familial low HDL, and which factors contribute to the inflammatory parameters.
Method. High‐sensitivity C‐reactive protein (hsCRP), soluble intercellular adhesion molecule‐1 (sICAM‐1), vascular cell adhesion molecule‐1 (sVCAM‐1), and sE‐selectin were measured in 91 subjects with low HDL‐cholesterol from 41 low‐HDL families and in 112 normolipidemic controls with comparable age‐ and gender distribution. Presence of the features of the metabolic syndrome (MetS) was recorded.
Results. sVCAM‐1, sICAM‐1, sE‐selectin, and hsCRP were significantly higher in low‐HDL subjects than in the controls (sVCAM‐1: 560±147 ng/mL versus 496±95 ng/mL, P = 0.001; sICAM‐1: 247±60 ng/mL versus 215±47 ng/mL, P<0.001; sE‐selectin: 52±20 ng/mL versus 44±16 ng/mL, P = 0.022; and hsCRP: 1.73±2.05 mg/L versus 0.85±1.10 mg/L, P<0.001). Low‐HDL subjects had increased body mass index (BMI) and waist, and elevated insulin and triglyceride levels. Adhesion molecules and hsCRP increased according to the number of the features of the MetS.
Conclusions. The presence of the MetS in subjects with familial low HDL‐cholesterol aggravates the low‐grade inflammation and endothelial activation, and ultimately may add to the higher susceptibility for atherosclerotic disease in these individuals.
| Abbreviations | ||
| ANOVA | = | analysis of variance |
| apo | = | apolipoprotein |
| BMI | = | body‐mass index |
| CAM | = | cellular adhesion molecule |
| CHD | = | coronary heart disease |
| DBP | = | diastolic blood pressure |
| EDTA | = | ethylenediaminetetraacetate |
| eNOS | = | endothelial nitric oxide synthase |
| ELISA | = | enzyme‐linked immunosorbent assays |
| FCHL | = | familial combined hyperlipidemia |
| hsCRP | = | high‐sensitivity C‐reactive protein |
| HDL‐C | = | high‐density lipoprotein cholesterol |
| HOMA | = | IR homeostasis model assessment for insulin resistance |
| Lp | = | lipoprotein |
| LDL | = | low‐density lipoprotein |
| MetS | = | metabolic syndrome |
| PAI‐1 | = | plasminogen activator inhibitor‐1 |
| sE‐selectin | = | soluble E‐selectin |
| sICAM‐1 | = | soluble intercellular adhesion molecule‐1 |
| sVCAM‐1 | = | soluble vascular cell adhesion molecule‐1 |
| SBP | = | systolic blood pressure |
| TC | = | total cholesterol |
| TG | = | triglyceride |
| TNF‐α | = | tumor necrosis factor‐alpha |
Introduction
Both epidemiological and clinical studies provide strong evidence that low plasma high‐density lipoprotein cholesterol (HDL‐C) concentration is a major risk factor for coronary heart disease (CHD). Genetically determined low serum HDL (familial low HDL or hypoalphalipoproteinemia) is a common familial lipoprotein disorder Citation1. Among men with CHD before the age of 60 years, this disorder has a prevalence of 18%–39% Citation1. Most patients with low HDL‐C have clustering of multiple cardiovascular risk factors and features of the metabolic syndrome (MetS) such as obesity, hypertension, elevated serum triglycerides (TGs) and small, dense low‐density lipoprotein (LDL) particles Citation1,2. Insulin resistance, as measured by surrogate markers as homeostasis model assessment for insulin resistance (HOMA IR), fasting hyperglycemia, or hyperinsulinemia, has been linked to chronic, low‐grade inflammation of the endothelium in several studies Citation3–6, but whether low HDL‐C per se independently aggravates inflammation is uncertain. Inflammation decreases HDL‐C, and has a major influence on the remodeling of HDL particles in the circulation Citation7,8.
HDL promotes the efflux of free cholesterol from cells and its transport to the liver by the reverse cholesterol transport Citation9. HDL also prevents oxidation of LDL particles and protects the arterial wall against oxidative damage Citation10. One of the earliest events in atherogenesis is the adhesion of monocytes to endothelial cells, through cell‐surface expression of cellular adhesion molecules (CAMs), namely vascular cell adhesion molecule‐1 (VCAM‐1), intercellular adhesion molecule‐1 (ICAM‐1), and E‐selectin Citation11. CAMs are partly released from the endothelial surface to the circulation, and the serum concentrations of the soluble forms serve as markers for their endothelial expression Citation12. The cytokine‐induced expression of CAMs is inhibited by HDL in a concentration dependent manner Citation13. Thus, HDL can inhibit the interaction between monocytes and the endothelium, and prevent the recruitment of circulating leukocytes to sites of vascular injury. Calabresi et al. Citation14 first reported the elevated plasma levels of soluble ICAM (sICAM) and sVCAM in Italian subjects with low HDL‐C. Subsequently, hypercholesterolemia, hypertriglyceridemia Citation15, and insulin resistance Citation5,Citation16 have been linked to elevated sCAM levels.
