Abstract
Pulmonary arterial hypertension is a disease of the small pulmonary arteries characterized by vascular narrowing and increased pulmonary vascular resistance, which eventually leads to right ventricular failure. Vasoconstriction, vascular proliferation, remodeling of the pulmonary vessels, and thrombosis are all contributing factors to the increased vascular resistance seen in this disease. Pulmonary arterial hypertension develops as a sporadic disease (idiopathic), as an inherited disorder (familial), or in association with certain conditions (collagen vascular diseases, portal hypertension, human immunodeficiency virus infection, congenital systemic‐to‐pulmonary shunts, ingestion of drugs or dietary products, or persistent fetal circulation). The pathogenesis of pulmonary arterial hypertension is a complicated, multifactorial process. It seems doubtful that any one factor alone is sufficient to activate the necessary pathways leading to the development of this disease. Rather, clinically apparent pulmonary arterial hypertension most likely develops after a second insult occurs in an individual who is already susceptible owing to genetic factors, environmental exposures, or acquired disorders. Currently, there is no cure for pulmonary arterial hypertension but several novel therapeutic options are now available that can improve symptoms and increase survival.
| Abbreviations | ||
| 5‐HT | = | 5‐hydoxytryptamine |
| 6MWT | = | 6‐minute walk test |
| ALK‐1 | = | activin‐like kinase type‐1 |
| ANA | = | anti‐nuclear antibody |
| BMPR2 | = | bonemorphogenetic proteinreceptor type‐2 |
| cGMP | = | cyclic guanosinemonophosphate |
| CPET | = | cardiopulmonary exercise test |
| CT | = | computerized tomography |
| CXR | = | chest X‐ray |
| ECG | = | electrocardiogram |
| ET‐1 | = | endothelin‐1 |
| ETA | = | endothelin‐A |
| ETB | = | endothelin‐B |
| HIV | = | human immunodeficiency virus |
| IV | = | intravenous |
| NO | = | nitric oxide |
| NYHA | = | New York Heart Association |
| PASP | = | pulmonary artery systolic pressure |
| PCH | = | pulmonary capillary hemangiomatosis |
| PE | = | pulmonary embolism |
| PVOD | = | pulmonary veno‐occlusive disease |
| RAP | = | right atrial pressure |
| RHC | = | right heart catheterization |
| RVSP | = | right ventricular systolic pressure |
| TGF‐β | = | transforming growth factor‐beta |
| V/Q | = | ventilation‐perfusion |
| VEGF | = | vascular endothelium growth factor |
| VIP | = | vasoactive intestinal peptide |
| vasoactive | = | intestinal peptide |
| WHO | = | World Health Organization |
Introduction
Pulmonary arterial hypertension is a disease of the small pulmonary arteries (muscular pulmonary arteries and arterioles) characterized by vascular narrowing and increased pulmonary vascular resistance, which eventually leads to right ventricular failure. Vasoconstriction, vascular proliferation, remodeling of the pulmonary vessels, and thrombosis all contribute to the increased vascular resistance seen in this disease Citation1–3. Pulmonary arterial hypertension refers to conditions that share common isolated elevations in pulmonary arterial pressure. Hemodynamically, it is defined by a sustained mean pulmonary arterial pressure greater than 25 mmHg at rest or greater than 30 mmHg with exercise. With the exception of an increase in mean pulmonary‐capillary wedge pressure with pulmonary veno‐occlusive disease (PVOD) or pulmonary capillary hemangiomatosis (PCH), pulmonary arterial hypertension patients typically have a normal mean pulmonary‐capillary wedge pressure and left end‐diastolic pressure or left atrial pressure (<15 mmHg) Citation4–6.
Clinical classification
Previously, pulmonary hypertension was classified as either primary or secondary based on the presence or absence of identifiable causes or risk factors Citation7,8. It has become clear, however, that certain conditions formerly categorized as secondary pulmonary hypertension have histopathological lesions and clinical features resembling those observed in primary pulmonary hypertension. For this reason, in 1998 the World Health Organization (WHO) developed a revised clinical classification system for pulmonary hypertension. Based on pathological features, mechanism of disease, and response to therapeutic intervention, pulmonary hypertensive diseases were placed into one of five categories: pulmonary arterial hypertension, pulmonary venous hypertension, pulmonary hypertension associated with hypoxemia, pulmonary hypertension due to chronic thrombotic or embolic disease, and a miscellaneous category () Citation9,10.
Table I. The Revised World Health Organization Classification of Pulmonary Hypertension*
Based on the revised classification system, pulmonary arterial hypertension develops as sporadic disease (idiopathic), as an inherited disorder (familial), or in association with certain conditions (collagen vascular diseases, portal hypertension, human immunodeficiency virus (HIV) infection, congenital systemic‐to‐pulmonary shunts, ingestion of drugs or dietary products, or persistent fetal circulation) Citation3,Citation11,12. At the 2003 Third World Symposium on Pulmonary Hypertension, PVOD and PCH were included as a subset of pulmonary arterial hypertension, since the pathologic changes observed in the pulmonary arteries of PCH and PVOD patients were similar to those seen in other forms of pulmonary artery hypertension Citation3,Citation13,14.
Key messages
Pulmonary arterial hypertension is a disease of the small pulmonary arteries (muscular pulmonary arteries and arterioles) characterized by vascular narrowing and increased pulmonary vascular resistance, which eventually leads to right ventricular failure.
Vasoconstriction, vascular proliferation, remodeling of the pulmonary vessels, and thrombosis are all contributing factors to the increased vascular resistance seen in this disease.
The pathogenesis of pulmonary arterial hypertension is a complicated, multifactorial process, and one factor alone is not sufficient to activate the necessary pathways leading to the development of this disease. Rather, clinically apparent pulmonary arterial hypertension most likely develops after a second insult occurs in an individual who is already susceptible owing to genetic factors, environmental exposures, or acquired disorders.
Currently, there is no cure for pulmonary arterial hypertension but several novel therapeutic options are now available that can improve symptoms and increase survival.
Cellular and molecular pathobiology
The primary vascular changes leading to increased vascular resistance in pulmonary arterial hypertension are vasoconstriction, remodeling of the pulmonary vascular wall, and thrombosis. Pulmonary vasoconstriction, one of the earliest components of the pulmonary hypertensive process, is most likely due to an abnormal expression of vascular smooth muscle potassium channels or endothelial dysfunction resulting from hypoxia, shear stress, or inflammation. Excessive vasoconstriction causes an alteration in cell proliferation, coagulation, and production of growth factors and vasoactive substances. Chronic impairment in the production of these vascular cell mediators ultimately results in a homeostatic imbalance that affects vascular tone and promotes vascular remodeling Citation2,Citation15.
In the normal pulmonary vasculature, cell mediators such as prostacyclin (prostaglandin I2), vasoactive intestinal peptide (VIP), and nitric oxide (NO) act as potent vasodilators and inhibitors of platelet aggregation and smooth muscle cell proliferation Citation16–20. In addition to exerting a direct vasoconstrictor effect, endothelin‐1 (ET‐1) stimulates cellular proliferation of vascular smooth muscle cells, induces fibrosis, and acts as a pro‐inflammatory mediator by enhancing the expression of adhesion molecules Citation21–24. Two endothelin‐receptors, endothelin‐A (ETA) and endothelin‐B (ETB) have been identified Citation23–25. Activation of ETA receptors induces vasoconstriction and proliferation of vascular smooth muscle cells, while ETB receptors mediate pulmonary endothelin clearance and induce vasodilation through increased production of nitric oxide and prostacyclin Citation25.
Dysfunctional pulmonary artery endothelial cells, such as those observed in individuals with idiopathic pulmonary arterial hypertension, have decreased production of prostacyclin Citation16 and endogenous nitric oxide Citation17,Citation26 with increased production of endothelin‐1 Citation27–29. This imbalance promotes vasoconstriction and proliferation of smooth muscle cells in the pulmonary arteries. In addition, pulmonary artery smooth muscle cells in idiopathic pulmonary arterial hypertension have a strong proliferative response to serum serotonin (5‐hydroxytryptamine (5‐HT)) owing to an overexpression of the 5‐HT transporter Citation30,31. In several studies, individuals with idiopathic pulmonary arterial hypertension were found to have decreased levels of VIP in the serum and lungs Citation18, elevated levels of plasma serotonin Citation31, reduced expression of prostacyclin and nitric oxide synthase Citation17,Citation32, and upregulation of ETB receptors Citation33,34 on smooth muscle cells, all of which contribute to the general vasoconstrictor response. Overall, the homeostatic imbalance that occurs in pulmonary arterial hypertension favors vasoconstriction, endothelial cell proliferation, and thrombosis.
At the histological level, disorganized proliferation of endothelial cells lead to the formation of plexiform lesions (i.e., focal proliferation of endothelial channels lined by myofibroblasts, smooth muscle cells, and connective tissue matrix). Defects in growth suppressive genes, including transforming growth factor‐beta (TGF‐β) receptor‐2, have been reported in plexiform lesions in patients with idiopathic pulmonary arterial hypertension. Owing to an abnormal angiogenic response to hypoxia, endothelial cells within the plexiform lesions tend to overexpress vascular endothelium growth factor (VEGF) and its receptor, VEGF receptor‐2, which leads to clonal expansion of these endothelial cells and their aborted attempt at forming tubes Citation2,Citation35–37.
