Abstract
The extracellular signal‐regulated kinase (ERK) signaling pathway is a major determinant in the control of diverse cellular processes such as proliferation, survival, differentiation and motility. This pathway is often up‐regulated in human tumors and as such represents an attractive target for the development of anticancer drugs. Because of its multiple roles in the acquisition of a complex malignant phenotype, specific blockade of the ERK pathway is expected to result in not only an anti‐proliferative effect but also in anti‐metastatic and anti‐angiogenic effects in tumor cells. Recently potent small‐molecule inhibitors targeting the components of the ERK pathway have been developed. Among them, BAY 43‐9006 (Raf inhibitor), and PD184352, PD0325901 and ARRY‐142886 (MEK1/2 inhibitors) have reached the clinical trial stage. We briefly discuss the possibility that combination of ERK pathway inhibitors (cytostatic agents) and conventional anticancer drugs (cytotoxic agents) provides an excellent basis for the development of new chemotherapeutic strategies against cancer.
| Abbreviations | ||
| ATF2 | = | activating transcription factor 2 |
| Bad | = | bcl‐2 associated death promoter |
| Bcl‐2 | = | B cell leukemia‐2 |
| Bim | = | bcl‐2 interacting mediator of cell death |
| CAS | = | Crk‐associated substrate |
| Crk | = | CT10 regulator of kinase |
| ECM | = | extracellular matrix |
| EGFR | = | epidermal growth factor receptor |
| ERK1/2 | = | extracellular signal‐regulated kinase 1/2 |
| FAK | = | focal adhesion kinase |
| FGFR | = | fibroblast growth factor receptor |
| Grb2 | = | growth factor receptor‐bound protein 2 |
| HIF‐1α | = | hypoxia inducible factor‐1α |
| MAP2/4 | = | microtubule associated protein 2/4 |
| MAPK | = | mitogen‐activated protein kinase |
| MAPKK | = | MAPK kinase |
| MAPKKK | = | MAPKK kinase |
| MEK1/2 | = | MAP kinase/ERK kinase 1/2 |
| MEKK | = | MEK kinase |
| MLCK | = | myosin light chain kinase |
| MNK1/2 | = | MAP kinase interacting kinase 1/2 |
| PDGFR | = | platelet derived growth factor receptor |
| PKC | = | protein kinase C |
| PLC‐γ | = | phospholipase C‐γ |
| Rsk | = | ribosomal S6 kinase |
| RTK | = | receptor tyrosine kinase |
| Shc | = | src homologous and collagen |
| SOS | = | son of sevenless |
| Stat | = | signal transducers and activators of transcription |
| VEGFR | = | vascular endothelial growth factor receptor |
Introduction
The extracellular signal‐regulated kinase (ERK) pathway, also called the 42‐/44‐kDa mitogen‐activated protein kinase (MAPK) pathway, is activated in a variety of cell types by diverse extracellular stimuli and is among the most thoroughly studied of signaling pathways that connect different membrane receptors to the nucleus Citation1–3. Activation of the ERK pathway involves the guanosine triphosphate (GTP)‐loading of Ras at the plasma membrane, and the sequential activation of a series of protein kinases (). Initially, activated Ras recruits the Raf family of kinases such as Raf‐1 to the plasma membrane, a key step in a complex activation process that is not yet fully resolved. Raf‐1 acts as a MAPK kinase kinase and activates MAPK/ERK kinase 1 and 2 (MEK1/2; also called MAPK kinase1/2) by serine phosphorylation. MEK1/2, dual‐specificity protein kinases, then catalyze the phosphorylation of ERK1 and ERK2 (p44 MAPK and p42 MAPK, respectively) on tyrosine and threonine residues. When activated, ERK1/2 phosphorylate various downstream substrates involved in a multitude of cellular responses such as cell proliferation, cell differentiation, cell survival and cell motility Citation1–3.
Figure 1. Schematic representation of the extracellular signal‐regulated kinase (ERK) pathway focusing on its converging function of diverse intracellular signals. The mitogen‐activated protein (MAP) kinase cascade is composed of three sequential kinases: MAP kinase kinase kinase (MAPKKK), MAP kinase kinase (MAPKK), and MAP kinase (MAPK). ERK1/2 phosphorylate a variety of nuclear, cytosolic and cytoskeletal targets. Integrin‐focal adhesion kinase (FAK) pathway, which is activated by adhesion of integrins to specific extracellular matrix (ECM) molecules, is another important signaling to fully activate the ERK pathway Citation72,73. Activation of the ERK pathway is most often associated with cell proliferation, cell survival and cell migration. Well‐characterized inhibitors of Raf and MEK1/2 are shown. Several negative regulators of this pathway exist, such as MAP kinase phosphatases (MKPs/DUSPs) and Sprouty proteins. Expression of MKPs and Sprouty proteins is induced in an ERK‐dependent manner, and thus these proteins participate in the negative feedback regulatory loop of the ERK pathway (not shown in this Figure; see text).
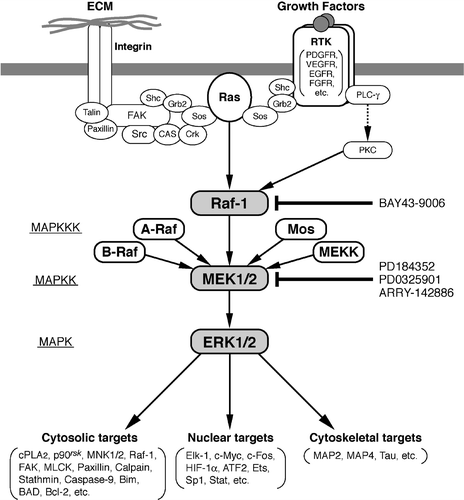
Very precise spatio‐temporal control mechanisms for intracellular pathways have evolved to ensure homeostasis in multicellular organisms. Such mechanisms have been particularly well studied for ERK signaling Citation4. Accordingly, inappropriate activation of these pathways would result in the induction of several refractory diseases. In fact, aberrant activation of the ERK pathway has been shown to be an essential feature common to many types of human tumors Citation5–7. Thus, interest in the components of the ERK signaling pathway as attractive targets for cancer chemotherapy has exploded in the past few years Citation8–10.
In this review, the current status of inhibitors of the ERK pathway will be presented, with focus on the potential application of small‐molecule inhibitors of Raf‐1 and MEK1/2 as anticancer drugs. Recent data from clinical trials indicate that targeted inhibition of the ERK signaling pathway has evolved as a viable approach for anticancer therapy.