HDL‐C and its major protein component apolipoprotein (apo) A‐I are reduced in inflammatory conditions Citation7. Decreased HDL‐C and low‐grade inflammation are components of the MetS, which associates strongly with cardiovascular disease Citation17,18. In the present study, we measured the levels of high‐sensitivity C‐reactive protein (hsCRP) and serum concentrations of sICAM‐1, sVCAM‐1 and sE‐selectin in 91 individuals with familial low HDL, and in 112 control subjects with comparable age‐ and gender distribution. The parameters of lipid metabolism and the presence of the features of the MetS were recorded in all study subjects. We hypothesized that accumulation of the features of the MetS could cause the increase in the markers of endothelial activation, such as sCAMs or hsCRP, in Finnish individuals with the familial low‐HDL trait.
Key messages
Finnish subjects with familial low high‐density lipoprotein (HDL) have increased serum concentrations of the soluble adhesion molecules including intercellular adhesion molecule‐1 (sICAM‐1), vascular cell adhesion molecule‐1 (sVCAM‐1), and sE‐selectin, and an increased level of the inflammatory acute phase protein C‐reactive protein (CRP).
The majority of the individuals with low HDL‐cholesterol (HDL‐C) have a complex phenotype that resembles the metabolic syndrome (MetS), characterized by elevated levels of fasting insulin, triglyceride and apoB, and increased body mass index (BMI) and waist/hip ratio.
The presence of the metabolic syndrome predicts the increased levels of high‐sensitivity CRP and sCAMs in these subjects; therefore, we suggest that the chronic inflammatory process on the vascular endothelium, due to the accumulation of the features of the MetS predisposes the low‐HDL subjects to an increased risk of an atherosclerotic disease.
Materials and methods
Study subjects
Study subjects were members of well characterized families with low HDL and premature CHD Citation19. Inclusion criteria for the probands of these families were as follows: 1) Age 30–60 years; 2) HDL‐C level below the 10th age‐sex specific Finnish population percentile Citation20 (<0.9 mmol/L in men and <1.1 mmol/L in women); 3) CHD verified by either coronary angiography (at least 50% stenosis in one or more coronary arteries) or myocardial infarction. Additional lipid criteria for the probands were as follows: 1) Total cholesterol (TC) <6.3 mmol/L in men and <6.0 mmol/L in women; and 2) TG <2.3 mmol/L in both genders. Exclusion criteria for the probands were Type 1 or 2 diabetes, hepatic or renal diseases, untreated hypothyreosis, familial hypercholesterolemia, and body mass index (BMI) >30 kg/m2. All family members of the probands who had HDL‐C below the 10th percentile levels were considered affected. At least one first‐degree family member was required to have HDL‐C below the 10th age‐sex specific percentile criteria, in order to have at least two affected subjects in each family. Altogether 91 affected family members from 41 low‐HDL families comprised the low‐HDL group. There were 29 subjects within the low‐HDL group who had CHD. The control group comprised 112 individuals, including healthy volunteers and normolipidemic spouses from the low‐HDL families, with similar age‐ and gender distribution with the low‐HDL group. The controls were required to have no signs of CHD, and no lipid lowering medication. Those within the low‐HDL group who took any lipid lowering medication discontinued that for 4 weeks prior to blood sampling. Subjects on hormone replacement therapy or with chronic inflammatory disorders were excluded from the study.
Each study subject gave written informed consent prior to participating in the study. All samples were collected in accordance with the Helsinki declaration. The ethics committee of the Helsinki University Central Hospital approved the study design.
