Abstract
Celiac disease (CD) is characterized by a chronic immune reaction in the small intestine to the gluten proteins that are present in a (Western) daily diet. Besides the well known involvement of the HLA class II histocompatibility antigen (HLA)‐DQ2.5 and ‐DQ8 heterodimers (encoded by particular combinations of the HLA‐DQA1 and ‐DQB1 gene) in CD and the minor contribution of the CTLA‐4 gene, recently the myosin IXB (MYO9B) gene has also been found to be genetically associated. This review covers the general aspects of CD as well as current insight into important molecular aspects. We evaluate the role of susceptibility genes in CD by following gluten along its path from ingestion to uptake in the body, which leads us through the three aspects of CD's pathology. The first is the presence of gluten in the lumen of the intestine, where it is broken down by several enzymes. The second is the intestinal barrier through which gluten peptides pass. The third is the reaction of the immune system in response to gluten peptides, in which both the innate and the adaptive immune systems play a role. Our main conclusion, based on the current genetic and functional studies, is that we should look for causal genes in the barrier function as well as in the immune systems.
| Abbreviations | ||
| ASCA | = | anti‐Saccharomyces cerevisiae antibodies |
| CARD | = | caspase recruitment domain |
| CD | = | celiac disease |
| DH | = | dermatitis herpetiformis |
| RCD | = | refractory CD |
| EATL | = | enteropathy‐associated T cell lymphoma |
| RR | = | relative risk |
| MYO9B | = | myosin IXB |
| CTLA‐4 | = | cytotoxic T lymphocyte‐associated 4 |
| PREP | = | prolyl endopeptidase |
| PGPEPI | = | pyroglutamyl‐peptidase I |
| tTG | = | tissue transglutaminase |
| IBD | = | inflammatory bowel disorders |
| MYLKalias MLCK | = | myosin light chain kinase |
| ROCK | = | Rho kinase |
| TNF | = | tumor necrosis factor |
| MICA | = | MHC class I polypeptide‐related chain A |
| IEL | = | intraepithelial lymphocytes |
Introduction
An inflammatory disorder in response to gluten
Celiac disease (CD) is characterized by a chronic immune reaction in the small intestine to the gluten proteins that are present in a (Western) daily diet. Gluten proteins are storage proteins present in wheat, barley and rye, and are needed to maintain the processing quality of food derivates such as bread, pasta, and cookies; they are also widely used to bind and thicken sauces. CD mostly occurs in Westernized populations where gluten‐derived products form a large part of the diet. The involvement of the HLA‐DQ2.5 and ‐DQ8 heterodimers (encoded by particular combinations of the HLA‐DQA1 and ‐DQB1 gene) in CD is well known since most of the patients carry these molecules.
This review covers the current insight into important aspects of CD and evaluates the role of susceptibility genes in CD by following the gluten molecules from the lumen of the small intestine, passing through the epithelial barrier of the intestine and eliciting the immune response (see also Figure ).
Figure 1 Overview of the path taken by gluten. a) Gluten enters the lumen of the small intestine and is cleaved by several enzymes. b) It subsequently passes through the enterocytes in the intestinal barrier, which is impaired in celiac disease (CD) patients; myosin IXB (MYO9B) might play a role here by affecting the tight junctions. Enterocytes express the MICA ligand that binds to the NKG2D receptor on the intraepithelial lymphocytes (IEL). Besides this, enterocytes also produce IL‐15 capable of stimulating IELs and T cells. c) Both the innate and the adaptive immune system play a role. 1) Adaptive immune response: gluten is deamidated by tissue transglutaminase (tTG) in the lamina propria and elicits an immune response in genetically susceptible individuals who are also HLA‐DQ2.5 and/or ‐DQ8 positive. 2) Innate immune response: gluten induces the antigen‐presenting cell (APC) to produce Il‐15, which stimulates the production and interaction of the MICA ligand and the NKG2D receptor. IL‐15 also stimulates the adaptive immune response.