Key messages
The extracellular signal‐regulated kinase (ERK) signaling pathway is a major determinant in the control of proliferation, survival, and differentiation.
ERK pathway is often up‐regulated in human tumors and as such represents an attractive target for anticancer drugs development.
Combining ERK pathway inhibitors with conventional anticancer drugs represents a promising new chemotherapeutic strategy against cancer.
Rationale for targeting the ERK signaling pathway to treat cancer
Constitutive activation of ERK1/2 in human cancers
Aberrant activation of signal transducing proteins has been linked to cancer. In this respect, ERK1/2 are activated by a wide variety of mitogenic stimuli that interact with structurally distinct receptors and thus represents a convergence point for the majority, if not all, of the mitogenic signaling pathways Citation1–3. Thus, aberrant activation of any signaling molecule that functions upstream of ERK1/2 () would culminate in the constitutive activation of these kinases, leading to tumorigenesis. Accordingly, over‐expression or activating mutations of epidermal growth factor (EGF) receptors (frequently detected in cancers of the lung, breast, colon, ovary, bladder, etc.) Citation11,12, activating mutations of ras (cancers of the pancreas, colon, lung, etc.) Citation13,14, or activation mutations of raf (melanomas, cancers of the colon, ovary, thyroid, etc.) Citation15,16 have been detected, with concomitant activation of MEK1/2 and ERK1/2 in the majority of cases.
Down‐regulation of negative regulators of the ERK pathway, such as mitogen‐activated protein (MAP) kinase phosphatases (MKPs) and Sprouty proteins, would also result in the constitutive activation of ERK1/2. MKPs are dual‐specificity phosphatases (DUSPs) that dephosphorylate phosphothreonine and phosphotyrosine residues within MAP kinases and inactivate them: among MKPs/DUSPs, MKP‐3/DUSP6 is highly specific for ERK1/2 inactivation Citation17. Sprouty proteins bind to several signaling molecules, such as Grb2, Sos and Shp2, and interfere in their interaction with other signaling molecules, leading to the interruption of the ERK pathway Citation18. Expression of MKPs/DUSPs and Sprouty proteins is induced in an ERK‐dependent manner, and thus these proteins participate in the negative feedback regulatory loop of the ERK pathway Citation17,18. Recently, suppressed expression of Sprouty1 in prostate cancer Citation19 or Sprouty1/2 in breast cancer Citation20, and down‐regulation of MKP1/DUSP1 in ovarian cancer Citation21 have been reported; constitutive activation of ERK1/2, however, has not been shown in these cases.
Thus far, no activating mutation in MEK1/2 or ERK1/2 has been detected in human cancers. In this respect, we have shown that constitutive activation of ERK1/2 is always associated with the activation of MEK1/2 in tumor cell lines as well as in primary tumor tissues. Furthermore, co‐activation of MEK1/2 and Raf‐1 has been detected in almost all the tumor cells analyzed Citation4. These results suggest that the constitutive activation of ERK1/2 in tumor cells is not due to the aberrant activation of ERK1/2 or MEK1/2, but is the consequence of activation mutation of some of the signaling molecules that function upstream of MEK1/2. Because of the key role of MEK1/2 and ERK1/2 in the transduction of cell signaling, any mutation in these molecules might not be tolerable for cell survival Citation22.
Specific inhibition of the ERK pathway is expected to effectively intercept aberrant mitogenic signals in cancer cells
As discussed above, the major cause of constitutive activation of the ERK pathway in tumor cells is inappropriate activation of signaling molecules that stand upstream of the pathway, e.g. growth factor receptor tyrosine kinases (RTKs) and Ras. Accordingly, specific inhibitors of the EGF receptor tyrosine kinase are currently being developed and examined for therapeutic application as anticancer drugs; these include Gefitinib, Erlotinib, Lapatinib and IMC‐C225 Citation23. Recently, Gefitinib has been approved in several countries, including Japan and the United States, for the treatment of non‐small‐cell lung cancer (NSCLC). Sensitivity of cancer cells to Gefitinib depends on the somatic mutations within the EGF receptor kinase domain, but not on the over‐expression of the EGF receptor Citation12.
The signaling activity of Ras is crucially dependent on its association with the inner face of the plasma membrane, for which the key post‐translational modification that places a farnesyl group on a cysteine residue near the C‐terminus of Ras is required. This modification is catalyzed by farnesyltransferase (FTase). Accordingly, FTase inhibitors have been developed as anti‐Ras compounds; these include FTI‐276, FTI‐2148, L‐739,750, BZA‐2B and SCH66336 Citation24,25. Recently, however, the antitumor activity of these FTase inhibitors has been suggested to not depend simply on the inhibition of Ras but also to depend on the inhibition of other farnesylated proteins. Furthermore, FTase inhibitor inhibition of the Rab geranyltransferase activity has recently been reported Citation26. Crucial targets of FTase inhibitors for exhibiting the antitumor activity remain to be elucidated.
The components of the ERK pathway, such as Raf‐1 and MEK1/2, represent excellent targets for the development of anticancer drugs: because of the converging function of these signaling molecules (), their specific inhibition is expected to quite effectively intercept a wide variety of mitogenic signals. In fact, a MEK inhibitor PD98059, for example, totally inhibits the proliferation of HT1080 fibrosarcoma cells in which the ERK pathway is constitutively activated as a result of activation mutation of N‐rasCitation27. PD98059 also inhibits the proliferation of PC9 NSCLC cells, in which a deletion mutation in the kinase domain of the EGF receptor has been detected to induce its aberrant activation. Mutated gene products with elevated activity themselves do not necessarily represent the most suitable therapeutic targets.
Inhibition of the ERK pathway is expected to result in anti‐metastatic as well as anti‐angiogenic effects in cancer cells
Cell motility is a fundamental process that is required during normal embryonic development, wound repair, inflammatory response and tumor metastasis Citation28. In this respect, the ERK signaling pathway has been shown to be involved in the regulation of cell motility. For example, the ERK pathway plays an essential role in the induction of epithelial cell motility in response to hepatocyte growth factor (HGF) Citation29. HGF‐induced activation of the ERK pathway is linked to the expression of the matrix metalloproteinase (mmp)‐9 gene, and MMP‐9 activity is required for the induction of cell motility via the degradation of the extracellular matrix Citation30. Elevated expression of MMPs is associated with increased metastatic potential in many tumor cells, and inhibition of MMP activity results in the reduction of tumor invasion and metastasis Citation31. Transfection of a constitutive active form of MEK1 induces increased expression of MMP2/9 and confers metastatic potential to NIH3T3 cells Citation32. Alternatively, implication of the ERK pathway in the activation of the cell's motility machinery has been reported, in which ERK1/2 phosphorylates and enhances the myosine light‐chain kinase (MLCK) activity leading to increased MLC phosphorylation and enhanced cell migration Citation33.