Pathogenesis
The pathogenesis of pulmonary arterial hypertension is a complicated, multifactorial process. It seems doubtful that any one factor alone is sufficient to activate the necessary pathways leading to the development of this vascular disease. Rather, clinically apparent pulmonary arterial hypertension most likely develops after a second insult occurs in an individual who is already susceptible owing to genetic factors, environmental exposures, or acquired disorders. Thus, an understanding of the interaction of these risk factors is critical to identifying possible pathogenic mechanisms and defining the spectrum of disease Citation38.
Environmental risk factors
Environmental factors, such as use of appetite‐suppressant or stimulant drugs and chronic hypoxia, may be one step in the pathway leading to the development or progression of pulmonary arterial hypertension. Idiopathic pulmonary arterial hypertension was 7.5 times more likely to occur in individuals using anorexigenic agents, such as fenfluramine, for greater than 6 months. Although the incidence of pulmonary arterial hypertension increases with the duration of use of fenfluramine, increased pulmonary arterial pressure was observed after only a limited exposure time, approximately 3–4 weeks Citation39–42. It has also been suggested that stimulant use, including methamphetamine and cocaine, may be associated with an increased risk of pulmonary arterial hypertension, but no conclusive studies have confirmed that these drugs are clearly risk factors Citation43. Although chronic hypoxia is probably not a crucial risk factor for the development of pulmonary arterial hypertension, it may contribute to disease progression through the mechanism of pulmonary arterial vasoconstriction and subsequent vascular remodeling Citation44,45.
Genetic risk factors
The familial form of pulmonary arterial hypertension occurs in approximately 6% of all cases Citation7. Recently, mutations in two receptors of the TGF‐β receptor family have been identified in the majority of cases of inherited (familial) pulmonary arterial hypertension Citation46. The TGF‐β family consists of the TGF‐β isoforms, the bone morphogenetic proteins, activins, and growth and differentiation factors. These molecules have diverse roles in a wide variety of physiological processes, such as smooth muscle cell proliferation, differentiation, immunity, and inflammation Citation47,48.
Mutations in the gene encoding for the bone morphogenetic protein receptor type‐2 (BMPR2), which normally modulates the growth and survival of vascular smooth muscle cells Citation49, is present in approximately 50%–60% of familial cases of pulmonary arterial hypertension and in 10%–30% of idiopathic or sporadic cases. Although some of the sporadic cases may be associated with de novo mutations, the majority are likely due to familial transmission of a truncated or point mutation with a low penetrance. The gene encoding for BMPR2 has been localized to chromosome 2q33, and more than 45 different mutations have been identified in patients with familial pulmonary arterial hypertension Citation50–53. Inheritance occurs in an autosomal dominant pattern with highly variable and incomplete expression, female predominance, and genetic anticipation (i.e., with each successive generation, clinical disease develops earlier and in a more severe form) Citation54. Mutations in BMPR2 seem to be fairly specific for idiopathic pulmonary arterial hypertension, but a few cases have been observed in individuals with anorexigen associated pulmonary arterial hypertension and pulmonary veno‐occlusive disease. Thus far, no other forms of pulmonary arterial hypertension have been associated with a BMPR2 mutation Citation55,56. BMPR2 mutations are most likely insufficient to cause vascular disease by themselves as only approximately 20% of disease gene carriers develop clinical disease. This observation re‐emphasizes the importance of other genetic or environmental factors in conferring susceptibility to pulmonary hypertension Citation53. A second mutation in the activin‐like kinase type‐1 (ALK‐1) receptor has been found in a minority of patients with hereditary hemorrhagic telangiectasia and co‐existent pulmonary arterial hypertension Citation57.
Serotonin (5‐HT), a neuropeptide produced in the pulmonary vasculature and stored in platelets, is one of the best‐studied modifiers of pulmonary hypertension Citation53. Anorexigens interact with the serotonin transporter and cause release of 5‐HT from platelets, thereby increasing free circulating levels and inhibiting its reuptake Citation58,59. Studies have revealed that patients with pulmonary arterial hypertension exhibit elevated plasma levels of serotonin and that exposure to fenfluramine derivatives results in an increased risk for the development of pulmonary arterial hypertension. A single gene on chromosome 17q11.2 encodes for the 5‐HT transporter. A functional polymorphism in the gene's promoter region, which allows for increased expression of the transporter and serotonin‐dependent vascular smooth cell proliferation, is more prevalent in individuals with idiopathic pulmonary arterial hypertension than controls. Additionally, the polymorphism contributes to differences in the level of hypoxia‐induced 5‐HT expression and may affect susceptibility to hypoxic pulmonary hypertension Citation21,Citation60. This gene polymorphism for the serotonin transporter may either serve as an important modifier for the development of clinically apparent pulmonary arterial hypertension or as a factor conveying susceptibility to pulmonary hypertension.
Associated conditions
Several associated or coexisting conditions have been identified with an increased prevalence of pulmonary arterial hypertension. Some of the strongest evidence exists for the following conditions: scleroderma, infection with HIV or human herpesvirus, portal hypertension, thrombocytosis, hemoglobinopathies, and hereditary hemorrhagic telangiectasia (Osler‐Weber‐Rendu disease) Citation3.
Histologic changes, including the development of constrictive vascular lesions (i.e., concentric laminar intimal thickening), have been observed in patients with pulmonary arterial hypertension and limited systemic sclerosis (i.e., sclerodactyly, digital ulcers, and often pulmonary fibrosis) Citation11,Citation61. These findings are especially prevalent in patients with the CREST variant (calcinosis cutis, Raynaud's phenomenon, esophageal dysfunction, sclerodactyly, and telangiectasia). Additionally, in the presence of associated Raynaud's phenomenon (cold temperature‐induced vasospasm of the digital arteries causing numbness, pallor, and pain), pulmonary arterial hypertension may occur in patients with systemic lupus erythematosus, mixed connective tissue disease, and rheumatoid arthritis. Thus, the vascular changes seen in pulmonary arterial hypertension may, in part, be similar in pathogenesis to those that occur in Raynaud's phenomenon Citation62–64.
Patients infected with HIV have an incidence of pulmonary arterial hypertension of 0.5%, which is approximately 6–12 times the incidence observed in the general population. The development of pulmonary arterial hypertension is independent of CD‐4 cell count, but does seem to depend on the length of the HIV‐related infection. Since HIV does not infect endothelial cells, the mechanism of disease development is unknown Citation65,66. Recently, an association of human herpesvirus‐8, the causative agent of Kaposi's sarcoma, with the plexiform lesions of idiopathic pulmonary hypertension has been observed Citation67,68.
Hemodynamic studies have demonstrated that pulmonary arterial hypertension exists in 2%–5% of patients with cirrhosis and in approximately 6% of patients undergoing assessment for liver transplant. The mechanism of disease development is unknown, but increased risk occurs with increased duration of cirrhosis. Pulmonary artery hypertension is unlikely to occur in the setting of cirrhosis without portal hypertension, but may occur in patients with portal hypertension in the absence of hepatic cirrhosis. In the latter setting, vasoactive substances may play a role in the pathogenesis of the disease Citation69–71.
Pulmonary arterial hypertension has been observed in patients with asplenia, hemoglobinopathies, and myeloproliferative disorders. Pulmonary arterial hypertension, a well known complication of sickle cell disease, significantly reduces survival and is the cause of death in approximately 3% of these patients. Plexiform lesions, which develop as a result of endothelial cell injury in patients with idiopathic pulmonary arterial hypertension, have been observed in patients with sickle cell disease. Increased shear stress from deformed erythrocytes as they move through the pulmonary vasculature has been proposed as a mechanism for endothelial injury; in addition, nitric oxide bioavailability is reduced in these patients Citation72–74. In a similar manner, patients with asplenia may be at an increased risk for development of pulmonary arterial hypertension, as abnormal erythrocytes remain in the circulation longer and may serve as a nidus for platelet activation in these patients Citation75. The pulmonary lesions of idiopathic pulmonary arterial hypertension and/or thrombotic pulmonary arteriopathy have been found in some patients with beta‐thalassemia Citation76. The association of pulmonary hypertension and chronic myeloproliferative disorders may result from splenectomy, portal hypertension, chemotherapy‐induced pulmonary veno‐occlusive disease, and pulmonary parenchymal infiltration with hematopoietic cells Citation77,78.
Hereditary hemorrhagic telangiectasia or Osler‐Weber‐Rendu disease is an autosomal dominant disorder characterized by the presence of multiple arteriovenous malformations, particularly in the pulmonary, hepatic, and cerebral circulations. Mutations in two genes encoding for TGF‐β receptors, endoglin and ALK‐1, have been linked to the development of pulmonary arterial hypertension in these patients. Occlusions of small arteries, typical of idiopathic pulmonary arterial hypertension, occur in approximately 15% of cases of Osler‐Weber‐Rendu disease Citation79,80.
Clinical presentation and diagnosis
Symptoms and detection
Although symptoms of pulmonary arterial hypertension are often nonspecific and may mimic common disorders, most patients are diagnosed as a result of an evaluation that was initiated because of a symptomatic complaint. Exertional dyspnea is the most commonly encountered symptom of pulmonary arterial hypertension, although symptoms such as fatigue or weakness, angina, syncope, peripheral edema, or ascites may also be present in patients. The disorder is occasionally discovered during screening of an asymptomatic high‐risk population. Screening with Doppler echocardiography is advised in individuals with a known genetic mutation that is associated with pulmonary arterial hypertension, or if the individual under consideration has a first‐degree relative with idiopathic pulmonary arterial hypertension. In addition, individuals with scleroderma, congenital heart disease, systemic‐to‐pulmonary shunts, or portal hypertension undergoing evaluation for liver transplant should be screened Citation7.