Experimental methods
Venous blood samples were obtained after an overnight fast. Serum and ethylenediaminetetraacetate (EDTA) plasma were separated by centrifugation and stored at ‐80°C until analysis. Serum TC and TG were determined with an automated Cobas Mira analyzer (Hoffman‐La Roche, Basel, Switzerland) by enzymatic methods (Hoffman‐La Roche). LDL cholesterol (LDL‐C) was calculated by using the Friedewald formula Citation21. Concentrations of apoA‐I, apoA‐II and apoB were measured by immunoturbidometric methods with commercial kits (for apoA‐I: Wako Chemicals GmbH, Germany; for apoA‐II: National Public Health Institute, Helsinki, Finland; and for apoB: Orion Diagnostica, Espoo, Finland). Concentration of lipoprotein AI (LpA‐I) particles was measured by differential electroimmunoassay (Sebia, Issy‐les‐Moulienaux, France) Citation22. The concentration of LpAI‐AII particles was calculated by subtracting the concentration of LpA‐I from the total concentration of apoA‐I in serum. HDL‐C, HDL2–C and HDL3–C were separated by ultracentrifugation Citation23. Plasma glucose concentrations were analyzed by glucose dehydrogenase method (Precision‐G Blood Glucose Testing System, Medisense, Abbott, IL, USA). Serum‐free insulin concentration was measured by radioimmunoassay (Phadeseph Insulin Ria, Pharmacia & Upjohn, Uppsala, Sweden). The HOMA IR was calculated from the fasting plasma glucose and serum insulin concentrations as follows: fasting insulin (µU/mL) × fasting glucose (mmol/L) / 22.5 Citation24. Serum sICAM‐1, sVCAM‐1 and sE‐selectin were measured by using commercially available monoclonal antibody‐based enzyme‐linked immunosorbent assays (ELISAs: R&D Systems, Minneapolis, MN, USA). hsCRP was determined by a commercial kit from Medix Biochemica, Kauniainen, Finland. Subjects with hsCRP levels over 10 mg/L were excluded. Each participant filled in a standard questionnaire on medication, drinking, and smoking habits. The smoking status of the subjects was categorized into two groups: current or former smokers, and those who had never smoked. Hypertension was defined as self‐reported use of antihypertensive medications, or systolic blood pressure (SBP) ⩾140 mmHg, and diastolic blood pressure (DBP) ⩾90 mmHg.
Statistical methods
Statistical comparisons of clinical and biomedical parameters were performed with SPSS 11.0 for Windows (SPSS Inc., Chicago, IL, USA). Values represent mean±SD for continuous variables, and frequencies or percentages for categorical variables. P‐value <0.050 was considered significant (two‐tailed). Variables with skewed distribution were log10 transformed before the analyses. Independent Samples T‐test was used to assess differences between the groups. Adjustment for age, gender, BMI and smoking was performed by treating these factors as covariates in the general linear model univariate analysis, and saving the non‐standardized residual value as the adjusted variable. The analysis of variance (ANOVA) with Bonferroni post hoc test was used to test differences between more than two groups. The frequency distribution between the groups was compared with the Chi‐square test. Bivariate correlations were calculated using the Pearson correlation analysis. Multivariate stepwise linear regression analysis was performed to test which factors determine the hsCRP levels in the low‐HDL subjects. hsCRP was used as a dependent variable, and all variables in were chosen as independent variables for the stepwise linear regression model. HOMA IR, glucose, and insulin were not included in the same model, since these were highly intercorrelated. The independent variables were removed from the model until the best fitting model with the maximum multiple R2 was achieved.
The MetS was defined according to the Adult Treatment Panel (ATP) III Citation17: Abdominal obesity (waist circumference >102 cm in men and >88 cm in women), TG >1.7 mmol/L in both genders, HDL‐C<1.03 mmol/L in men and <1.29 mmol/L in women, blood pressure ⩾130/85 mmHg, and fasting glucose ⩾6.1 mmol/L in both genders. Subjects fulfilling three or more of these determinants are considered to have the MetS Citation17.
Results
Subject characteristics
Clinical and biochemical characteristics of the study subjects are presented in . Age and gender distribution were comparable between low‐HDL subjects and control subjects. Low‐HDL subjects had increased BMI and waist circumference, and they had higher levels of TG, apoB, LDL‐C, fasting insulin, and HOMA IR than the control subjects. By definition, the low‐HDL subjects had lower levels of total HDL‐C, HDL2–C and HDL3–C than the controls. These differences remained significant also when men and women were analyzed separately (data not shown). The percentage of hypertensive patients was higher in low‐HDL subjects than in controls (49% versus 17%, P<0.001), as was the percentage of patients on aspirin treatment (36% versus 2%, P<0.001, respectively).
Table I. Clinical and biochemical characteristics of the study subjects.