New blood vessel formation (angiogenesis) is another fundamental process required for efficient tumor growth and metastatic dissemination. An important master regulator of angiogenesis is the hypoxia‐inducible factor 1α (HIF‐1α), which regulates the expression of critical pro‐angiogenic genes, such as the vascular endothelial growth factor (VEGF) Citation34,35. Several studies suggest that HIF‐1α function is regulated by MAP kinases. Specifically it has been shown that ERK1/2 phosphorylates HIF‐1α and Sp1 directly, a double action inducing transcriptional expression of VEGF Citation35–37. In addition, sustained activation of the ERK pathway has been reported to be required for basic fibroblast growth factor‐induced angiogenesis Citation38.
All these observations indicate that, in addition to the anti‐proliferative effect, specific inhibition of the ERK pathway is expected to result in anti‐metastatic as well as anti‐angiogenic effects in tumor cells. In accordance with these possibilities, specific blockade of the ERK pathway, for example, has been shown to inhibit the disruption of cell‐cell contact and motility required for the metastatic process in colon tumor cells Citation39 and, furthermore, to inhibit the invasiveness of tumor cells through the down‐regulation of MMP‐3/‐9/‐14 and CD44 Citation40.
Specific inhibitors of the ERK pathway
Raf inhibitors
The major cause of constitutive activation of the ERK pathway in human tumors is the result of activating mutations in Raf, Ras or some other signaling molecules upstream of Ras Citation4. In this respect, B‐Raf has been reported to be mutated in 70% of malignant melanomas Citation15, in 33% of papillary thyroid carcinomas Citation41, and in lower frequencies in other cancers Citation42. Approximately 90% of the mutations involve the replacement of Val599 with a glutamic acid residue, which lies within the kinase activation loop. Recent crystal structure studies of wild‐type and mutant B‐Rafs indicate that V599E mutation acts to destabilize the inactive conformation of the protein kinase, favoring the activated state of the enzyme Citation43. Expression of V599EB‐Raf in cultured melanocytes induces the constitutive activation of the ERK pathway and tumorigenicity in nude mice Citation44. Furthermore, small interfering‐RNA knockdown of V599EB‐Raf, but not Raf‐1, abrogates the transformed phenotype of WM793 human melanoma cells as assessed by colony formation in soft agar Citation45, indicating that such activated B‐Raf is an attractive target for cancer chemotherapy. Although several small‐molecule Raf inhibitors have been reported, only one of them, BAY 43‐9006, has reached the clinical trial stage ().
Figure 2. Specific inhibitors of the extracellular signal‐regulated kinase (ERK) pathway. Raf inhibitor: BAY 43‐9006. MEK inhibitors: PD98059, U0126, PD184352 (CI‐1040), PD0325901, ARRY‐14886.
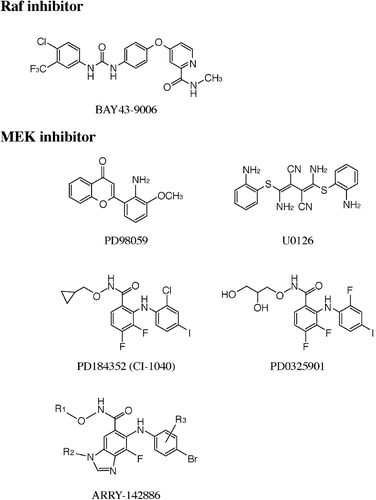
BAY 43‐9006 was identified as a potent inhibitor of Raf‐1 by screening many thousands of chemical compounds using a combination of an in vitro Raf kinase biochemical assay and a tumor cell‐based mechanistic assay Citation46. BAY 43‐9006, a novel bi‐aryl urea, inhibits Raf‐1 kinase activity in vitro with an IC50 value of 6 nM, B‐Raf (wild‐type) (IC50, 22 nM), and V599EB‐Raf (IC50, 38 nM) Citation47. The crystal structure of the B‐Raf/BAY 43‐9006 complex revealed that the inhibitor binds in the adenosine triphosphate (ATP) pocket and interacts with residues of the kinase activation loop of Raf proteins. This interaction prevents the activation loop and the catalytic residues from adopting a conformation that is competent to bind and phosphorylate substrates Citation43. BAY 43‐9006 does not directly inhibit other protein kinases (ERK1, MEK1, EGF receptor, insulin‐like growth factor (IGF) receptor‐1, c‐Met, HER2, Akt, etc.), except that it inhibits certain pro‐angiogenic receptor tyrosine kinases (RTKs), such as VEGF receptor‐2 (IC50 value of 90 nM), VEGF receptor‐3 (IC50, 20 nM), Flt‐3 (IC50, 58 nM), c‐Kit (IC50, 68 nM), and platelet‐derived growth factor (PDGF) receptor‐β (IC50, 57 nM) Citation47.
BAY 43‐9006 exhibits broad‐spectrum antitumor activity in human tumor xenograft models of colon, pancreas, breast and non‐small‐cell lung origin with mutation in B‐Raf or K‐Ras. The anti‐tumor efficacy of BAY 43‐9006 is closely associated with the inhibition of ERK activation in many, but not all, xenograft models. In contrast, significant inhibition of neovascularization is observed in all the cases examined. Certainly this inhibition reflects the suppression of VEGF expression as a direct consequence of inhibition of the ERK pathway Citation35 and in addition could also be the result of VEGF‐R2 inhibition. These results indicate that BAY 43‐9006 functions as a novel dual‐action Raf kinase and VEGF receptor inhibitor that prevents tumor growth by combining two anticancer activities: inhibition of tumor cell proliferation and tumor angiogenesis Citation47.
BAY 43‐9006, an orally active compound, is now undergoing clinical evaluation. Results of a recent phase I trial have shown that BAY 43‐9006 is generally well tolerated by patients with advanced refractory solid tumors Citation48. The drug‐related toxicities were mostly mild to moderate, and of a gastrointestinal and dermatologic nature, i.e. diarrhea and rash. Among the 45 patients treated continuously with BAY 43‐9006 at doses of ⩾100 mg b.i.d., 1 patient had a partial response (hepatocellular carcinoma), 25 patients had stable disease (colorectal carcinoma (14 patients), hepatocellular carcinoma (4 patients), etc.), with 8 patients lasting >6 months and 5 patients for >12 months Citation49. Phase II/III trials are currently being carried out in patients with several different tumor types, including melanoma, renal‐cell, colorectal and hepatocellular carcinomas ().