A diagnostic strategy of testing should be employed to determine if the patient's symptoms are due to pulmonary arterial hypertension and, if so, the underlying etiology. The initial assessment in detecting pulmonary arterial hypertension should include a physical examination, chest X‐ray (CXR), electrocardiogram (ECG), and Doppler echocardiography. Initial physical exam findings include a prominent and widely split S2 with an increased P2 and a diastolic murmur of pulmonic regurgitation. The physical examination findings also typically include a left parasternal lift, a pansystolic murmur of tricuspid regurgitation, a right ventricular S3, jugular vein distension, hepatomegaly, peripheral edema, ascites, and cool extremities once pulmonary arterial hypertension has progressed to right ventricular failure. Radiographic signs of pulmonary arterial hypertension and/or right ventricular failure include right ventricular enlargement (anterior displacement of the right ventricle into the retrosternal space on the lateral view) and enlarged (>17 mm) main and hilar pulmonary arterial shadows with peripheral pruning (attenuation of peripheral vascular markings). An ECG often provides further evidence of hemodynamically significant pulmonary arterial hypertension, such as right ventricular hypertrophy and strain, and right atrial dilation. Right ventricular hypertrophy is present in 87% and right atrial axis deviation is present in 79% of individuals with idiopathic pulmonary arterial hypertension Citation7,Citation81.
Despite the use of these testing modalities, it is not uncommon for Doppler echocardiography to be the first test to suggest the diagnosis pulmonary arterial hypertension Citation82. Doppler echocardiography estimates the pulmonary artery systolic pressure (PASP), which is equivalent to the right ventricular systolic pressure (RVSP) in absence of pulmonary outflow obstruction. The RVSP is determined by measuring the systolic regurgitant tricuspid flow velocity (ν), estimating the right atrial pressure (RAP), and then applying these findings to the modified Bernoulli equation (RVSP = 4ν2 + RAP). Detecting pulmonary arterial hypertension using PASP as determined by Doppler echocardiography has a reported sensitivity ranging from 0.79 to 1.00 and a specificity of 0.60 to 0.98 Citation82–85. Most studies report a high correlation (0.57–0.93) between Doppler echocardiography and right heart catheterization (RHC) measurements of PASP Citation84. Ultimately, a RHC is needed for the definitive diagnosis of pulmonary arterial hypertension, as well as the assessment of its severity and prognosis, and as a guide to therapy. Confirmation by RHC is warranted prior to beginning an extensive evaluation to determine an underlying etiology or prognosis.
Etiology
Certain tests are essential in the evaluation of pulmonary arterial hypertension to establish an underlying etiology, determine severity and prognosis, and choose an initial treatment strategy (). Initial serological testing should include complete blood count including a platelet count, liver function tests, antinuclear antibody titers to screen for autoimmune collagen vascular disorders, HIV serologies, and antiphospholipid antibodies, specifically anticardiolipin antibodies and the lupus anticoagulant. Although approximately 40% of patients with idiopathic pulmonary arterial hypertension have a positive but low antinuclear antibody (ANA) titer (⩾1:80 dilutions) with a nonspecific pattern, any patient with rheumatologic symptoms and/or substantially elevated ANA titers should be referred to a rheumatologist for further testing Citation7,Citation86.
Figure 1. Guideline for diagnosis and determination of the etiology of pulmonary arterial hypertension(adapted from Rubin et al. Citation100). ANA = anti‐nuclear antibody; CHD = coronary heart disease; CT = computerized tomography; CTD = connective tissue disorder; ECG = electrocardiogram; HIV = human immunodeficiency virus; LFTs = liver function tests; LHC = left heart catheterization; PAH = pulmonary arterial hypertension; PE = pulmonary embolism; PFTs = pulmonary function tests; RAE = right atrial enlargement; RHC = right heart catheterization; RV: right ventricle; RVE = right ventricular enlargement; RVSP = right ventricular systolic pressure; TRV: tricuspid regurgitant velocity; V/Q = ventilation‐perfusion.
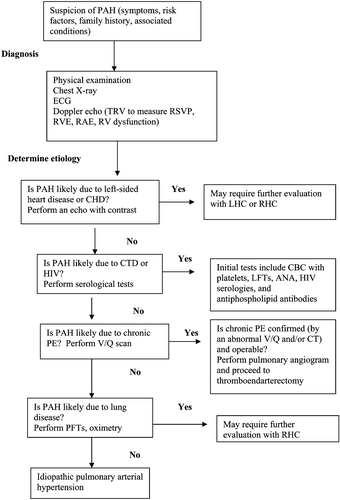
Pulmonary function testing should be performed to exclude or characterize the contribution of underlying obstructive or restrictive disease to the overall pathology of pulmonary arterial hypertension. Pulmonary function testing also helps quantify the extent of hypoxemia (potentially due to ventilation‐perfusion mismatching, right‐to‐left intracardiac shunting, or intrapulmonary shunting), diffusion capacity abnormalities (potentially due to systemic sclerosis), and restrictive disease (potentially due to idiopathic pulmonary arterial hypertension or chronic thromboembolic pulmonary arterial hypertension) Citation7,Citation87–89. As hypoxemia is a potent vasoconstrictor, all patients with unexplained pulmonary arterial hypertension should have an assessment of oxygen saturation during sleep and exercise to exclude significant obstructive sleep apnea or hypopnea. More than 75% of patients with idiopathic pulmonary arterial hypertension who do not have sleep apnea will, however, also have episodes of nocturnal hypoxemia Citation90,91. Obstructive sleep apnea may be a contributing factor to mild pulmonary arterial hypertension, but alone does not explain severe pulmonary arterial hypertension.
Since chronic thromboembolic pulmonary hypertension is a potentially curable cause of pulmonary arterial hypertension, ventilation‐perfusion (V/Q) lung scintigraphy or some form of pulmonary angiography should be performed in all individuals in whom chronic pulmonary embolism (PE) is suspected. Perfusion scanning allows differentiation between thromboembolic disease and primary pulmonary perfusion defects. The V/Q scan in patients with chronic PE typically shows at least one segmental perfusion defect. If the V/Q scan result is equivocal and the physician has a high level of suspicion for chronic PE, or if the physician needs to define further a suspected embolism, then it is prudent to order additional testing, such as contrast‐enhanced spiral (or helical) computerized tomography (CT), high‐resolution CT, magnetic resonance angiography, or pulmonary angiography Citation92–95. Spiral CT has a sensitivity of >85%–90% for the detection of central pulmonary embolism, which is visualized as complete occlusion of the pulmonary arteries, eccentric filling defects, recanalization, stenosis, or webs. High‐resolution CT provides detailed views of the lung parenchyma and may allow for diagnosis of pulmonary fibrosis in association with pulmonary arterial hypertension Citation96,97, and has supplanted V/Q scanning at many institutions. Pulmonary angiography is the definitive test for the diagnosis and preoperative assessment of patients with thromboembolic pulmonary hypertension Citation98.
Assessment of disease severity
After diagnosis and classification of pulmonary arterial hypertension, assessment of functional exercise capacity should be undertaken to evaluate disease severity. The two most commonly used exercise tests for patients with pulmonary hypertension are the 6‐minute walk test (6MWT) and cardiopulmonary exercise testing (CPET) with gas exchange measurements. These tests determine baseline functional capacity, which is used initially to guide therapy and assess prognosis, and, later in the course of the disease, to follow the response to therapy.
For the 6MWT, patients are asked to walk as far as possible in 6 minutes. This sub‐maximal exercise test determines functional capacity for patients who are incapable of undergoing maximal exercise testing. In patients with idiopathic pulmonary arterial hypertension, the 6MWT result is predictive of survival and correlates inversely with New York Heart Association (NYHA)/WHO functional status. A recent study reported an 18% mortality risk reduction for each additional 50 m walked during the 6MWT Citation99–101. CPET is a measure of ventilation and pulmonary gas exchange during exercise, and often provides an objective evaluation of functional capacity that is independent of effort. The peak oxygen consumption (VO2) or the anaerobic or lactic acid threshold during submaximal exercise provides an estimate of the magnitude of exercise limitation in individuals with pulmonary arterial hypertension Citation102.
Natural history and survival
Prior to the development of recent therapies, the clinical course of idiopathic pulmonary arterial hypertension followed a fairly rapid progression from right ventricular failure to death. Early, large prospective studies revealed 1, 3, and 5‐year survival rates of 68%–77%, 40%–56%, and 22%–38%, respectively. Patients with connective tissue disease‐associated pulmonary arterial hypertension tend to have a worse prognosis, whereas patients with pulmonary arterial hypertension due to congenital systemic‐to‐pulmonary shunts tend to have a slower progression of disease and longer survival. Indicators of a poor prognosis include a history of right heart failure, NYHA/WHO functional class III or IV (), elevated right atrial pressure, decreased cardiac output, elevated pulmonary vascular resistance, and low mixed venous oxygen saturation Citation81,Citation103.