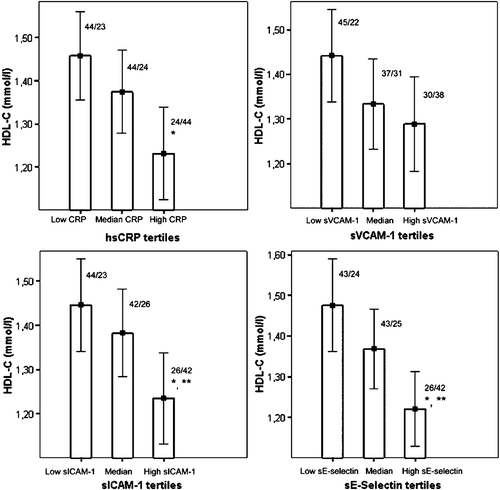
The concentrations of sVCAM‐1, sICAM‐1, sE‐selectin and hsCRP were significantly higher in the low‐HDL group as compared to the controls (). After adjustment for age, gender, BMI, TGs and smoking, the between‐group differences in sVCAM‐1 remained statistically significant. The between‐group comparisons of serum sCAM and hsCRP concentrations were also performed after the exclusion of 29 subjects with CHD (). The remaining low‐HDL subjects (n = 62) had significantly higher levels of sVCAM‐1, sICAM‐1, sE‐selectin, and hsCRP than the control subjects. Next, all study subjects were allocated into three groups according to the tertiles of hsCRP, sVCAM‐1, sICAM‐1, and sE‐selectin levels (). HDL‐C was significantly decreased in the highest tertiles of sE‐selectin, sICAM‐1, and hsCRP, as compared to the lowest tertiles.
Table II. Serum levels of soluble cell adhesion molecules and hsCRP in low‐HDL subjects and in controls.
Figure 1. The bars indicate the mean HDL‐C levels in the combined study group when divided into subgroups according to levels of serum hsCRP, sVCAM‐1, sE‐selectin, and sICAM‐1. Error bars show the 95% Confidence Interval of mean. The numbers above the bars indicate the frequency distribution of controls and low‐HDL subjects in each tertile ((n)controls/(n)low‐HDL subjects). *Significant difference when compared to the lowest tertile (P<0.01); **Significant difference when compared to the lowest tertile, with adjustment for age, gender, BMI and smoking (P<0.05). Between the lowest and highest hsCRP tertiles: P<0.001; between the lowest and the highest sE‐selectin tertiles: P<0.001 (non‐adjusted), and P=0.047, (adjusted for age, gender, BMI and smoking); between the lowest and highest sICAM‐1 tertiles: P=0.005 (non‐adjusted), and P=0.047, (adjusted for age, gender, BMI and smoking).
The percentage of smokers was higher among low‐HDL subjects than in controls (56% versus 26%, P<0.001). In the whole group, smokers (n = 80) had significantly higher levels of sICAM‐1 and sE‐selectin than the non‐smokers (n = 123): for sICAM‐1, 246±62 ng/mL versus 219±48 ng/mL, P = 0.001; and for sE‐selectin, 53±19 ng/mL versus 45±18 ng/mL, P = 0.002, respectively. The levels of sVCAM‐1 and hsCRP were not significantly different between smokers and non‐smokers. Within the low‐HDL group alone, the levels of sCAM or hsCRP were not significantly different between smokers and non‐smokers.
Correlation analyses between the levels of sCAMs and hsCRP and other variables
Results of the correlation analyses are presented in . The correlation analyses were carried out in the low‐HDL group alone. hsCRP showed significant positive correlation with age, BMI, waist circumference, insulin, HOMA IR, and apoB. sVCAM‐1 showed significant negative associations with apoA‐II and LpAI‐AII, and positive associations with age, SBP and LpA‐I. sE‐selectin showed positive associations with BMI, waist, insulin, HOMA IR, and apoA‐II. sICAM‐1 was inversely associated with glucose and male gender. No significant associations existed between HDL‐C and sCAMs or hsCRP in the low‐HDL subjects alone. Due to the recruitment strategy, the range of HDL‐C distributions was narrow within the low‐HDL group, limiting the possibility to detect significant results in the correlation analysis. However, the combining of the low‐HDL group and control group expanded the range of HDL‐C so that significant association was observed between HDL‐C and hsCRP (r = −0.292, P<0.001), between HDL‐C and sE‐selectin (r = −0.220, P = 0.002), between HDL‐C and sICAM‐1 (r = −0.256, P<0.001), and between HDL‐C and sVCAM‐1 (r = −0.154, P = 0.028).
In the low‐HDL subjects, the multivariate model for hsCRP resulted in the highest R2 of 0.251, with F‐value of 13.755 (P<0.001). BMI and age entered the final model, and the highest standardized coefficient was observed for BMI (r = 0.455, P<0.001), with 95% confidence interval from 0.033 to 0.081.
Table III. Pearson correlation coefficients for the relationships between serum levels of hsCRP and sCAMs and clinical and biochemical parameters in 91 low HDL subjects.