Table I. Clinical trials of ERK pathway inhibitors.
MEK1/2 inhibitors
MEK1/2 have extremely unique characteristics among the components of the ERK pathway. Although Raf‐1 is the major activator of MEK1/2, these kinases are activated also by several other kinases, such as Mos, A‐Raf, and B‐Raf. On the other hand, no substrates for MEK1/2 have been identified other than ERK1 and ERK2. In addition, MEK1/2 are dual‐specificity kinases that phosphorylate both tyrosine and threonine residues, e.g. MEK1/2 sequentially phosphorylates ERK1 at 185Tyr and then at 183Thr. Furthermore, MEK1/2 stand at the focal point of many mitogenic signaling pathways to integrate them into the ERK pathway. All these very prominent features, i.e. unusually restricted and unique substrate specificities and the integrating role of mitogenic signaling pathways, highlight MEK1/2 as an excellent target for the development of inhibitors against the ERK pathway Citation8–10. Currently, specific and potent MEK1/2 inhibitors have been developed ().
PD98059, the first pharmacological inhibitor of MEK1/2, was identified by screening a compound library with an in vitro cascade assay that measured the inhibition of ERK1 activation by a constitutive active mutant MEK1 recombinant protein Citation50. Meanwhile, U0126 was discovered by screening a total of 40,000 compounds in a cell‐based reporter assay that measured the inhibition of phorbol ester‐stimulated AP‐1 transactivation Citation51. Because of their pharmaceutical limitations, PD98059 and U0126 have not been tested in clinical trials but have become widely used reagents to elucidate the role of the ERK pathway in a variety of biological processes in in vitro cell culture systems. For example, PD98059 and U0126 have been shown to completely suppress the proliferation of RPMI‐SE (renal cell carcinoma) and HT1060 (fibrosarcoma) cells in which the ERK pathway is constitutively activated, through a mechanism that involves the up‐regulation of p27Kip1, association of p27Kip1 with cyclin E‐cyclin‐dependent kinase (CDK) 2 complexes, concomitant inhibition of cyclin E‐CDK2 kinase activity, and consequent decrease in the phosphorylation state of the retinoblastoma protein. PD98059 also induces a modest apoptotic response in these tumor cells Citation27. These observations have strengthened the idea that the ERK pathway really represents an attractive target for the development of anticancer drugs. Subsequently, PD184352 (CI‐1040), an orally active MEK inhibitor with enhanced bioavailability, has been synthesized Citation39.
The majority of protein kinase inhibitors that have so far been developed are competitive with ATP and interact within the ATP binding site of the respective protein kinase. In this respect, PD98059, U0126 and PD184352 do not compete with ATP and thus represent unique protein kinase inhibitors. Such a peculiar characteristic may allow PD98059, U0126 and PD184352 to work as extremely specific inhibitors of MEK1/2; none of these compounds significantly inhibit the activity of a large panel of protein kinases that include ERK1, c‐Jun N‐terminal kinase (JNK) 1 and p38 MAP kinases in an in vitro assay Citation52. However, different experimental conditions employed in each study, e.g. usage of purified recombinant enzymes versus immunoprecipitated enzymes, wild‐type enzymes (activated) versus constitutively active mutant enzymes, or alternatively in vitro versus cell‐based assays, have introduced some confusion regarding the precise mechanism of action of PD98059, U0126 and PD184352 as MEK inhibitors. In this respect, results showing that the concentration of these compounds required to inhibit the activation of MEK1 in cells is 100–200‐fold lower than that required to inhibit the MEK1 activity in vitroCitation52 have led to propose that these inhibitors exert their effects on cells by an indirect mechanism in that they suppress the activation of MEK1/2 by upstream kinases, but do not inhibit MEK1/2 activity directly.
Recently, crystal structures of MEK1 and MEK2 have been determined as ternary complexes with Mg‐ATP and PD184352‐like inhibitors, proposing a novel, non‐competitive mechanism for protein kinase inhibition Citation53. The structures reveal that MEK1 and MEK2 each have a unique inhibitor‐binding site located in an interior hydrophobic pocket adjacent to, but not overlapping with, the Mg‐ATP‐binding site. The binding of PD184352‐like inhibitors induces several conformational changes in unphosphorylated MEK1 and MEK2 that lock them into closed and catalytically inactive species. It could be possible that such conformational changes also make MEK1 and MEK2 unphosphorylatable by upstream kinases. Notably, the PD184352‐like MEK inhibitor binding‐site is located in a region where the sequence homology to other protein kinases is quite low. With the exception of MEK2 (100% identical) and mitogen protein kinase kinase (MKK)5 (∼81% identical), all other protein kinases share low sequence identity (60%–70%) with MEK1 in the inhibitor‐binding site, which may explain why PD184352‐like MEK inhibitors are exceptionally specific for MEK1, MEK2 and MKK5 (although to a much lesser extent), but do not inhibit many other protein kinases.
PD184352 has been shown to exhibit a prominent growth inhibitory effect on human colon tumor xenografts in vivo (colon 26 and HT‐29 carcinomas in which the ERK pathway is constitutively activated) when given orally to mice every 12 hours for 2 weeks; under such conditions, very importantly, phosphorylation (activation) of ERK1/2 is efficiently suppressed. PD184352 also decreased the invasiveness as well as motility of HT‐29 cells Citation39. This study revealed, however, that PD184352 is metabolically rather unstable; although tumors excised after PD184352 treatment showed complete suppression of ERK1/2 phosphorylation through 6 hours, phosphorylation of ERK1/2 started to return by 12 hours and attained control levels by 24 hours after a single oral dose of the inhibitor.
PD184352 has been evaluated in phase I and phase II trials in patients with advanced solid tumors. A phase I study in 77 patients revealed that continuous administration of PD184352 (800 mg b.i.d.) with food for 21 days repeated every 28 days was well tolerated with a safety profile consisting primarily of mild diarrhea, asthenia, rash, nausea and vomiting. Under such conditions, tumor tissues obtained from patients showed markedly reduced levels of phosphorylated ERK1/2 (median 73%; range 46%–100%), indicating that PD184352 did affect its target in humans. One partial response lasting 355 days was achieved in a patient with pancreatic cancer, and 19 patients with a variety of tumors including non‐small‐cell lung, breast and colon cancer achieved stable disease lasting a median of 5.5 months (range 4–17 months) Citation54. On the basis of these encouraging phase I results, a phase II study has been carried out in 67 patients with advanced breast, colon, non‐small‐cell lung, and pancreatic cancer. However, results of this phase II trial were rather disappointing; no complete or partial response was observed, but stable disease lasting a median of 4.4 months (range 4–18 months) was confirmed in eight patients Citation55. Such insufficient efficacy has provided little support for further investigation of PD184352 in cancer treatment: the main difficulty seems to come from its pharmaceutical limitations such as poor solubility, high metabolic clearance and low bioavailability.