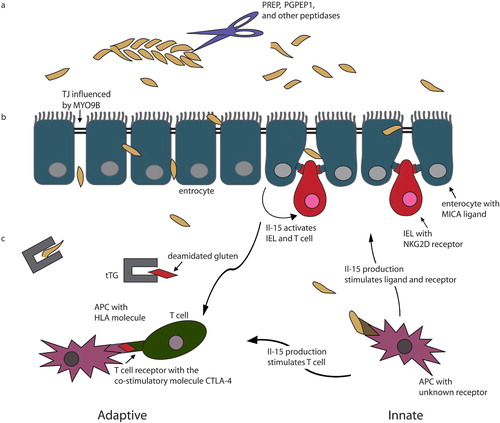
General overview of CD
Clinical features and treatment
Until recently CD was considered to be a pediatric intestinal disorder, seen mostly in young children who had just started a gluten‐containing diet.
The intestinal problems seen in CD patients comprise chronic diarrhea, growth retardation, weight loss, abdominal pain, vomiting, bloating, distention, and constipation (reviewed in Rewers et al.) Citation1. CD is now being recognized more and more in older patients who do not present with all the clinical features or who are developing more of these features at a later age. Their symptoms are more systemic, and the disease is now more likely to present as a multiorgan disorder, including bone problems, ataxia, carditis, reproductive problems and skin manifestations like dermatitis herpetiformis Citation2. These older patients may present with either active or silent CD (reviewed in Dewar et al.) Citation2. The more systemic presentation of active CD leads to these patients not always being recognized or properly diagnosed. The silent cases are unaware of their disease due to a lack of symptoms, or they may have only vague symptoms which often remain undiagnosed. They are mostly identified during the screening of large populations to determine prevalence, or of other risk groups like index patients' family members.
Over the last 25 years it was observed that the frequency of patients presenting with diarrhea at diagnosis has gradually dropped from 91% to 37% Citation3. The duration of symptoms before diagnosis has also decreased, from 11 years to 4 years, nonetheless this is just the average duration and 4 years is still a long period, meaning that there are many patients who have remained undiagnosed for a long time.
The only existing treatment for CD is a life‐long gluten‐free diet (GFD), which reverses the damage in the intestine so that the Marsh III stage (Marsh stages, see diagnosis and Table ) gradually changes to a Marsh 0 stage, the clinical symptoms disappear, and the risk of complications diminishes. The normalization in the intestine is slower in adults than in young children, who often reach the Marsh 0 stage in a few months, while adults may take up to a few years Citation4.
Features observed in CD patients are an impaired intestinal barrier, less regular tight junctions and altered sugar absorption ratios. These features are mostly seen in patients on a gluten‐containing diet, although the functionality of the intestinal barrier does not recover completely in patients on a gluten‐free diet who regain a normalized intestine Citation5–7. Patients with dermatitis herpetiformis (DH, discussed below) who show no histological damage in the intestine may also have an impaired intestinal barrier Citation8.
One of the systemic manifestations of CD is DH generally seen as a cutaneous manifestation of CD (reviewed in Zone et al.) Citation9. Most DH patients have varying degrees of lesions in the small intestine and react to gluten. DH and CD run in families and often co‐occur in individuals. The best treatment is a gluten‐free diet, although most patients only treat their skin problems using medicine and neglect the underlying intestinal problems caused by gluten.
A small proportion (<7%) of CD patients do not respond to a strict GFD Citation4. This group of CD patients (generally referred to as refractory CD (RCD) patients) is split into RCD type I and RCD type II patients and was defined by Daum et al. as individuals with a persisting villous atrophy with crypt hyperplasia and increased intraepithelial lymphocytes (IEL) in spite of being on a strict GFD for more than 12 months, or those whose severe and persisting symptoms necessitate intervention independent of the duration of their GFD Citation10. The RCD type II group (<0.3% of the total CD population) shows abnormal IELs, with abnormal surface markers in small intestinal biopsies, like the expression of intracytoplasmic CD3e and the lack of several classical T cell markers, e.g. CD8 and CD4. A proportion of this group develops enteropathy‐associated T cell lymphoma (EATL).