The distribution of the sCAMs and hsCRP in relation to the features of the MetS
Next, all study subjects were allocated into three groups according to the features of the MetS. All low‐HDL subjects had some MetS features: 47 low‐HDL subjects (52%) had 1–2 MetS features and the remaining 44 low‐HDL subjects (48%) had 3 or more features, i.e. they fulfilled the diagnostic criteria for the MetS. The majority of the control subjects, 64 (57%), had no features of the MetS, while 46 of controls (41%) had 1–2 features, and 2 (2%) had 3 or more features of the MetS (). The levels of sICAM‐1, sVCAM‐1, sE‐selectin, and hsCRP increased according to the increased number of the MetS features. After adjustment for age, gender, BMI and smoking, the difference between those who had three or more MetS features and no MetS features remained significant for hsCRP, sICAM‐1 and sVCAM‐1. Within the low‐HDL group, those 44 subjects who had the MetS had significantly higher hsCRP level than the 47 subjects with 1–2 MetS features (2.29±2.25 mg/L versus 1.21±1.71 mg/L, P<0.001, respectively).
Table IV. Serum levels of hsCRP, sE‐selectin, sVCAM‐1, sICAM‐1 and HDL fraction subgroups categorized according to the presence of the features of the MetS.
Discussion
The cell‐surface expression of CAMs in the vascular endothelium promotes the adhesion of monocytes to the vessel wall, and their accumulation in the arterial intima Citation11. The recruitment of inflammatory cells due to the increased CAM expression is seminal in the development of vascular injury. Thus, the levels of circulating CAMs may predict the risk of cardiovascular events. In the present study, we measured the sCAMs and hsCRP in subjects with familial low HDL. These subjects are members of Finnish families where the proband has premature CHD and low HDL, collected with strict criteria and a well defined phenotype for low HDL‐C Citation19. Our main observation was that the individuals with familial low HDL had higher levels of sVCAM‐1, sICAM‐1, sE‐selectin, and hsCRP than the control subjects with similar age‐ and gender distribution. sCAMs and CRP were significantly increased in the low‐HDL group also after the exclusion of patients with CHD. The observed increase in the inflammatory parameters was closely related to the MetS.
Epidemiological studies have established a link between infection, inflammation and atherosclerosis Citation11. Measurement of the inflammatory biomarkers can add to the predictive value achieved by assessment of plasma lipid levels alone Citation18. Increased CRP, an independent risk factor for cardiovascular disease Citation25, may also act as a mediator of atherosclerosis Citation26. Interestingly, it is localized in infarcted myocardium and in atherosclerotic lesions in human tissue samples, and it has been found to induce the cellular expression of ICAM‐1, VCAM‐1, E‐selectin, and monocyte chemoattractant protein‐1 in vitroCitation26,27. CRP also induces the in vitro expression and activity of plasminogen activator inhibitor‐1 (PAI‐1) and reduces the expression and activity of endothelial nitric oxide synthase (eNOS) Citation29,30. Therefore, it persuades vasoconstriction, LDL oxidation, and atherothrombosis. Recently, Wadham et al. Citation28 demonstrated that reconstituted HDLs can inhibit the CRP‐induced expression of CAMs in endothelial cells. Thereby HDL may counteract the CRP‐mediated proinflammatory activity in the circulation.
Macrophages, endothelial cells and vascular smooth muscle cells express VCAM‐1, ICAM‐1 and E‐selectin Citation12. Cytokines up‐regulate this expression as a response to the presence of CHD risk factors such as smoking, hypertension, hypercholesterolemia, hypertriglyceridemia, and insulin resistance. The capability of HDL to inhibit the cytokine‐induced CAM expression is potentially highly antiatherogenic Citation13. HDL inhibits the CAM expression by suppressing the tumor necrosis factor‐alpha (TNF‐α)‐stimulated sphingosine kinase activity in endothelial cells Citation29. The phospholipid composition of HDL modulates the inhibition of VCAM‐1 expression, and HDL‐associated lysosphingolipids, including sphingosylphosphorylcholine and lysosulfatide, reduce the TNF‐α‐induced E‐selectin expression Citation30,31. Recently, Calabresi et al. suggested that HDL has an in vivo effect on CAM expression, as increasing HDL‐C levels by fenofibrate treatment resulted in reduction in the sCAM levels Citation14. However, fibric acid derivatives may have a direct inhibitory effect on the expression of VCAM‐1 and ICAM‐1 in endothelial cells, through the activation of peroxisome‐proliferator‐activated receptor‐alpha Citation32.