In order to improve several of the pharmaceutical limitations of PD184352 described above, its structure has been modified by replacement of the cyclopropylmethoxy group with a (R)‐dihydroxypropoxy group and the 2‐chloro substituent with a 2‐fluoro substituent on the second aromatic ring to make a second‐generation MEK inhibitor, PD0325901 Citation56. PD0325901 has markedly superior pharmacological and biopharmaceutical properties, which include a more than 50‐fold increased potency against MEK (IC50 values of 1 nM in in vitro assay and 0.43 nM in a cell‐based assay: those of PD184352 are 17 nM and 53 nM, respectively), much improved oral bioavailability, and longer duration of target suppression. Prominent anti‐cancer activity of PD0325901 has been demonstrated for a broad spectrum of human tumor xenografts. PD0325901 is currently being evaluated in Phase I/II clinical trials.
Recently, ARRY‐142886, a benzimidazole derivative, has been reported to potently inhibit MEK1/2 with IC50 values of 12 nM in an in vitro assay and 8 nM in a cell‐based assay Citation57,58. The mechanism of inhibition is non‐competitive with respect to ATP, which could ensure its strict specificity against MEK1/2 as in the case for other MEK inhibitors. ARRY‐14886, an orally bioavailable compound with low metabolic clearance, has been shown to exhibit promising anti‐tumor efficacy in several human xenograft models, which include colon, pancreas, breast and non‐small‐cell lung carcinomas and melanoma with V599EB‐raf or K‐/N‐ras mutations Citation59. Importantly, tumor growth inhibition correlates well with decreased phospho‐ERK levels. ARRY‐14886 has recently entered clinical trials.
So far, no specific and potent inhibitors against ERK1/2 have been reported.
Future perspectives
After initial active interest, ERKs are now becoming highly promising therapeutic targets for the development of anticancer drugs. Thus, a variety of inhibitors against the ERK pathway, especially small‐molecule compounds that selectively inhibit Rafs or MEK1/2, have been developed. Among them, orally active PD0325901 and ARRY‐142886 (second‐generation MEK inhibitors with much improved pharmaceutical properties) and BAY‐43‐9006 (it targets both Rafs and VEGFRs) have been chosen as promising clinical candidates to be validated for their therapeutic efficacy in patients with advanced solid tumors. Clearly, those who are expected to benefit from treatment with these inhibitors are patients with tumors in which constitutive activation of the ERK pathway is detected. In vitro analysis has indicated that tumor cells with higher ERK activation levels are more susceptible to MEK inhibitors for their proliferation Citation27. Analysis of biopsy samples, or alternatively tumor samples obtained at the time of surgery, for phospho‐ERK immunostaining would have to be mandatory to accurately select candidate patient populations that would benefit from this therapeutic strategy.
Inhibitors of the ERK pathway can be classified as ‘cytostatic’ but not as cytotoxic anticancer drugs. Such cytostatic agents may, by selectively inhibiting the activity of their respective target, suppress the growth of tumor cells in which abnormal activation of the corresponding target molecule occurs, but they by themselves may not ‘kill’ tumor cells. In fact, although specific blockade of the ERK pathway by treatment with PD98059 completely suppressed the growth of tumor cells in which the pathway is constitutively activated, by itself it showed only a modest effect on the induction of apoptosis Citation27. Furthermore, as the inhibitory activity of the ERK pathway inhibitors described above is reversible, their removal would induce the re‐initiation of tumor cell proliferation; the majority of tumor cells are just ‘resting’ but not dying in the presence of such cytostatic inhibitors.
In accordance with this idea, results of preclinical trials suggest that continuous exposure to BAY43‐9006 or PD0184352 is required for tumor growth inhibition: tumor growth seems suspended while the drug is present but returns to baseline rates when the inhibitor is withdrawn. Furthermore, although continuous dosing of PD184352 or BAY 43‐9006 resulted in stable disease in a significant number of patients with advanced solid tumors, no complete response was observed, and only a few patients had a partial response Citation49,Citation54,55. Although these inhibitors are generally well tolerated even under such continuous dosing conditions, some strategy to improve their anticancer activity would promise great therapeutic benefit for many tumor patients.
Combination therapy is becoming a rule, particularly in cancer treatment. It has generally been driven by safety considerations, i.e. combination of cytotoxic agents with non‐overlapping toxicities. In this respect, MEK inhibitors have been suggested to enhance the lethal action of diverse cytotoxic anticancer agents such as ara‐C (a deoxycytidine analogue which inhibits DNA replication), cisplatin (a DNA‐reactive agent which induces intrastrand crosslinks), paclitaxel/taxotere (taxane derivatives which stabilize microtubules) and vinblastine/vincristine (vinca alkaloids which bind to tubulin subunits and inhibit tubulin polymerization) Citation60. In this context, all these conventional chemotherapeutic drugs have been shown to induce the activation of the ERK pathway as well as the JNK/p38 MAPK pathways. It has generally been suggested that activation of the ERK pathway is associated with anti‐apoptotic processes while activation of the JNK/p38 MAPK pathways is linked to pro‐apoptotic processes. It has therefore been proposed that the balance between the ERK pathway and JNK/p38 MAPK pathways determines the fate of cells after various stresses Citation61,62. The ERK pathway has been linked to, for example, the phosphorylation of B‐cell leukemia (Bcl)‐2 that contributes to cell survival Citation63, the suppression of the apoptotic effect of Bcl‐2 associated death agonist (BAD) Citation64, the accumulation of the p53 tumor suppressor protein Citation65, and the up‐regulation of the anti‐apoptotic protein myeloid cell leukemia (MCL)‐1 Citation66. Thus, although the precise mechanism by which a combination of MEK inhibitors and cytotoxic drugs induces enhanced anticancer effects are largely unknown, specific interruption of the putatively cytoprotective ERK pathway could, by shifting the balance between pro‐ and anti‐apoptotic signaling, enhance the lethal actions of established cytotoxic anticancer drugs.