Table II. World Health Organization Functional Classification of Pulmonary Arterial Hypertension*
Treatment
General measures
Currently, there is no cure for pulmonary arterial hypertension, but several therapeutic options targeted to specific molecular mechanisms are now available. Treatment has vastly improved over the past decade and now not only offers patients symptom relief, but also often prolongs their survival. The mainstay of current therapy consists of the use of a combination of agents, which may include supplemental oxygen, diuretics, anticoagulants, calcium‐channel blockers, prostanoids, endothelin‐receptor antagonists, phosphodiesterase‐5 inhibitors, or interventional and surgical procedures. The etiology and severity of pulmonary arterial hypertension determine the choice of therapeutic approach.
Anticoagulation with warfarin in patients with idiopathic pulmonary arterial hypertension is based on two small studies—one retrospective Citation104 and one prospective Citation105—that reported improved 3‐year survival rates of 21% to 49% Citation104 and 31% to 47% Citation105. There is no evidence of a difference in anticoagulation efficacy for different functional classes of pulmonary arterial hypertension, and the use of anticoagulation is controversial in patients with pulmonary arterial hypertension due to scleroderma or congenital heart disease. Current consensus suggests a target INR of 1.5–2.5 for idiopathic pulmonary arterial hypertension patients on anticoagulation Citation106,107. Patients with evidence of right heart failure frequently benefit from the use of diuretics. The exact class and dose of diuretic is left to the discretion of the treating physician since trial data are lacking. The use of digoxin in patients with isolated right ventricular failure remains controversial since digoxin appears to be most beneficial in patients with cor pulmonale or atrial fibrillation. Short‐term intravenous use of digoxin in idiopathic disease may improve cardiac output and decrease the circulating levels of norepinephrine, although studies have not been conducted to evaluate the effect of long‐term administration Citation108,109. There are no consistent data concerning long‐term use of supplemental oxygen in pulmonary arterial hypertension, but since chronic hypoxemia may cause vasoconstriction, supplemental oxygen is routinely used to maintain oxygen saturation above 90% Citation110.
Pregnancy and labor place an increased demand on the cardiopulmonary system in patients with pulmonary hypertension who have a mortality risk of 30%–50%; thus, pregnancy is contraindicated in these patients. Safe and effective contraception is, therefore, recommended in all women of childbearing age with pulmonary hypertension. Usually, either dual barrier method contraception or progesterone‐only oral contraceptives, in women without a history of thromboembolic disease, are the recommended forms of birth control for these patients. Estrogen‐containing contraceptives are not recommended as they may lead to an increased risk of venous thromboembolism Citation10,Citation110.
Figure 2. Treatment algorithm for pulmonary arterial hypertension(adapted from Galie et al. Citation149). CCB = calcium‐channel blocker; NYHA = New York Heart Association; PDE‐5 = phosphodiesterase‐5; WHO = World Health Organization.
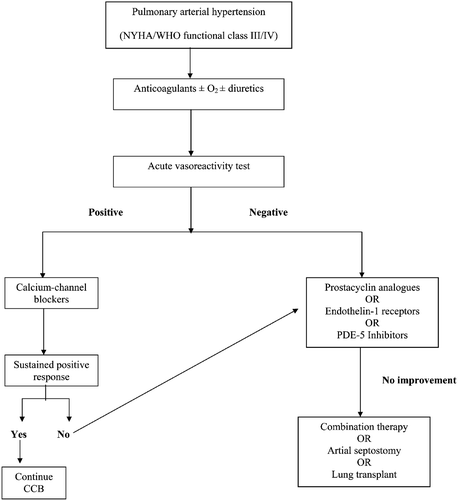
Calcium‐channel blockers
Pulmonary artery vasoconstriction may contribute to the pathogenesis of pulmonary arterial hypertension Citation2,Citation105. Studies have shown that patients with idiopathic pulmonary arterial hypertension who respond to short‐acting vasodilators have improved survival with long‐term use of calcium‐channel blockers. An acute vasodilator challenge within the catheterization laboratory using short‐acting agents, such as intravenous epoprostenol, adenosine, or inhaled nitric oxide, identifies patients who may benefit from long‐term use of a calcium‐channel blocker. The development of acute pulmonary edema suggests the presence of pulmonary veno‐occlusive disease or pulmonary capillary hemangiomatosis and is a contraindication to chronic vasodilator therapy. Agents with negative inotropic effects, such as verapamil, should be avoided. A positive short‐term response to vasodilators is defined as at least a 10 mmHg decrease in mean pulmonary arterial pressure to 40 mmHg or less, with normal or increased cardiac output. Only approximately 10%–15% of patients with idiopathic pulmonary arterial hypertension will meet these criteria, and, of those patients started on long‐term therapy, only approximately one‐half will maintain sustained hemodynamic (measured at 3 months) and functional (NYHA/WHO functional class I or II) improvement Citation106–108. The effects of high‐dose calcium‐channel blockers on other types of pulmonary arterial hypertension have not yet been clearly demonstrated Citation109.
Prostanoids
Chronic intravenous (IV) epoprostenol, chronic subcutaneous treprostinil, inhaled iloprost, and oral beraprost are the primary prostacyclins that have been studied and are currently being used as treatment in patients with pulmonary arterial hypertension. The efficacy of IV epoprostenol has been studied in several clinical trials for the treatment of idiopathic and scleroderma‐associated pulmonary arterial hypertension. Epoprostenol improves symptoms, exercise capacity, and hemodynamic parameters in both conditions, as well as improves survival in patients with the idiopathic form Citation110–112. A survival benefit depends on the severity of disease at baseline and the response to 3 months of epoprostenol therapy Citation113. The beneficial effects of this therapy may be sustained for several years in many patients with idiopathic pulmonary arterial hypertension Citation113–116. Improvements with epoprostenol have also been reported in patients who had pulmonary arterial hypertension associated with congenital left‐to‐right shunts, portal hypertension, and HIV infection Citation117–119. Owing to its short half‐life, epoprostenol requires constant IV infusion, and abrupt or inadvertent discontinuation leads to rebound worsening of pulmonary hypertension and symptomatic deterioration. Dose‐dependent side effects include flushing, headache, jaw and lower extremity muscular pain, diarrhea, nausea, and rash Citation111,Citation113,Citation116. Epoprostenol is approved for patients with idiopathic or scleroderma‐associated pulmonary arterial hypertension in NYHA/WHO functional class III or IV, but its use is usually reserved for patients with advanced disease who have failed oral therapy.
Owing to the complications associated with long‐term IV therapy, other formulations of prostacyclins have been studied more recently. Treprostinil is a stable prostacyclin analogue that is approved for administration subcutaneously or intravenously in patients with NYHA/WHO functional class II, III, or IV. The side effects are similar to epoprostenol with the exception of pain at the infusion site. Improvements in exercise capacity and symptoms with a reduction in clinical events were observed in patients treated with treprostinil for 3 months Citation120. The orally active prostacyclin analogue, beraprost, has been shown to increase exercise capacity in idiopathic pulmonary arterial hypertension patients at 3 months without affecting hemodynamic variables or survival. A long‐term study did not find that improvement was present at 9 or 12 months. Currently, beraprost is only approved in Japan Citation121,122. Iloprost, an inhaled prostacyclin analogue, has been shown to improve exercise capacity, hemodynamic variables, and NYHA/WHO functional class. Side effects included coughing, flushing, headache, and syncope. Owing to the drug's short half‐life, six to nine inhalations are required per day; it has recently been approved for use in patients in NYHA/WHO functional class III or IV Citation123–125.
Endothelin‐receptor antagonists
Bosentan, an orally active dual endothelin‐receptor antagonist, has been studied in patients with idiopathic and scleroderma‐associated pulmonary arterial hypertension. This agent blocks both ETA and ETB receptor activity. Two large, multicenter trials of bosentan have shown improvement in cardiopulmonary hemodynamic variables, NYHA/WHO functional class, exercise capacity, and time to clinical worsening; this agent did, however, cause asymptomatic dose‐dependent hepatic aminotransferase elevations in some patients. Elevations up to eight times the upper limit of normal occurred in 3% and 7% of patients taking the 125 mg dose and 250 mg dose, respectively. Potential additional side effects include anemia, edema, teratogenicity, male infertility, and testicular atrophy. This oral medication has been approved for use in patients with pulmonary arterial hypertension in the NYHA/WHO function class III or IV. Liver function tests need to be performed monthly owing to this potential of liver toxicity Citation126,127. Sitaxsentan and ambrisentan, selective orally active ETA receptor antagonists, have shown improvement in exercise capacity and hemodynamics in preliminary trials, but additional studies are needed to address the long‐term benefits Citation128.
Nitric oxide and phosphodiesterase inhibitors
Nitric oxide, a potent vasodilator platelet inhibitor, and antiproliferative agent, acts on the vascular smooth muscle cells through stimulation of guanylyl cyclase, which increases the production of intracellular cyclic guanosine monophosphate (cGMP). The vasodilatory effects of intracellular cGMP are relatively short‐lived because of its rapid degradation by phosphodiesterases. Phosphodiesterase‐5 gene expression and activity is increased in the pulmonary vasculature in patients with chronic pulmonary hypertension Citation129–131. Sildenafil, an erectile dysfunction medication, is a highly specific phosphodiesterase‐5 inhibitor, and studies have shown that its short‐term effects include reduction of pulmonary arterial pressure and augmentation of the effects of inhaled nitric oxide, intravenous iloprost, and inhaled prostacyclin. Recently, sildenafil has been approved for use in patients with pulmonary arterial hypertension Citation132–134.