Our findings in Finnish subjects with familial low HDL are complementary to those reported by Calabresi et al., who showed that the levels of sICAM‐1 and sE‐selectin are elevated in subjects with low HDL and elevated TG, BMI, and glucose, as well as in subjects with isolated low‐HDL trait Citation14. However, hsCRP levels or the presence of the MetS were not examined in this previous study. The present study is the first comprehensive analysis of hsCRP, sICAM‐1, sVCAM‐1 and sE‐selectin in subjects with familial low HDL‐C, and of the role of the insulin resistance and different HDL apolipoproteins in relation to the inflammatory markers. We observed that the subjects fulfilling the diagnostic criteria for the MetS had the highest levels of sICAM‐1, sVCAM‐1, sE‐selectin, and hsCRP. The strongest independent predictor of serum hsCRP level among the low‐HDL subjects was BMI, emphasizing the association between inflammation and obesity.
Previous reports have established the positive association between increased body fatness and increased plasma CRP and sCAM levels, both in healthy subjects Citation3,Citation33 as well as in insulin resistant individuals Citation4,5,Citation34. In the correlation analyses, positive associations were observed between sE‐selectin and hsCRP, and fasting insulin as well as HOMA IR. In fact, previous reports have connected elevated sE‐selectin to insulin resistance, hyperglycemia, and hyperinsulinemia, suggesting that high sE‐selectin level could have a specific relationship to the diabetic vascular changes, and not to atherosclerosis per se Citation5,Citation16. In our study, the subjects with familial low HDL presented a complex phenotype that is commonly observed in familial combined hyperlipidemia (FCHL) or in the metabolic syndrome, a phenotype characterized by elevated fasting insulin levels, as well as higher BMI, TGs, and apoB, despite that these factors were taken into account in the proband selection. The affected subjects also had major rearrangement in the HDL subpopulation profile towards the smaller particles; they had markedly decreased HDL2‐C, LpA‐I and apoA‐I, which is typically seen in patients with hypertriglyceridemia. The observation that low HDL‐C share many phenotypic features with FCHL and insulin resistance suggests a partially overlapping molecular pathogenesis behind these traits. This was also supported by our previous pooled data analysis of the genome scans for low HDL‐C and FCHL, which identified three shared chromosomal regions for a qualitative HDL‐C trait on 8q23.1, 16q23.3, and 20q13.32 Citation19. In turn, the known molecular defects in candidate genes including ApoAI/CIII/AIV cluster, apoA‐II, and ABCA1 have been tested and excluded Citation19, Citation35. Interestingly, the loci on 16q24.1‐24.2 and 20q13.11 have previously shown association with elevated TGs, insulin resistance, and Type 2 diabetes Citation36–38. Therefore, clinical phenotypes and presumably the genetic regulation of the familial low HDL, the FCHL, and the metabolic syndrome overlap. This is such a fundamental characteristic of our low‐HDL family members that it might not be feasible to separate the pure ‘low HDL’ effect from this study sample, in a similar manner that can be done when selecting individuals just on a basis of low HDL‐C, with no requirement of familial predisposition or the presence of premature CHD in the family. Therefore, it is even more important to elucidate the markers of endothelial activation and, thereby, predictors of vascular injury in these subjects. Our results show that the subjects with familial low HDL have indications of chronic inflammatory process on the vascular endothelium, which potentially predispose them to an increased risk of an atherosclerotic disease. Thus, in these subjects, low HDL‐C level may substitute for a surrogate marker of the pro‐atherogenic disturbances, and thereby may even precede the manifestations of decreased insulin sensitivity, namely the metabolic syndrome or type 2 diabetes.
In summary, subjects with familial low HDL exhibited increases of sICAM‐1, sVCAM‐1, sE‐selectin and hsCRP. The clinical phenotype of the familial low‐HDL trait overlaps with the MetS. In these subjects, the features of the MetS coexist with low‐grade inflammation, as indicated by increased levels of the inflammatory marker hsCRP. Our major finding is that the insulin resistance predisposes subjects with familial low HDL to increased endothelial activation, increased inflammatory cytokine expression and, thereby, to increased risk for atherosclerotic disease.
Acknowledgements
We thank the study subjects for participating in this study. Virve Naatti, Helinä Perttunen‐Nio and Hannele Hilden are thanked for their excellent technical assistance. This work was supported by grants from the Finnish Cardiovascular Research Foundation, Special State Grants for health science research, The Finnish Medical Society Duodecim, The Paulo Foundation, Aarne Koskelo Foundation and Maud Kuistila Foundation.