More specifically, enhancement of the therapeutic efficacy of Taxol by PD184352 has recently been reported in human heterotransplanted non‐small‐cell lung cancer tumor xenografts in vivoCitation67. Also, a combination of MEK inhibitors and microtubule‐depolymerizing agents (such as vincristine and TZT‐1027) has markedly enhanced the apoptosis‐inducing activity of the latter cytotoxic agents not only in in vitro culture of tumor cells but also in human colon tumor xenografts in vivoCitation68. Furthermore, striking pro‐apoptotic synergism was achieved when PD0325901 was combined with very low concentrations of histone deacetylase (HDAC) inhibitors such as trichostatin A and HC‐toxin; under such conditions, HDAC inhibitors by themselves did not exhibit significant apoptosis‐inducing activity Citation69. Importantly, such enhancing effects of MEK inhibitors on these cytostatic anticancer drugs have been observed only in tumor cells in which constitutive activation of the ERK pathway is detected.
Malignant behavior of the majority of human tumors is driven by the accumulation of several genetic and epigenetic aberrations but not by a single defect, suggesting that targeting a single signal transduction pathway may not be sufficiently efficacious. For example, constitutive activation of not only the ERK pathway (the major proliferation signaling Citation1–3) but also the PI3 kinase/Akt pathway (the major survival signaling Citation70,71) is detected in several tumor cells such as T24 bladder carcinoma cells. Thus, combination of multiple signal‐transduction inhibitors, e.g. ERK pathway inhibitors and PI3 kinase/Akt pathway inhibitors, represents another potential strategy to treat human cancers. Although all signal‐transduction inhibitors are classified as ‘cytostatic’ and not cytotoxic, complementary blockade of respective signaling pathway by these inhibitors could result in synergistic therapeutic efficacy.
Another promising alternative will be to combine ERK pathway inhibitors with anti‐angiogenic agents. However, precise temporal and sequential delivery protocols will have to be developed like treating first with anti‐ERKs and finishing with anti‐VEGF agents.
The ERK pathway surely represents a prime target for mechanism‐based therapeutic approaches for advanced solid tumors, and can now be clinically targeted by highly selective small‐molecule inhibitors such as BAY 43‐9006, PD0325901 and ARRY‐142886. From a practical point of view, combination of cytostatic ERK pathway inhibitors and cytotoxic anticancer drugs, or alternatively other cytostatic signal‐transduction inhibitors, would provide an excellent basis for the development of new chemotherapeutic strategies for cancer. As a matter of fact, the most promising potential benefit of ERK pathway inhibitors could be in such combination chemotherapy. These are no doubt very exciting times in the field of molecular targeted therapies, as a number of potent ERK pathway inhibitors get put to the real test.
Acknowledgements
We would like to thank Dr Christiane Brahimi‐Horn for critical reading of the manuscript, and all the colleagues in our laboratories for helpful discussion. This work was supported in part by Grants‐in‐aid for Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MK), and from the CNRS, the French Ministry of Education and Research, the Agency for Cancer Research (JP).
References
- Pages G., Lenormand P., L'Allemain G., Chambard J. C., Meloche S., Pouyssegur J. Mitogen‐activated protein kinases p42mapk and p44mapk are required for fibroblast proliferation. Proc Natl Acad Sci 1993; 90: 8319–23
- Seger R., Krebs E. G. The MAPK signaling cascade. FASEB J 1995; 9: 726–35
- Lewis T. S., Shapiro P. S., Ahn N. G. Signal transduction through MAP kinase cascades. Adv Cancer Res 1998; 74: 49–139
- Pouyssegur J., Lenormand P. Fidelity and Spatio‐temporal control in MAP kinase (ERKs) signaling. Eur J Biochem 2003; 270: 3291–9
- Hoshino R., Chatani Y., Yamori T., Tsuruo T., Oka H., Yoshida O., et al. Constitutive activation of the 41‐/43‐kDa mitogen‐activated protein kinase signaling pathway in human tumors. Oncogene 1999; 18: 813–22
- Gioeli D., Mandell J. W., Petroni G. R., Frierson HF J. r., Weber M. J. Activation of mitogen‐activated protein kinase associated with prostate cancer progression. Cancer Res 1999; 59: 279–84
- Oka H., Chatani Y., Hoshino R., Ogawa O., Kakehi Y., Terachi T., et al. Constitutive activation of mitogen‐activated protein (MAP) kinases in human renal cell carcinoma. Cancer Res 1995; 55: 4182–7
- English J. M., Cobb M. H. Pharmacological inhibitors of MAPK pathway. Trend Pharmacol Sci 2002; 23: 40–5
- Kohno M., Pouyssegur J. Pharmacological inhibitors of the ERK signaling pathway: application as an anticancer drugs. Prog Cell Cycle Res 2003; 5: 219–24
- Sebolt‐Leopold J. S., Herrera R. Targeting the mitogen‐activated protein kinase cascade to treat cancer. Nature Rev Cancer 2004; 4: 937–47
- Arteaga C. L. Epidermal growth factor receptor dependence in human tumors: more than just expression?. Oncologist 2002; 7: 31–9
- Paez J. G., Janne P. A., Lee J. C., Tracy S., Greulich H., Gabriel S., et al. EGFR mutations in lung cancer: correlation with clinical response to Gefitinib therapy. Science 2004; 304: 1497–500
- Cox A. D., Der C. J. Ras family signaling: therapeutic targeting. Cancer Biol Ther 2002; 1: 599–606
- Downward J. Targeting RAS signaling pathways in cancer therapy. Nature Rev. Cancer 2003; 3: 11–2
- Davies H., Bignell G. R., Cox C., Stephens P., Edkins S., Clegg S., et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417: 949–54
- Wan P. T. C., Garnett M. J., Roe S. M., Lee S., Niculescu‐Duvaz D., Good V. M., et al. Mechanism of activation of the RAF‐ERK signaling pathway by oncogenic mutations of B‐RAF. Cell 2004; 116: 855–67