CD also co‐occurs with other autoimmune disorders in families and in patients Citation11, suggesting an overlap in causative genes between different disorders.
Diagnosis
Serological screening is often used as the first step in the diagnosis of CD, measuring the endomysial antibodies and tissue transglutaminase antibodies; the latter is the autoantibody in CD (see review by Schuppan et al.) Citation12. A diagnosis of CD is made when a person's symptoms meet the revised ESPGHAN criteria Citation13. The main diagnostic criteria are a positive biopsy (Marsh III A, B, or C) while on a gluten‐containing diet, and a full clinical remission when the patient is on a gluten‐free diet. Duodenal biopsies are classified according to Marsh who defined four stages, the last of which was later divided into three substages (see Table ) Citation14, Citation15. Positive antibodies that disappear when the patient is on a gluten‐free diet add to the diagnosis.
If there is any doubt about the initial diagnosis, extra steps should be taken. A doubtful diagnosis may occur more often in children under the age of 2 years, who may also suffer from other causes of enteropathy (like giardiasis, cows' milk sensitive enteropathy, and postenteritis syndrome). A biopsy on a gluten‐free diet should be taken—this should show a normalized histology—and then they should be given a gluten challenge after 2 years (or longer) on a gluten‐free diet. The gluten challenge should not be given before the age of 6, and it needs to be followed by a biopsy, which should again show a Marsh III classification Citation13.
Prevalence and genetics
The prevalence of CD in the general population ranges from 0.5% to 1.26% and is estimated to be on average 1%, based on serology screenings in unselected populations (systematically reviewed by Dube et al.) Citation16. These numbers have also been found in the USA, where CD was long thought to be a rare disorder Citation17, Citation18. The prevalence in patients' family members ranges from 2.8% to 22.5% depending on the study methods (i.e. serology and/or biopsy) and on the groups tested (e.g. first/second degree family members, two affected patients already known in the family) Citation16. This implies an increased familial clustering of ∼10%, resulting in a relative risk (RR) of 10 for the disorder and suggesting a role for genetic factors. The prevalence was further corroborated by two large population‐based twin studies in the Italian population, which found a concordance rate in dizygotic twins of around 20% and in monozygotic twins of around 85% Citation19, Citation20. The increase in concordance between twins and the fact that monozygotic twins do not have a 100% concordance rate suggests the involvement of environmental factors in addition to genetic factors.
A complex genetic disorder
CD is a complex genetic disorder involving multiple genes as well as environmental factors, of which the most important is gluten. For the genetic part of CD we expect to find multiple genes since inheritance of CD does not follow a Mendelian pattern. We assume a model comprising a major gene (the HLA gene that will be discussed below) and several low‐risk genes (such as myosin IXB (MYO9B) and cytotoxic T lymphocyte‐associated 4 (CTLA‐4)) Citation21–26. Discovering the gene(s) in complex disorders is a daunting task and past progress has been slow, but due to modern techniques more and more causative genes will now be found (Box 1; Table ). The loci presented in are reviewed in detail by van Heel et al. Citation27.
HLA gene has been the only known genetic variant for over 30 years
The involvement of the HLA complex located in the major histocompatibility complex (MHC) region on chromosome 6, which predisposes to CD, has been known for over 30 years Citation28–30. Via the observed involvement of HLA‐A8, HLA‐DW3 (now HLA‐DR3) and HLA‐DQ in CD Citation28, Citation29, Citation31–33, the HLA‐DQA1 and ‐DQB1 genes were shown to be involved and were the first genes found to be genetically and repeatedly associated to CD Citation34–37. The HLA‐DQA1 and ‐DQB1 genes are in a region of high linkage disequilibrium and together form heterodimers, some forms of which are associated to CD. The estimated contribution of the HLA region on developing CD is around 40% Citation38, Citation39, meaning the remaining 60% of genetic factors involved in CD was unknown.