References
- Genest J. J., Martin‐Munley S. S., McNamara J. R., Ordovas J. M., Jenner J., Myers R. H., et al. Familial lipoprotein disorders in patients with premature coronary artery disease. Circulation 1992; 85: 2025–33
- Rashid S., Uffelman K. D., Lewis G. F. The mechanism of HDL lowering in hypertriglyceridemic, insulin‐resistant states. J Diabetes Complications 2002; 16: 24–8
- Yudkin J. S., Stehouwer C. D., Emeis J. J., Coppack S. W. C‐reactive protein in healthy subjects: associations with obesity, insulin resistance, and endothelial dysfunction: a potential role for cytokines originating from adipose tissue?. Arterioscler Thromb Vasc Biol 1999; 19: 972–8
- Festa A., D'Agostino R., Jr., Howard G., Mykkanen L., Tracy R. P., Haffner S. M. Chronic subclinical inflammation as part of the insulin resistance syndrome: the Insulin Resistance Atherosclerosis Study (IRAS). Circulation 2000; 102: 42–7
- Leinonen E., Hurt‐Camejo E., Wiklund O., Hulten L. M., Hiukka A., Taskinen M. R. Insulin resistance and adiposity correlate with acute‐phase reaction and soluble cell adhesion molecules in type 2 diabetes. Atherosclerosis 2003; 166: 387–94
- Lee W. Y., Park J. S., Noh S. Y., Rhee E. J., Sung K. C., Kim B. S., et al. C‐reactive protein concentrations are related to insulin resistance and metabolic syndrome as defined by the ATP III report. Int J Cardiol 2004; 97: 101–6
- Van Lenten B. J., Navab M., Shih D., Fogelman A. M., Lusis A. J. The role of high‐density lipoproteins in oxidation and inflammation. Trends Cardiovasc Med 2001; 11: 155–61
- Tietge U. J., Maugeais C., Lund‐Katz S., Grass D., deBeer F. C., Rader D. J. Human secretory phospholipase A2 mediates decreased plasma levels of HDL cholesterol and apoA‐I in response to inflammation in human apoA‐I transgenic mice. Arterioscler Thromb Vasc Biol 2002; 22: 1213–8
- Oram J. F. HDL apolipoproteins and ABCA1: partners in the removal of excess cellular cholesterol. Arterioscler Thromb Vasc Biol 2003; 23: 720–7
- Barter P. J., Nicholls S., Rye K. A., Anantharamaiah G. M., Navab M., Fogelman A. M. Antiinflammatory Properties of HDL. Circ Res 2004; 95: 764–72
- Ross R. Atherosclerosis—an inflammatory disease. N Engl J Med 1999; 340: 115–26
- Blankenberg S., Barbaux S., Tiret L. Adhesion molecules and atherosclerosis. Atherosclerosis 2003; 170: 191–203
- Cockerill G. W., Huehns T. Y., Weerasinghe A., Stocker C., Lerch P. G., Miller N. E., et al. Elevation of plasma high‐density lipoprotein concentration reduces interleukin‐1‐induced expression of E‐selectin in an in vivo model of acute inflammation. Circulation 2001; 103: 108–12
- Calabresi L., Gomaraschi M., Villa B., Omoboni L., Dmitrieff C., Franceschini G. Elevated soluble cellular adhesion molecules in subjects with low HDL‐cholesterol. Arterioscler Thromb Vasc Biol 2002; 22: 656–61
- Lundman P., Eriksson M. J., Silveira A., Hansson. L. O., Pernow J., Ericsson C. G., et al. Relation of hypertriglyceridemia to plasma concentrations of biochemical markers of inflammation and endothelial activation (C‐reactive protein, interleukin‐6, soluble adhesion molecules, von Willebrand factor, and endothelin‐1). Am J Cardiol 2003; 91: 1128–31
- Kowalska I., Straczkowski M., Szelachowska M., Kinalska I., Prokop J., Bachorzewska‐Gajewska H., et al. Circulating E‐selectin, vascular cell adhesion molecule‐1, and intercellular adhesion molecule‐1 in men with coronary artery disease assessed by angiography and disturbances of carbohydrate metabolism. Metabolism 2002; 51: 733–6
- Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). JAMA 2001; 285: 2486–97
- Ridker P. M., Buring J. E., Cook N. R., Rifai N. C‐reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8‐year follow‐up of 14 719 initially healthy American women. Circulation 2003; 107: 391–7
- Soro A., Pajukanta P., Lilja H. E., Ylitalo K., Hiekkalinna T., Perola M., et al. Genome scans provide evidence for low‐HDL‐C loci on chromosomes 8q23, 16q24.1‐24.2, and 20q13.11 in Finnish families. Am J Hum Genet 2002; 70: 1333–40
- Vartiainen E., Jousilahti P., Alfthan G., Sundvall J., Pietinen P., Puska P. Cardiovascular risk factor changes in Finland, 1972–1997. Int J Epidemiol 2000; 29: 49–56