- Theodosiou A., Ashworth A. MAP kinase phosphatases. Genome Biol 2002; 3: Reviews 3009
- Kim H. J., Bar‐Sagi D. Modulation of signaling by Sprouty: a developing story. Nature Rev Mol Cell Biol 2004; 5: 441–50
- Kwabi‐Addo B., Wang J., Erdem H., Vaid A., Castro P., Ayala G., et al. The expression of sprouty1, an inhibitor of fibroblast growth factor signal transduction, is decreased in human prostate cancer. Cancer Res 2004; 64: 4728–35
- Lo T. L., Yusoff P., Fong C. W., Guo K., McCaw B. J., Phillips W. A., et al. The Ras/mitogen‐activated protein kinase pathway inhibitor and likely tumor suppressor proteins, sprouty1 and Sprouty2 are deregulated in breast cancer. Cancer Res 2004; 64: 6127–36
- Manzano R. G., Montuenga L. M., Dayton M., Dent P., Kinoshita I., Vicent S., et al. CL100 expression is down‐regulated in advanced epithelial ovarian cancer and its re‐expression decreases its malignant potential. Oncogene 2002; 21: 4435–47
- Cobb M. H., Goldsmith E. J. How MAP kinases are regulated. J Biol Chem 1995; 270: 14843–6
- Herbst R. S., Fukuoka M., Baselga J. Gefitinib—a novel targeted approach to treating cancer. Nature Rev Cancer 2004; 4: 956–65
- Sebti S. M., Der C. J. Searching for the elusive targets of farnesyltransferase inhibitors. Nature Rev Cancer 2003; 3: 945–51
- Zhu K., Hamilton A. D., Sebti S. M. Farnesyltransferase inhibitors as anticancer agents: current status. Curr Opin Investig Drugs 2003; 4: 1428–35
- Lackner M. R., Kindt R. M., Carroll P. M., Brown K., Cancilla M. R., Chen C., et al. Chemical genetics identifies Rab geranylgeranyl transferase as an apoptotic target of farnesyl transferase inhibitors. Cancer Cell 2005; 7: 325–36
- Hoshino R., Tanimura S., Watanabe K., Kataoka T., Kohno M. Blockade of the extracellular signal‐regulated kinase pathway induces marked G1 cell cycle arrest and apoptosis in tumor cells in which the pathway is constitutively activated: up‐regulation of p27Kip1. J Biol Chem 2001; 276: 2686–92
- Lauffenburger D. A., Horwitz A. F. Cell migration: a physically integrated molecular process. Cell 1996; 84: 359–69
- Tanimura S., Chatani Y., Hoshino R., Sato M., Watanabe S., Kataoka T., et al. Activation of the 41/43·kDa mitogen‐activated protein kinase signaling pathway is required for hepatocyte growth factor‐induced cell scattering. Oncogene 1998; 17: 57–65
- Tanimura S., Nomura K., Ozaki K., Tsujimoto M., Kondo T., Kohno M. Prolonged nuclear retention of activated extracellular signal‐regulated kinase 1/2 is required for hepatocyte growth factor‐induced cell motility. J Biol Chem 2002; 277: 28256–64
- Reddy K. B., Nabha S. M., Atanaskova N. Role of MAP kinase in tumor progression and invasion. Cancer Metastasis Rev 2003; 22: 395–403
- Welch D. R., Sakamaki T., Pioquinto R., Leonard T. O., Goldberg S. F., Hon Q., et al. Transfection of Constitutively Active Mitogen‐activated Protein/Extracellular Signal‐regulated Kinase Kinase Confers Tumorigenic and Metastatic Potentials to NIH3T3 Cells. Cancer Res 2000; 60: 1552–6
- Klemke R. L., Cai S., Giannini A. L., Gallagher P. J., de Lanerolle P., Cheresh D. A. Regulation of cell motility by mitogen‐activated protein kinase. J Cell Biol 1997; 137: 481–92
- Semenza G. L. Targeting HIF‐α for cancer therapy. Nature Rev Cancer 2003; 3: 721–32
- Pages G., Pouyssegur J. Transcriptional regulation of the Vascular Endothelial Growth Factor gene—a concert of activating factors. Cardiovasc Res 2005; 65: 564–73
- Richard D. E., Berra E., Gothie E., Roux D., Pouyssegur J. p42/p44 mitogen‐activated protein kinases phosphorylate hypoxia‐inducible factor 1α (HIF‐1α) and enhance the transcriptional activity of HIF‐1. J Biol Chem 1999; 274: 2631–7
- Brahimi‐Horn C., Mazure N., Pouyssegur J. Signalling via the hypoxia‐inducible factor 1α requires multiple posttranslational modifications. Cell Signal 2005; 17: 1–9
- Eliceiri B. P., Klemke R., Stromblad S., Cheresh., D. A. Integrin αvβ3 requirement for sustained mitogen‐activated protein kinase activity during angiogenesis. J Cell Biol 1998; 140: 1255–63
- Sebolt‐Leopold J. S., Dudley D. T., Herrera R., Van Becelaera K., Wiland A., Gowan R. C., et al. Blockade of the MAP kinase pathway suppresses growth of colon tumors in vivo. Nature Med 1999; 5: 810–6
- Tanimura S., Asato K., Fujishro S., Kohno M. Specific blockade of the ERK pathway inhibits the invasiveness of tumor cells: down‐regulation of matrix metalloproteinase‐3/‐9/‐14 and CD44. Biochem Biophys Res Commun 2003; 304: 801–6
- Kimura E. T., Nikiforova N. M., Zhu Z., Knauf J. A., Nikiforov Y. E., Fagin J. A. High prevalence of BRAF mutations in thyroid cancer: genetic evidence for constitutive activation of the RET/PTC‐RAS‐BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res 2003; 63: 1454–7
- Rajagopalan H., Bardelli A., Lengauer C., Kinzler K. W., Vogelstein B., Velculescu V. E. Tumorigenesis: RAF/RAS oncogenes and mismatch‐repair status. Nature 2002; 418: 934
- Wan P. T. C., Garnett M. J., Roe S. M., Lee S., Niculescu‐Duvaz D., Good V. M., et al. Mechanism of activation of the RAF‐ERK signaling pathway by oncogenic mutations of B‐RAF. Cell 2004; 116: 855–67
- Wellbrock C., Ogilvie L., Hedley D., Karasarides M., Martin J., Niculescu‐Duvaz D., et al. V599EB‐RAF is an oncogene in melanocytes. Cancer Res 2004; 64: 2338–42
- Hingorani S. R., Jacobetz M. A., Robertson G. P., Herlyn M., Tuveson D. A. Suppression of BRAFV599E in human melanoma abrogates transformation. Cancer Res 2003; 63: 5198–202
- Lyons J. F., Wilhelm S., Hibner B., Bollag G. Discovery of a novel Raf kinase inhibitor. Endocr Relat Cancer 2001; 8: 219–25
- Wilhelm S. M., Carter C., Tang L. Y., Wilkie D., McNabola A., Rong H., et al. BAY 43‐9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004; 64: 7099–109