Recently a second gene, MYO9B on chromosome 19, was identified in the Dutch population Citation40; it may explain some 20% of the genetic risk factors involved in the Dutch CD population. Replication in other populations is needed to determine the true relative risk. At the moment there is no positive replication, which might be due to lack of power and overestimation of the genetic risk (a problem that is often seen in genetic studies; see Box 1 for a more extensive discussion of genetics studies, with MYO9B as an example) Citation41–43. Besides this, the CTLA‐4 gene that plays a role in several immune disorders is also involved in CD, although it probably only accounts for a few percent of the genetic variation Citation21–26. Since we do not know how these three genes interact, it is difficult to determine the exact genetic space left for other possible genes.
Molecular pathogenesis of CD
The molecular aspects of CD can be divided into three parts; these will be elaborated in the rest of this review.
The main environmental factor involved in CD is gluten. Following its path from ingestion to uptake in the body leads us through three aspects of the pathology of CD. The first is the presence of gluten in the lumen of the intestine, where it is broken down by several enzymes into smaller peptides. The second is the intestinal barrier through which gluten peptides pass; it is the divide between the body and the external world. The third is the reaction of the immune system in response to gluten peptides. The genes that have been implicated in CD (Table ), and the ones still to be found, may be involved in all these aspects and reveal more about the molecular basis of CD.
Key messages
Gluten proteins are resistant to enzymatic breakdown, leading to peptides that may evoke an immune response in genetically susceptible individuals.
The intestinal barrier is impaired in celiac disease patients and this affects its protective function against pathogens and the selective uptake of nutrients. Myosin IXB was found to be genetically involved in the impaired barrier.
Both the innate and the adaptive immune response play a role in celiac disease and the genetic involvement of the HLA‐DQ2.5 and ‐DQ8 molecules as well as the CTLA‐4 gene has been shown.
Gluten in the lumen of the small intestine
Gluten consists of multiple immunogenic peptides
Gluten is a mixture of gliadin and glutenin proteins, which, in turn, are mixtures of α‐, β‐, γ‐, ω‐gliadins, low molecular weight glutenins, and high molecular weight glutenins, each having allelic variants resulting in a huge number of functional and nonfunctional proteins Citation44. A subset of these glutens, the α‐ and γ‐gliadins, low molecular weight glutenins, and high molecular weight glutenins, can give rise to potential toxicity. Gluten proteins and their homologues are present in different grains: wheat, rye and barley. Pasta wheat consists of a combination of two genomes giving rise to the AABB tetraploids, while bread wheat consists of a combination of three genomes, giving rise to the AABBDD hexaploids. Especially the D genome contains the toxic gluten epitopes. Some cultivars of the pasta wheat (tetraploid AABB genome) contain no toxic gluten epitopes. This implies that it is possible to find or modify cultivars that lack the toxic epitopes, have baking qualities and might be safe for CD patients. Several of the existing wheat varieties have been tested for their potential toxicity, and they show differences in toxic T cell responses Citation45–47. One example of a naturally occurring, nontoxic wheat variety is Teff Citation48.
Several enzymes play a role in gluten break down
Dietary proteins are broken down by pepsin in the stomach, secreted pancreatic proteases and carboxypeptidases. Then exo‐ and endopeptidases located in the brush border membrane of the epithelial layer of the small intestine continue the digestion into mono‐, di‐ and tripeptides that can then be transported across the epithelial cells into the lamina propria.