Interventional and surgical therapies
Different interventional and surgical techniques have been developed over the past 20 years to treat pulmonary arterial hypertension. The main interventions include atrial septostomy, pulmonary endarterectomy, and lung transplantation. Graded balloon‐dilation atrial septostomy is indicated in patients with persistent right ventricular heart failure and/or recurrent syncope who have failed conventional medical therapy and have no other therapeutic options or are awaiting lung transplantation Citation135,136. Atrial septostomy creates a right‐to‐left shunt that increases cardiac output, augments systemic oxygen transport, reduces afterload, and improves right ventricular failure. This procedure has been performed mainly in patients with the idiopathic form of pulmonary arterial hypertension with an overall procedure‐related mortality of 5.4% Citation137. In most studies, exercise endurance and the symptoms and signs of right ventricular heart failure improve after atrial septostomy, but the impact on long‐term survival is unknown Citation137,138.
Chronic thromboembolic pulmonary hypertension results from unresolved acute pulmonary embolism. It is characterized by intraluminal thrombus organization and fibrous stenoses that obstruct branches of the pulmonary artery, which leads to an elevation of pulmonary artery pressure and vascular resistance, and right ventricular heart failure Citation96,97. After the diagnosis of chronic thromboembolic pulmonary hypertension is established, the decision for surgical intervention is determined by the degree of functional impairment, the severity of pulmonary arterial pressure, and the accessibility of the lesions. A minimum of 3 months of anticoagulation is needed prior to surgery. The operation involves a true endarterectomy in which the organized and incorporated fibrous obstructive tissue is removed from the pulmonary artery bed Citation138. The surgery can potentially cure the patient if successful, but the operative mortality risk is between 5% and 24% Citation92. Studies have reported an improvement in pulmonary artery pressures, cardiac output, oxygenation, and functional class after surgery with a 5‐year survival rate of 75%–80% Citation139.
For patients who do not respond to conventional therapy, transplantation should be considered. Since the waiting time for transplantation is usually of long duration, patients who have not responded to intravenous epoprostenol therapy as determined by right heart catheterization have a guarded prognosis and should proceed to transplantation evaluation Citation113,Citation115. Serial 6‐minute walk tests and cardiopulmonary exercise testing may be useful in determining the appropriate timing for transplantation. Patients with a 6‐minute walk distance of <332 m have a 1‐year mortality of 40%, and patients with a peak oxygen uptake ⩽10.4 mL/kg/min and a peak systolic blood pressure ⩽120 mmHg have a 1‐year mortality of 50% and 70%, respectively. Only 23% of patients survive for 1 year if both peak oxygen uptake and peak blood pressure values are reduced Citation99,Citation140.
Transplantation procedures currently being performed in patients with pulmonary arterial hypertension include single lung, bilateral lung, and heart‐lung transplant. No randomized trials have been performed to evaluate the relative efficacy of different transplant procedures, so significant debate and practice variability exist between individual centers Citation141,142. Single lung transplantation is more readily available, an easier procedure to perform, and usually requires less ischemic and cardiopulmonary bypass time, but has an increased chance of V/Q mismatch and reperfusion injury Citation141,Citation143. Yet, despite the potentially large ventilation‐perfusion imbalance with single lung transplantation, early 30‐day survival rates have been similar to those observed for bilateral transplantation. Although not statistically significant, increased posttransplantation survival rates have been observed in bilateral lung transplantation recipients registered in the International Society for Heart and Lung Transplantation database. This survival advantage in posttransplant idiopathic pulmonary arterial patients was observed at all time points up to 9 years. Initially by providing better overall lung function, bilateral transplantation may be protective against common manifestations of chronic rejection. A survival benefit of heart‐lung transplant over single or bilateral transplant has not been clearly elucidated Citation144–146.
An elevated bilirubin, the need for supplemental oxygen prior to transplantation, or an abnormally increased creatinine are all independent risk factors for mortality at 1 and 5 years after lung transplantation. In addition, donor and recipient age and a pretransplant pulmonary artery pressure >40 mmHg are also associated with an increased 5‐year mortality. Survival data from the International Society for Heart and Lung Transplantation show that patients with idiopathic pulmonary arterial hypertension and Eisenmenger syndrome have the highest perioperative mortality rates and the lowest 3‐month survival rates among lung transplant recipients. Most likely, an increased need for cardiopulmonary bypass leading to a higher risk of hemorrhagic complications and early graft dysfunction, in addition to early postoperative hemodynamic instability, account for the disparity in survival Citation141,Citation144,Citation146. The major limitation to long‐term survival after lung transplantation is death due to chronic rejection, which manifests as obliterative bronchiolitis. Any injury to the lung allograft, such as infection, rejection, ischemia, or reperfusion injury will likely contribute to development of obliterative bronchiolitis. Patients undergoing single lung transplantation may have higher rates of reperfusion edema and injury resulting in prolonged mechanical ventilation and protracted intensive care stays Citation143,Citation145,Citation148.
Conclusion
Although pulmonary arterial hypertension is an uncommon disorder, its high morbidity and mortality make its early diagnosis crucial. Symptoms of pulmonary arterial hypertension are often nonspecific and may mimic common disorders, but the diagnosis should be considered in all patients without traditional cardiovascular risk factors who present with symptoms of dyspnea and fatigue. Only recently have there been major advances in the understanding of the mechanisms of disease development and in the treatment of pulmonary arterial hypertension, but gaps in our knowledge still remain.
Acknowledgements
The authors wish to thank Stephanie Tribuna for expert secretarial assistance.
References
- Runo J. R., Loyd J. E. Primary pulmonary hypertension. Lancet 2003; 361: 1533–44
- Voelkel N. F., Tuder R. M., Weir E. K. Pathophysiology of primary pulmonary hypertension. Primary pulmonary hypertension., L. J Rubin, S Rich, editors. Marcel Dekker, New York 1997; p. 83–129, In
- Simonneau G., Galie N., Rubin L. J., Langleben D., Seeger W., Domenighetti G., et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004; 43: 5S–12S
- Rubin L. J. Primary pulmonary hypertension. N Engl J Med 1997; 336: 111–7
- McGoon M., Gutterman D., Steen V., Barst R., McCrory D. C., Fortin T. A., et al. Screening, early detection and diagnosis of pulmonary arterial hypertension: ACCP evidence‐based clinical practice guideline. Chest 2004; 126((Suppl))14S–34S
- Gaine S. P., Rubin L. J. Primary pulmonary hypertension. Lancet 1998; 352: 719–25
- Rich S., Dantzker D. R., Ayres S. M., Bergofsky E. H., Brundage B. H., Detre K. M., et al. Primary pulmonary hypertension. A national prospective study. Ann Intern Med 1987; 107: 216–23
- Barst R. J. Medical therapy of pulmonary hypertension: an overview of treatment and goals. Clin Chest Med 2001; 22: 509–15
- Fishman A. P. Clinical classification of pulmonary hypertension. Clin Chest Med 2001; 22: 385–91
- Humbert M., Nunes H., Sitbon O., Parent F., Herve P., Simonneau G. Risk factors for pulmonary arterial hypertension. Clin Chest Med 2001; 22: 459–75
- Pietra G. G., Capron F., Stewart S., Leone O., Humbert M., Robbins I. M., et al. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol 2004; 43: 25S–32S
- Pietra G. C. The pathology of primary pulmonary hypertension. Primary Pulmonary Hypertension: Lung Biology in Health and Disease., L. J Rubin, S Rich, editors. Marcel Dekker, New York, NY 1997; p. 19–61, In
- Mandel J., Mark E. J., Hales C. A. Pulmonary veno‐occlusive disease. Am J Respir Crit Care Med 2000; 162: 1964–73