- Friedewald W. T., Levy R. I., Fredrickson D. S. Estimation of the concentration of low‐density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem 1972; 18: 499–502
- Parra H. J., Mezdour H., Ghalim N., Bard J. M., Fruchart J. C. Differential electroimmunoassay of human LpA‐I lipoprotein particles on ready‐to‐use plates. Clin Chem 1990; 36: 1431–5
- Taskinen M. R., Kuusi T., Helve E., Nikkila E. A., Yki‐Jarvinen H. Insulin therapy induces antiatherogenic changes of serum lipoproteins in noninsulin‐dependent diabetes. Arteriosclerosis 1988; 8: 168–77
- Matthews D. R., Hosker J. P., Rudenski A. S., Naylor B. A., Treacher D. F., Turner R. C. Homeostasis model assessment: insulin resistance and beta‐cell function from fasting plasma glucose and insulin concentrations in man. Diabetologia 1985; 28: 412–9
- Ridker P. M., Rifai N., Rose L., Buring J. E., Cook N. R. Comparison of C‐reactive protein and low‐density lipoprotein cholesterol levels in the prediction of first cardiovascular events. N Engl J Med 2002; 347: 1557–65
- Pasceri V., Willerson J. T., Yeh E. T. Direct proinflammatory effect of C‐reactive protein on human endothelial cells. Circulation 2000; 102: 2165–8
- Pasceri V., Cheng J. S., Willerson J. T., Yeh E. T., Cheng J. Modulation of C‐reactive protein‐mediated monocyte chemoattractant protein‐1 induction in human endothelial cells by anti‐atherosclerosis drugs. Circulation 2001; 103: 2531–4
- Wadham C., Albanese N., Roberts J., Wang L., Bagley C. J., Gamble J. R., et al. High‐density lipoproteins neutralize C‐reactive protein proinflammatory activity. Circulation 2004; 109: 2116–22
- Xia P., Vadas M. A., Rye K. A., Barter P. J., Gamble J. R. High density lipoproteins (HDL) interrupt the sphingosine kinase signaling pathway. A possible mechanism for protection against atherosclerosis by HDL. J Biol Chem 1999; 274: 33143–7
- Nofer J. R., Geigenmuller S., Gopfert C., Assmann G., Buddecke E., Schmidt A. High density lipoprotein‐associated lysosphingolipids reduce E‐selectin expression in human endothelial cells. Biochem Biophys Res Commun 2003; 310: 98–103
- Baker P. W., Rye K. A., Gamble J. R., Vadas M. A., Barter P. J. Phospholipid composition of reconstituted high density lipoproteins influences their ability to inhibit endothelial cell adhesion molecule expression. J Lipid Res 2000; 41: 1261–7
- Marx N., Sukhova G. K., Collins T., Libby P., Plutzky J. PPARalpha activators inhibit cytokine‐induced vascular cell adhesion molecule‐1 expression in human endothelial cells. Circulation 1999; 99: 3125–31
- Greenfield J. R., Samaras K., Jenkins A. B., Kelly P. J., Spector T. D., Gallimore J. R., et al. Obesity is an important determinant of baseline serum C‐reactive protein concentration in monozygotic twins, independent of genetic influences. Circulation 2004; 109: 3022–8
- Lemieux I., Pascot A., Prud'homme D., Almeras N., Bogaty P., Nadeau A., et al. Elevated C‐reactive protein: another component of the atherothrombotic profile of abdominal obesity. Arterioscler Thromb Vasc Biol 2001; 21: 961–7
- Lilja H. E., Soro A., Ylitalo K., Nuotio I., Viikari J. S., Salomaa V., et al. A candidate gene study in low HDL‐cholesterol families provides evidence for the involvement of the APOA2 gene and the APOA1C3A4 gene cluster. Atherosclerosis 2002; 164: 103–11
- Cederberg A., Gronning L. M., Ahren B., Tasken K., Carlsson P., Enerback S. FOXC2 is a winged helix gene that counteracts obesity, hypertriglyceridemia, and diet‐induced insulin resistance. Cell 2001; 106: 563–73
- Yanagisawa K., Hingstrup Larsen L., Andersen G., Drivsholm T., Cederberg A., Westergren R., et al. The FOXC2 ‐512C>T variant is associated with hypertriglyceridaemia and increased serum C‐peptide in Danish Caucasian glucose‐tolerant subjects. Diabetologia 2003; 46: 1576–80
- Ghosh S., Watanabe R. M., Valle T. T., Hauser E. R., Magnuson V. L., Langefeld C. D., et al. The Finland‐United States investigation of non‐insulin‐dependent diabetes mellitus genetics (FUSION) study. I. An autosomal genome scan for genes that predispose to type 2 diabetes. Am J Hum Genet 2000; 67: 1174–85