Unlike most dietary proteins, gluten is indigestible by most proteases due to its high proline rich content Citation49. The only gluten‐specific enzyme is prolyl endopeptidase (PREP), which cleaves peptide bonds at the C‐terminal side of proline residues Citation50. Other peptides can play a role in the subsequent digestion steps, like pyroglutamyl‐peptidase I (PGPEPI), which has been shown in vitro to cleave the indigestible parts of gluten, probably when L‐pyroglutamyl residues are being formed after the first cleavage steps Citation51. This cleavage leads to unstable peptides that are easily digestible.
It has been hypothesized that small aberrations in the function of one of the proteases could lead to longer peptides or an increased load of toxic gluten epitopes in the lumen or lamina propria Citation52. A decrease in gluten digestion by brush border enzymes has been observed in untreated CD patients when compared to controls Citation53. This resistance of gluten to enzymatic digestion might result from the changes in cell function of the intestine secondary to other pathological mechanisms, or it could be causal due to a genetic background that makes patients more vulnerable to gluten.
Following this, the two genes that encode for peptidase enzymes, PREP and PGPEP1, are also located in CD linkage regions (6q21–22 and MYO9B, respectively) and have been studied for their potential causal role in CD (Table ), but no association could be found Citation54, Citation55.
This suggests that if these enzymes are indeed involved in the less proper digestion of gluten, their role in CD is not causal but secondary, and it is rather the impaired structure of the intestine that influences the function of these enzymes.
Possible treatment with a bacterial form of PREP
The 33‐mer gliadin peptide that cannot be digested by gastric and pancreatic enzymes was shown to be digestible using a bacterial homologue (from Flavobacterium meningosepticum) of prolyl endopeptidase called PEP Citation52. Peptidase therapy might be a potential therapy, since this digestion resulted in a decrease of immunogenic gluten peptides, thereby diminishing the T cell response and the specificity to tissue transglutaminase (tTG, discussed below) Citation56.
The epithelial layer
Forms the barrier between the body and the external world
The body tissue is separated from bacterial‐filled lumen of the intestine by a single layer of cells, the enterocytes. The enterocytes have two major functions: to form a physical barrier as protection against unwanted antigens/pathogens (like microorganisms), and the selective uptake of nutrients. This important intestinal layer is more permeable in CD patients and some of their relatives, since tight junctions are less regular and sugar absorption ratios are altered Citation5–8. Normally this layer has an enormous surface area (equivalent to that of a soccer field) due to the folds in the villi and microvilli. This surface area is drastically reduced in CD patients. The stem cells in the crypt give rise to progenitor cells that move up along the crypt/villus axis, they lose their capacity to proliferate, and mature into differentiated enterocytes. As these cells after approximately 5 days become too old, unnecessary, or infected, they are shed into the lumen by an unknown but well orchestrated process that does not seem to involve apoptosis as a major or triggering factor. Interestingly, in spite of the constant cell shedding from this single epithelial cell layer, a study in mice has shown that its integrity does not seem to be impaired although gaps in this single cell layer can be seen for up to 1 hour Citation57. Tight junctions between the cells are one of the mechanisms to retain the barrier integrity, but there are also some undefined fluids seen in mice that fill the gaps until they are resolved.
Besides being a physical barrier, the enterocytes have several other functions, like uptake of small peptides, and defense against and cross‐talk with pathogens. The vast amount of bacteria and other microorganisms in the lumen of the intestine that can either be beneficial or pathogenic means there is a need for cross‐talk between them and the enterocytes Citation58. This cross‐talk is done partly by pattern recognition proteins (encoded for by Toll‐like receptors and caspase recruitment domain family genes (CARD)) leading to either a response against the pathogen or a tolerance to it (for more insight into the intestinal epithelial barrier see Ismail et al.) Citation58.
One of the peptides that have to pass the epithelial barrier is gluten. We do not know whether gluten peptides cross this barrier by a paracellular or transcellular mechanism, making it difficult to assess how the impaired barrier increases susceptibility to CD.