- Langleben D., Heneghan J. M., Batten A. P., Wang N. S., Fitch N., Schlesinger R. D., et al. Familial pulmonary capillary hemangiomatosis resulting in primary pulmonary hypertension. Ann Intern Med 1988; 109: 106–9
- Jeffery T. K., Morrell N. W. Molecular and cellular basis of pulmonary vascular remodeling in pulmonary hypertension. Prog Cardiovasc Dis 2002; 45: 173–202
- Christman B. W., McPherson C. D., Newman J. H., King G. A., Bernard G. R., Groves B. M., et al. An imbalance between the excretion of thromboxane and prostacyclin metabolites in pulmonary hypertension. N Engl J Med 1992; 327: 70–5
- Giad A., Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med 1995; 333: 214–21
- Petkov V., Mosgoeller W., Ziesche R., Raderer M., Stiebellehner L., Vonbank K., et al. Vasoactive intestinal peptide as a new drug for treatment of primary pulmonary hypertension. J Clin Invest 2003; 111: 1339–46
- Cox C. P., Linden J., Said S. I. VIP elevates platelet cyclic AMP (cAMP) levels and inhibits in vitro platelet activation induced by platelet‐activating factor (PAF). Peptides 1984; 5: 325–8
- Maruno K., Absood A., Said S. I. VIP inhibits basal and histamine‐stimulated proliferation of human airway smooth muscle cells. Am J Physiol 1995; 268: L1047–51
- Yanisagawa M., Kurihara H., Kimura S., Gotok A., Masaki T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988; 332: 411–5
- Hirata Y., Takagi Y., Fukuda Y., Marumo F. Endothelin is a potent mitogen for rat vascular smooth muscle cells. Artherosclerosis 1989; 78: 225–8
- Fagan K. A., McMutry I. F., Rodman D. M. Role of endothelin‐1 in lung disease. Respir Res 2001; 2: 90–101
- DiCarlo V. S., Chen S‐J., Meng Q. C., Durand J., Yano M., Chen Y. F., et al. ETA‐receptor antagonist prevents and reverses chronic hypoxia‐induced pulmonary hypertension in rat. Am J Physiol 1995; 269: L690–97
- Benigni A., Remuzzi G. Endothelin antagonists. Lancet 1999; 353: 133–8
- McQuillan L. P., Leung G. K., Marsden P. A., Kostyk S. K., Kourembanas S. Hypoxia inhibits expression of eNOS via transcriptional and posttranscriptional mechanisms. Am J Physiol 1994; 267: H1921–27
- Allen S. W., Chatfield B. A., Koppenhafer S. A., Schaffer M. S., Wolfe R. R., Abman S. H. Circulating immunoreactive endothelin‐1 in children with pulmonary hypertension: association with acute hypoxic pulmonary vasoreactivity. Am Rev Respir Dis 1993; 148: 519–22
- Giaid A., Yanagisawa M., Langleben D., Michael R. P., Levy R., Shennib H., et al. Expression of endothelin‐1 in the lungs of patients with pulmonary hypertension. N Engl J Med 1993; 328: 1732–9
- Vincent J. A., Ross R. D., Kassab J., Hsu J. M., Pinsky W. W. Relation of elevated plasma endothelin in congenital heart disease to increased pulmonary blood flow. Am J Cardiol 1993; 71: 1204–7
- Lee S. L., Wang W. W., Lanzillo J. J., Fanburg B. L. Serotonin produces both hyperplasia and hypertrophy of bovine pulmonary artery smooth muscle cells in culture. Am J Physiol 1994; 266: L46–52
- Herve P., Launay J. M., Scrobohaci M. L., Brenot F., Simonneau G., Petitpretz P., et al. Increased plasma serotonin in primary pulmonary hypertension. Am J Med 1995; 99: 249–54
- Tuder R. M., Cool C. D., Geraci M. W., Wang J., Abman S. H., Wright L., et al. Prostacyclin synthase expression is decreased in lungs from patients with severs pulmonary hypertension. Am J Respir Crit Care Med 1999; 159: 1925–32
- Dupuis J., Jasmin J. F., Prie S., Cernacek P. Importance of local production of endothelin‐1 and of the ETB receptor in the regulation of pulmonary vascular tone. Pulm Pharmacol Ther 2000; 13: 135–40
- Kuc R., Davenport A. Endothelin‐A receptors in human aorta and pulmonary arteries are downregulated in patients with cardiovascular disease: an adaptive response to increased levels of endothelin‐1?. J Cardiovasc Pharmacol 2000; 36 Suppl 1: S377–9
- Cool C. D., Stewart J. S., Werahera P., Miller G. J., Williams R. L., Voelkel N. F., et al. Three‐dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell‐specific markers. Evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth. Am J Pathol 1999; 155: 411–9
- Tuder R. M., Flook B. E., Voelkel N. F. Increased gene expression for VEGF and the VEGF receptors KDR/Flk and Flt in lungs exposed to acute or chronic hypoxia: modulation of gene expression by nitric oxide. J Clin Invest 1995; 95: 1798–807
- Yeager M. E., Halley G. R., Golpon H. A., Voelkel N. F., Tuder R. M. Microsatellite instability of endothelial cell growth and apoptosis genes within plexiform lesions in primary pulmonary hypertension. Circ Res 2001; 88: E2–11
- Humbert M., Morrell N. W., Archer S. L., Stenmark K. R., MacLean M. R., Lang I. M., et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43: 13S–24S
- Abenhaim L., Moride Y., Brenot F., Rich S., Benichou J., Kurz X., et al. Appetite‐suppressant drugs and the risk of primary pulmonary hypertension. N Engl J Med 1996; 335: 609–16
- Simonneau G., Fartoukh M., Sitbon O., Humbert M., Jargot J. L., Herve P. Primary pulmonary hypertension associated with the use of fenfluramine derivatives. Chest 1998; 114((Suppl3))195S–99S
- Mark E. J., Patalas E. D., Chang H. T., Evans R. J., Kessler S. C. Fatal pulmonary hypertension associated with short‐term use of fenfluramine and phenteramine. N Engl J Med 1997; 337: 602–6
- Rich S., Rubin L., Walker A. L., Schneeweiss S., Abenhaim L. Anorexigens and pulmonary hypertension in the United States: results from the Surveillance of North American Pulmonary Hypertension. Chest 2000; 117: 870–4
- Albertson T. E., Walby W. F., Derlet R. W. Stimulant‐induced pulmonary toxicity. Chest 1995; 108: 1140–9
- Dumas J. P., Bardou M., Goirand F., Dumas M. Hypoxic pulmonary vasoconstriction. Gen Pharmacol 1999; 33: 289–97
- Sweeney M., Yuan J. X. Hypoxic pulmonary vasoconstriction: role of voltage‐gated potassium channels. Respir Res 2000; 1: 40–8
- Loscalzo J. Genetic clues to the cause of primary pulmonary hypertension. N Engl J Med 2001; 345: 367–71
- Massague J., Chen Y. G. Controlling TGF‐beta signaling. Genes Dev 2000; 14: 627–44
- Shi Y., Massague J. Mechanisms of TGF‐beta signaling from cell membrane nucleus. Cell 2003; 113: 685–700
- Stewart D. J. Bone morphogenetic protein receptor‐2 and pulmonary arterial hypertension. Circ Res 2005; 96: 1033–5
- Lane K. B., Machado R. D., Pauciulo M. W., Thomson J. R., Philips J. A., 3rd., Loyd J. E., et al. Heterozygous germ‐line mutations in BMPR2, encoding a TGF‐β receptor, cause familial primary pulmonary hypertension. Nat Genet 2000; 26: 81–4
- Thomson J. R., Machado R. D., Pauciulo M. W., Morgan N. V., Humbert M., Elliot G. C., et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR‐II, a receptor member of the TGF‐β family. J Med Genet 2000; 37: 741–5
- Newman J. H., Wheeler L., Lane K. B., Loyd E., Gaddipati R., Philips J. A., 3rd., et al. Mutation in the gene for bone morphogenetic protein receptor II as a cause of primary pulmonary hypertension in a large kindred. N Engl J Med 2001; 345: 319–24
- Newman J. H., Trembath R. C., Morse J. A., Grunig E., Loyd J. E., Adnot S., et al. Genetic basis of pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43: 33S–39S
- Loyd J. E., Butler M. G., Foround T. M., Conneally P. M., Phillips J. R., Newman J. H. Genetic anticipation and abnormal gender ratio at birth in familial primary pulmonary hypertension. Am J Respir Crit Care Med 1995; 152: 93–7
- Humbert M., Deng Z., Simonneau G., Barst R. J., Sitbono O., Wolf M., et al. BMPR2 germline mutations in primary pulmonary hypertension associated with fenfluramine derivatives. Eur Respir J 2002; 20: 518–23
- Runo J. R., Vnencak‐Kones C. L., Prince M., Loyd J. E., Wheeler L., Robbins I. M., et al. Pulmonary veno‐occlusive disease caused by an inherited mutation in bone morphogenetic protein receptor II. Am J Respir Crit Care Med 2003; 167: 889–94
- Deng Z., Morse J. H., Slager S. L., Cuervo N., Moore K. J., Venetos G., et al. Familial primary pulmonary hypertension (gene PPH1) is caused by mutations in the bone morphogenetic protein receptor‐II gene. Am J Hum Genet 2000; 67: 737–44
- Eddahibi S., Humbert M., Fadel E., Raffestin B., Darmon M., Capron F., et al. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest 2001; 108: 1141–5
- MacLean M. R., Herve P., Eddahibi S., Adnot S. 5‐Hydroxytryptamine and the pulmonary circulation: receptors, transporters and relevance to pulmonary arterial hypertension. Br J Pharmacol 2000; 131: 161–8
- Eddahibi S., Chaouat A., Morrell N., Fadel E., Fuhrman C., Bugnet A. S., et al. Polymorphism of the serotonin transporter gene and pulmonary hypertension in chronic obstructive pulmonary disease. Circulation 2003; 108: 1839–44