- Clark J. W., Eder J. P., Ryan D., Lathia C., Lenz H‐J. Safety and pharmacokinetics of the dual action Raf kinase and vascular endothelial growth factor receptor inhibitor, BAY 43‐9006, in patients with advanced, refractory solid tumors. Clin Cancer Res 2005; 11: 5472–80
- Strumberg D., Richly H., Hilger R. A., Schleucher N., Korfee S., Tewes M., et al. Phase I clinical and pharmacokinetic study of the novel Raf kinase and vascular endothelial growth factor receptor inhibitor BAY 43‐9006 in patients with advanced refractory solid tumors. J Clin Oncol 2005; 23: 965–72
- Dudley D. T., Pang L., Decker S. J., Bridges A. J., Saltiel., A. R. A synthetic inhibitor of the mitogen‐activated protein kinase cascade. Proc Natl Acad Sci USA 1995; 92: 7686–9
- Favata M. F., Horiuchi K. Y., Manos E. J., Daulerio A. J., Stradley D. A., Feeser W. S., et al. Identification of a Novel Inhibitor of Mitogen‐activated Protein Kinase Kinase. J Biol Chem 1998; 273: 18623–32
- Davies S. P., Reddy H., Caivano M., Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J 2000; 351: 95–105
- Ohren J. F., Chen H., Pavlovsky A., Whitehead C., Zhang E., Kuffa P., et al. Structures of human MAP kinase kinase 1 (MEK1) and MEK2 describe novel noncompetitive kinase inhibition. Nature Struct Mol Biol 2004; 11: 1192–7
- LoRusso P. M., Adjei A. A., Varterasian M., Gadgeel S., Reid J., Mitchell D. Y., et al. Phase I and pharmacodynamic study of the oral MEK inhibitor CI‐1040 in patients with advanced malignancies. J Clin Oncol 2005; 23: 5281–93
- Rinehart J., Adjei A. A., LoRusso P. M., Waterhouse D., Hecht J. R., Natale R. B., et al. Multicenter phase II study of the oral MEK inhibitor, CI‐1040, in patients with advanced non‐small‐cell lung, breast, colon, and pancreatic cancer. J Clin Oncol 2004; 22: 4456–62
- Menon S. S., Whitfield L. R., Sadis S., Meyer M. B., Leopold J., Lorusso P. M., et al. Pharmacokinetics (PK) and pharmacodynamics (PD) of PD 0325901, a second generation MEK inhibitor after multiple oral doses of PD 0325901 to advanced cancer patients. Proc Am Soc Clin Oncol 2005; 23: 3066
- Lyssikatos J., Yeh T., Wallace E., Marsh V., Bernat B., Gross S., et al. ARRY‐14886, a potent and selective MEK inhibitor: I) ATP‐independent inhibition results in high enzymatic and cellular selectivity. Proc Am Assoc Cancer Res 2004; 45: 3888
- Yeh T., Walace E., Lyssikatos J., Winkler J. ARRY‐14886, a potent and selective MEK inhibitor: II) Potency against cellular MEK leads to inhibition of cellular proliferation and induction of apoptosis in cell lines with mutant Ras or B‐Raf. Proc Am Assoc Cancer Res 2004; 45: 3889
- Lee P., Wallace E., Yeh T., Poch G., Litwiler K., Pheneger T., et al. ARRY‐14886, a potent and selective MEK inhibitor: III) Efficacy against human xenograft models correlates with decreased ERK phosphorylation. Proc Am Assoc Cancer Res 2004; 45: 3890
- Dent P., Grant S. Pharmacologic interruption of the mitogen‐activated extracellular‐regulated kinase/mitogen‐activated protein kinase signal transduction pathway: Potential role in promoting cytotoxic drug action. Clin Cancer Res 2001; 7: 775–83
- Xia Z., Dickens M., Raingeaud J., Davis R. J., Greenberg M. E. Opposing effects of ERK and JNK‐p38 MAP kinases on apoptosis. Science 1995; 270: 1326–31
- Lavoie J. N., L'Allemain G., Brunet A., Muller R., Pouyssegur J. Cyclin D1 expression is regulated positively by the p42/p44MAPK and negatively by the p38/HOGMAPK pathway. J Biol Chem 1996; 271: 20608–16
- Deng X., Ruvolo P., Carr B., May W. S., Jr. Survival function of ERK1/2 as IL‐3‐activated, staurosporine‐resistant Bcl2 kinases. Proc Natl Acad Sci USA 2000; 97: 1578–83
- Hayakawa J., Ohmichi M., Kurachi H., Kanda Y., Hisamoto K., Nishio Y., et al. Inhibition of Bcl‐2 associated death agonist (BAD) phosphorylation either at serine 112 via extracellular signal‐regulated protein kinase cascade or at serine 136 via Akt cascade sensitizes human ovarian cancer cells to cisplatin. Cancer Res 2000; 60: 5988–94
- Persons D. L., Yazlovitskaya E. M., Pelling J. C. Effect of extracellular signal‐regulated kinase on p53 accumulation in response to cisplatin. J Biol Chem 2000; 275: 35778–85
- Townsend K. J., Trusty J. L., Traupman M. A., Eastman A., Craig R. W. Expression of the antiapoptotic MCL1 gene product is regulated by a mitogen activated protein kinase‐mediated pathway triggered through microtubule disruption and protein kinase C. Oncogene 1998; 17: 1223–34
- McDaid H. M., Lopez‐Barcons L., Grossman A., Lia M., Keller S., Román Pérez‐Soler R., et al. Enhancement of the therapeutic efficacy of taxol by the mitogen‐activated protein kinase kinase inhibitor CI‐1040 in nude mice bearing human heterotransplants. Cancer Res 2005; 65: 2854–60
- Watanabe K., Noda S., Iwashita K., Tanimura S., Ozaki K., Kohno M. Blockade of the extracellular signal‐regulated kinase pathway enhances the anti‐tumor activity of microtubule depolymerizing agents in tumor cells in which the pathway is constitutively activated. Proc Am Assoc Cancer Res 2002; 43: 2891
- Ozaki K., Minoda A., Kishikawa F., Kohno M. Blockade of the ERK pathway markedly sensitizes tumor cells to HDAC inhibitor‐induced cell death. Biochem Biophys Res Commun 2006; 339: 1171–7
- Vivanco I., Sawyers C. L. The phosphatidylinositol 3‐kinase–AKT pathway in human cancer. Nature Rev Cancer 2002; 489–501, 2
- Nicholson K. M., Anderson N. G. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal 2002; 14: 381–95
- Guo W., Giancotti F. G. Integrin signaling during tumour progression. Nature Rev Mol Cell Biol 2004; 5: 816–26
- Schlaepfer D. D., Mitra S. K. Multiple connections link FAK to cell motility and invasion. Curr Opin Genet Dev 2004; 14: 92–101