- Farber H. W., Loscalzo J. Pulmonary arterial hypertension. N Engl J Med 2004; 351: 1655–65
- Silver R. M. Scleroderma‐clinical problems: the lungs. Rheum Dis Clin North Am 1996; 22: 825–40
- Battle R. W., Davitt M. A., Cooper S. M., Buckley L. M., Leib E. S., Beglin P. A., et al. Prevalence of pulmonary hypertension in limited and diffuse scleroderma. Chest 1996; 110: 1515–9
- Peacock A. J. Primary pulmonary hypertension. Thorax 1999; 54: 1107–18
- Spiech R., Jenni R., Opravil M., Pfab M., Russi E. W. Primary pulmonary hypertension in HIV infection. Chest 1991; 100: 1268–71
- Farber H. W. HIV‐associated pulmonary hypertension. AIDS Clin Care 2001; 13: 53–9
- Weiss R. A., Whitby D., Talbot S., Kellam P., Boshoff C. Human herpesvirus 8 and Kaposi's sarcoma. J Natl Cancer Inst Monogr 1998; 23: 51–4
- Cool C. D., Rai P. R., Yeager M. E., Hernandez‐Saavedra D., Serls A. E., Bull T. M., et al. Expression of human herpesvirus 8 in primary pulmonary hypertension. N Engl J Med 2003; 349: 1113–22
- Yang Y. Y., Lin H. C., Lee W. C., Hou M. C., Lee F. Y., Chang F. Y., et al. Portopulmonary hypertension: distinctive hemodynamic and clinical manifestations. J Gastroenterol 2001; 36: 181–6
- Donovan C. L., Marcovitz P. A., Punch J. D., Bach D. S., Brown K. A., Lucey M. R., et al. Two‐dimensional and dobutamine stress echocardiography in the preoperative assessment of patients with end‐stage liver disease prior to orthotopic liver transplantation. Transplantation 1996; 61: 1180–8
- Molden D., Abraham J. L. Pulmonary hypertension: its association with hepatic cirrhosis and iron accumulation. Arch Pathol Lab Med 1982; 106: 328–31
- Castro O., Hoque M., Brown B. D. Pulmonary hypertension in sickle cell disease: cardiac catheterization results and survival. Blood 2003; 101: 1257–61
- Jison M. L., Gladwin M. T. Hemolytic anemia‐associated pulmonary hypertension of sickle cell disease and the nitric oxide/arginine pathway. Am J Resp Crit Care Med 2003; 168: 3–4
- Minter K. R., Gladwin M. T. Pulmonary complications of sickle cell anemia. A need for increased recognition, treatment, and research. Am J Resp Crit Care Med 2001; 164: 2016–9
- Hoeper M. M., Niedermeyer J., Hoffmeyer F., Flemming P., Fabel H. Pulmonary hypertension after splenectomy?. Ann Intern Med 1999; 130: 506–9
- Atichartakarn V., Likittanasombat K., Chuncharunee S., Chandanamattha P., Worapongpaiboon S., Angchaisuksiri P., et al. Pulmonary arterial hypertension in previously splenectomized patients with β‐thalassemic disorders. Int J Hematol 2003; 78: 139–45
- Marvin K., Spellberg R. D. Pulmonary hypertension secondary to thrombocytosis in a patient with myeloid metaplasia. Chest 1993; 103: 642–4
- Rostagno C., Prisco D., Abbate R., Poggesi L. Pulmonary hypertension associated with long‐standing thrombocytosis. Chest 1991; 99: 1303–5
- Trembath R. C., Thomson J. R., Machado R. D., Morgan N. V., Atkins C., Winship I., et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001; 345: 325–34
- Chaouat A., Coulet F., Favre C., Simonneau G., Weitzenblum E., Soubrier F., et al. Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax 2004; 59: 446–8
- Barst R. J., McGoon M., Torbicki A., Sitbon O., Krowka M. J., Olschewski H., et al. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43: 40S–47S
- Bossone E., Avelar E., Bach D. S., Gillespie B., Rubenfire M., Armstrong W. F. Diagnostic value of resting tricuspid regurgitation velocity and right ventricular ejection flow parameters for the detection of exercise‐induced pulmonary arterial hypertension. Int J Card Imaging 2000; 16: 429–36
- Ommen S. R., Nishimura R. A., Hurrell D. G., Klarich K. W. Assessment of right atrial pressure with 2‐dimensional and Doppler echocardiography: a simultaneous catheterization and echocardiographic study. Mayo Clin Proc 2000; 75: 24–9
- Denton C., Cailes J., Phillips G., Wells A., Black C., Du Bois R. Comparison of Doppler echocardiography and right heart catheterization to assess pulmonary hypertension in systemic sclerosis. Br J Rheumatol 1997; 36: 239–43
- Penning S., Robinson K., Major C., Garite T. A comparison of echocardiography and pulmonary artery catheterization for evaluation of pulmonary artery pressures in pregnant patients with suspected pulmonary hypertension. Am J Obstet Gynecol 2001; 184: 1568–70
- Rich S., Kieras K., Hart K., Groves B. M., Stobo J. D., Brundage B. H. Antinuclear antibodies in primary pulmonary hypertension. J Am Coll Cardiol 1986; 8: 1307–11
- Morris T. A., Auger W. R., Ysrael M. Z., Olson L. K., Channick R. N., Fedullo P. F., et al. Parenchymal scarring is associated with restrictive spirometric defects in patients with chronic thromboembolic pulmonary hypertension. Chest 1996; 110: 399–403
- Owens G. R., Fino G. J., Herbert D. L., Steen V. D., Medsger T. A., Jr., Pennock B. E., et al. Pulmonary function in systemic sclerosis. Comparison of CREST syndrome variant with diffuse scleroderma. Chest 1983; 84: 546–50
- Steen V., Medsger T. A., Jr. Predictors of isolated pulmonary hypertension in patients with systemic sclerosis and limited cutaneous involvement. Arthritis Rheum 2003; 48: 516–22
- Rafanan A. L., Golish J. A., Dinner D. S., Hague L. K., Arroliga A. C. Nocturnal hypoxemia is common in primary pulmonary hypertension. Chest 2001; 120: 849–9
- Atwood C. W., Jr., McCrory D., Garcia J. G., Abman S. H., Ahearn G. S. Pulmonary artery hypertension and sleep‐disordered breathing: ACCP evidence‐based clinical practice guidelines. Chest 2004; 126: 72S–7S
- Fedullo P. F., Auger W. R., Kerr K. M., Rubin L. J. Chronic thromboembolic pulmonary hypertension. N Engl J Med 2001; 345: 1465–72
- Azarian R., Wartski M., Collignon M. A., Parent F., Herve P., Sors H., et al. Lung perfusion scans and hemodynamics in acute and chronic pulmonary embolism. J Nuc Med 1997; 38: 980–3
- Worsley D. F., Palevsky H. I., Alavi A. Ventilation‐perfusion lung scanning in the evaluation of pulmonary hypertension. J Nucl Med 1994; 35: 793–6
- D'Alonzo G. E., Bower J. S., Dantzker D. R. Differentiation of patients with pulmonary and thromboembolic pulmonary hypertension. Chest 1984; 85: 457–61
- Moser K. M., Auger W. R., Fedullo P. F. Chronic major‐vessel thromboembolic pulmonary hypertension. Circulation 1990; 81: 1735–43
- Pruszczyk P., Torbicki A., Pacho R., Chellum M., Kuch‐Wocial A., Pusznski B., et al. Noninvasive diagnosis of suspected severe pulmonary embolism: transesophageal echocardiography vs. spiral CT. Chest 1997; 112: 722–8
- Jamieson S. W., Kapelanski D. P., Sakakibara N., Manecke G. R., Thistlethwaite P. A., Kerr K. M., et al. Pulmonary thromboendartectomy. Experience and results in 1,500 cases. Ann Thorac Surg 2003; 76: 1457–64
- Miyamoto S., Nagaya N., Satoh T., Kyotani S., Sakamaki F., Fujita M., et al. Clinical correlates and prognostic significance of six‐minute walk test in patients with primary pulmonary hypertension: comparison with cardiopulmonary exercise testing. Am J Respir Crit Care Med 2000; 161: 487–92
- Rubin L. J., Badesch D. B. Evaluation and management of the patient with pulmonary arterial hypertension. Ann Intern Med 2005; 143: 282–93
- Paciocco G., Martinez F., Bossone E., Pielsticker E., Gillespie B., Rubenfire M. Oxygen desaturation on the six‐minute walk test and mortality in untreated primary pulmonary hypertension. Eur Respir J 2001; 17: 647–52
- Hoeper M. M., Oudiz R. J., Peacock A., Tapson V. F., Haworth S. G., Frost A. E., et al. End points and clinical trial designs in pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43: 48S–55S
- Humbert M., Sitbon O., Simonneau G. Treatment of pulmonary arterial hypertension. N Engl J Med 2004; 351: 1425–36
- Fuster V., Steele P. M., Edwards W. D., Gersh B. J., McGoon M. D., Frye R. L. Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation 1984; 70: 580–7
- Rich S., Kaufmann E., Levy P. S. The effect of high doses of calcium‐channel blockers on survival in primary pulmonary hypertension. N Engl J Med 1992; 327: 76–81
- Rich S., Brundage B. H. High‐dose calcium channel‐blocking therapy for primary pulmonary hypertension: evidence for long‐term reduction in pulmonary arterial pressure and regression of right ventricular hypertrophy. Circulation 1987; 76: 135–41
- Raffy O., Azarian R., Brenot F., Parent F., Sitbon O., Petitpretz P., et al. Clinical significance of the pulmonary vasodilator response during short‐term infusion of prostacyclin in primary pulmonary hypertension. Circulation 1996; 93: 484–8
- Barst R. J., Maislin G., Fishman A. P. Vasodilator therapy for primary pulmonary hypertension in children. Circulation 1999; 99: 1197–208
- Nootens M., Kaufmann E., Rich S. Short‐term effectiveness of nifedipine in secondary pulmonary hypertension. Am J Cardiol 1993; 71: 1475–6
- Rubin L. J., Mendoza J., Hood M., McGoon M., Barst R., Williams W. B., et al. Treatment of primary pulmonary hypertension with continuous intravenous prostacyclin (epoprostenol). Results of a randomized trial. Ann Intern Med 1990; 112: 485–91
- Barst R. J., Rubin L. J., Long W. A., McGoon M. D., Rich S., Badesch D. B., et al. A comparison of continuous intravenous epoprostenol (prostacyclin) with conventional therapy for primary pulmonary hypertension. The Primary Pulmonary Hypertension Study Group. N Engl J Med 1996; 334: 296–302
- Badesch D. B., Tapson V. F., McGoon M. D., Brundage B. H., Rubin L. J., Wigley F. M., et al. Continuous intravenous epoprostenol for pulmonary hypertension due to the scleroderma spectrum of disease. A randomized, controlled trial. Ann Intern Med 2000; 132: 425–34
- Stibon O., Humbert M., Nunes H., Parent F., Garcia G., Herve P., et al. Long‐term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol 2002; 40: 780–8
- Barst R. J., Rubin L. J., McGoon M. D., Caldwell E. J., Long W. A., Levy P. S. Survival in primary pulmonary hypertension with long‐term continuous intravenous prostacyclin. Ann Intern Med 1994; 121: 409–15
- Shapiro S. M., Oudiz R. J., Cao T., Romano M. A., Beckmann X. J., Georgiou D., et al. Primary pulmonary hypertension: improved long‐term effects and survival with continuous intravenous epoprostenol infusion. J Am Coll Cardiol 1997; 30: 343–9
- McLaughlin V. V., Shillington A., Rich S. Survival in primary pulmonary hypertension: the impact of epoprostenol therapy. Circulation 2002; 106: 1477–82
- Rosenzweig E. B., Kerstein D., Barst R. J. Long‐term prostacyclin for pulmonary hypertension with associated congenital heart defects. Circulation 1999; 99: 1858–65
- Kuo P. C., Johnson L. B., Plotkin J. S., Howell C. D., Bartlett S. T., Rubin L. J. Continuous intravenous infusion of epoprostenol for the treatment of portopulmonary hypertension. Transplantation 1997; 63: 604–6
- Aquilar R. V., Farber H. W. Epoprostenol (prostacyclin) therapy in HIV‐associated pulmonary hypertension. Am J Respir Crit Care Med 2000; 162: 1846–50
- Simonneau G., Barst R. J., Galie N., Naeije R., Rich S., Bourge R. C., et al. Continuous subcutaneous infusion of treprostinil, a prostacyclin analogue, in patients with pulmonary arterial hypertension: a double‐blind randomized, placebo‐controlled trial. Am J Respir Crit Care Med 2002; 165: 800–4
- Galie N., Humbert M., Vachiery J. L., Vizza C. D., Kneussel M., Manes A., et al. Effects of beraprost sodium, an oral prostacyclin analogue, in patients with pulmonary arterial hypertension: a randomized, double‐blind, placebo‐controlled trial. J Am Coll Cardiol 2002; 39: 1496–502
- Barst R. J., McGoon M., McLauglin V., Tapson V., Rich S., Rubin L., et al. Baraprost therapy for pulmonary arterial hypertension. J Am Coll Cardiol 2003; 41: 2119–25
- Oleschewski H., Ghofrani H. A., Schmehl T., Winkleer J., Wilkens H., Hoper M. M., et al. Inhaled iloprost to treat severe pulmonary hypertension. An uncontrolled trial. German PPH Study Group. Ann Intern Med 2000; 132: 435–43
- Hoeper M. M., Schwarze M., Ehlerding S., Adler‐Schuermeyer A., Spiekerkoetter E., Niedermeyer J., et al. Long‐term treatment of primary pulmonary hypertension with aerosolized iloprost, a prostacyclin analogue. N Engl J Med 2000; 342: 1866–70
- Oleschewski H., Simonneau G., Galie N., Higenbottam T., Naeije R., Rubin L. J., et al. Inhaled iloprost for severe pulmonary hypertension. N Engl J Med 2002; 347: 322–9
- Channick R. N., Simonneau G., Sitbon O., Robbins I. M., Frost A., Tapson V. F., et al. Effects of the dual endothelin‐receptor antagonist bosentan in patients with pulmonary hypertension: a randomized placebo‐controlled study. Lancet 2001; 358: 1119–23
- Rubin L. J., Badesch D. B., Barst R. J., Galie N., Black C. M., Keogh A., et al. Bosentan therapy for pulmonary arterial hypertension. N Engl J Med 2002; 346: 896–903
- Barst R. J., Langleben D., Frost A., Horn E. M., Oudiz R., Shapiro S., et al. Sitaxsentan therapy for pulmonary arterial hypertension. Am J Respir Crit Care Med 2004; 169: 441–7
- Beavo J. A., Reifsynder D. H. Primary sequence of cyclic nucleotide phosphodiesterase isozymes and the design of selective inhibitors. Trends Pharmacol Sci 1990; 11: 150–5
- Ahn H. S., Foster M., Cable M., Pitts B. J., Sybertz E. J. Ca/CaM‐stimulated and cGMP‐specific phosphodiesterases in vascular and non‐vascular tissues. Adv Exp Med Biol 1991; 308: 191–7
- Braner D. A., Fineman J. R., Chang R., Soifer S. J. M&B 22948, a cGMP phosphodiesterase inhibitor is a pulmonary vasodilator in lambs. Am J Physiol 1993; 264: H252–8
- Michalakis E., Tymchak W., Lien D., Webster L., Hashimoto K., Archer S. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: comparison with inhaled nitric oxide. Circulation 2002; 105: 2398–403
- Lepore J. J., Maroo A., Pereira N. L., Ginns L. C., Dec G. W., Zapol W. M., et al. Effect of sildenafil on the acute pulmonary vasodilator response to inhaled nitric oxide in adults with primary pulmonary hypertension. Am J Cardiol 2002; 90: 677–80
- Wilkens H., Guth A., Konig J., Forestier N., Cremers B., Hermen B., et al. Effect of inhaled iloprost plus oral sildenafil in patients with primary pulmonary hypertension. Circulation 2001; 104: 1218–22
- Rothman A., Slansky M. S., Lucas V. W., Kashanie I. A., Shughneny R. D., Channick R. H. Atrial septostomy as a bridge to lung transplantation in patients with severe pulmonary hypertension. Am J Cardiol 1999; 84: 682–6
- Sandoval J., Rothman A., Pulido T. Atrial septostomy for pulmonary hypertension. Clin Chest Med 2001; 22: 547–60
- Sandoval J., Gaspar J., Pulido, Bautista E., Martinez‐Guerra M. L., Zeballos M., et al. Graded balloon dilation atrial septostomy in severe primary pulmonary hypertension. A therapeutic alternative for patients non‐responsive to vasodilator treatment. J Am Coll Cardiol 1998; 32: 297–304
- Jamieson S. W., Auger W. R., Fedullo P. F., Channick R. N., Kirett J. M., Tarazi R. Y., et al. Experience and results with 150 pulmonary thromboendarterectomy operations over a 29‐month period. J Thorac Cardiovasc Surg 1993; 106: 116–27
- Archibald C. J., Auger W. R., Fedullo P. F., Channick R. N., Kerr K. M., Jamieson S. W., et al. Long‐term outcome after pulmonary thromboendartectomy. Am J Respir Crit Care Med 1999; 160: 523–8
- Wensel R., Opitz C. F., Anker S. D., Winkler J., Hoffken G., Kleber F. X., et al. Assessment of survival in patients with primary pulmonary hypertension. Importance of cardiopulmonary exercise testing. Circulation 2002; 106: 319–24
- Doyle R. l., McCrory D., Channick R. N., Simmonneau G., Conte J. Surgical treatments/interventions for pulmonary arterial hypertension: ACCP evidence‐based clinical practice guidelines. Chest 2004; 126: 63S–71S
- Pielsticker E. J., Martinez F. J., Rubenfire M. Lung and heart‐lung transplant practice patterns in pulmonary hypertension centers. J Heart Lung Transplant 2001; 20: 1297–304
- Boujoukos A. J., Martich G. D., Vega J. D., Keenan R. J., Griffith B. P. Reperfusion injury in single lung transplant recipients with pulmonary hypertension and emphysema. J Heart Lung Transplant 1997; 16: 440–48
- Hertz M. I., Mohacsi P. J., Taylor D. O., Trulock E. P., Boucek M. M., Deng M. C., et al. The registry of the International Society for Heart and Lung Transplantation: twentieth official report. J Heart Lung Transplant 2003; 22: 610–15
- Whyte R. I., Robbins R. C., Altinger J., Barlow C. W., Doyle R. L., Theodore J., et al. Heart‐lung transplantation for primary pulmonary hypertension. Ann Thorac Surg 1999; 67: 937–41
- Bando K., Armitage J. M., Paradis I. L., Keenan R. J., Hardesty R. L., Konishi H., et al. Indications for and results of single, bilateral, and heart‐lung transplantation for pulmonary hypertension. J Thorac Cardiovasc Surg 1994; 108: 1056–65
- Klepetko W., Mayer E., Sandoval J., Trulock E., Vachiery J. L., Dartevelle P., et al. Interventional and surgical modalities of treatment for pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43: 73S–80S
- Sharples L. D., Tamm M., McNeil K., Higenbottam T. W., Stewart S., Wallwork J. Development of bronchiolitis obliterans syndrome in recipients of heart‐lung transplantation: early risk factors. Transplantation 1996; 61: 560–6
- Galie N., Seeger W., Naeije R., Simonneau G., Rubin L. J. Comparative analysis of clinical trials and evidence‐based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol 2004; 43: 81S–88S