An intriguing finding in CD patients is that the normally almost sterile proximal small intestine contains a high number of rod‐shaped bacteria on the mucosa Citation59. This feature is also seen in treated patients and seems to indicate that bacterial penetration and subsequent binding is promoted in CD patients. This might be due to an altered mucous/glycocalyx layer, although the origin of the process is unknown. Perhaps the increased stickiness of the epithelial layer to prolamin may irritate the epithelial surface, and prolamin is then mistaken for a pathogen giving rise to the immune system's unwanted reaction seen in CD patients.
Increased permeability of the epithelial barrier is seen in CD patients
CD patients show an impairment of the epithelial barrier but exactly how the damage is caused cannot be pinpointed to one mechanism. It was long thought that the immune response to gluten causes a self‐sustaining cycle of tissue damage and the release of tissue transglutaminase (tTG, discussed below) with subsequent repair, but enormous cell damage is not observed in the tissue and the reality is probably more complicated. The impairment could be due to an altered ratio of proliferation and differentiation of the enterocytes Citation60. The enterocytes might therefore lose some of their specialized functions. Another mechanism could be the increased permeability, due to genetic variants Citation5–8, seen in CD patients and some of their family members, which is greater in untreated patients but still present in treated patients and healthy relatives. The anti‐Saccharomyces cerevisiae antibodies (ASCA) that are seen as a molecular marker of intestinal permeability are present in one‐third of CD patients and partly disappear during a gluten‐free diet, suggesting the barrier recovers Citation61. The increased amounts of interferon‐γ (IFN‐γ) and TNF‐α seen in CD could also enhance increased permeability of the barrier Citation62.
Genetic association has been shown to MYO9B
Defining the exact mechanisms that make the epithelial barrier of CD patients more vulnerable will guide our search for causative factors. One of the genes involved in barrier impairment might be the recently discovered MYO9B gene, which is associated to CD in the Dutch population Citation40. Association of this gene has now also been observed with inflammatory bowel disorders (IBD) and especially with ulcerative colitis, which may share the same mechanism that is involved in barrier impairment Citation63. Although the function of this gene in general and its implication in CD is still undefined, the gene family and the domains provide some insight. MYO9B is a single‐headed myosin motor gene that, due to its actin binding domain, is able to bind to the actin filaments in cells and move along them Citation64, Citation65. MYO9B carries its own cargo, the Rho GTPase activation protein (Rho‐GAP) domain which regulates the Rho family GTPases, to its site of action. Rho family GTPases have two functions with respect to tight junctions; they regulate the junction assembly and the selectivity of the paracellular route in the enterocytes Citation66. A more active RhoA (guanosine triphosphate‐bound form) negatively regulates the tight junctions resulting in increased permeability. A more inactive form of RhoA (guanosine diphosphate‐bound form) decreases permeability, showing that a tight regulation of RhoA is important in the balance that the intestinal border needs to exert its two main functions of being a protective and a selective barrier. A recent report connects tTG (discussed below) to RhoA activation, and genetic variants in MYO9B might also influence its own capability to regulate Rho family proteins and therefore influence the actin filaments, tight junctions and cell shapes resulting in the leaky barrier seen in CD patients Citation40, Citation67.
MLCK and ROCK
A process that potentially had some overlap with MYO9B has been seen in IBD, where the gene myosin light chain kinase (MYLK, MLCK) has been shown to have a higher expression and activity in IBD patients, with the increase of expression correlating with the severity of the lesion Citation68. Patients with inactive disease and family members also show some increase of expression. MYLK is involved in myosin II activation and consequently in the functioning of the tight junctions Citation62. Here the proposed mechanism is either increased expression leading to a leakier barrier and reactions of the immune system, or an increase of expression due to cytokine‐signaling of pro‐inflammatory cytokines, like tumor necrosis factor (TNF), with the leaky barrier as a result. Both mechanisms could lead to a self‐sustaining cycle of barrier impairment and inflammatory responses. In addition, MYLK together with Rho kinase (ROCK) is involved in purse‐string wound healing Citation69. In the small intestine where there is constant cell shedding and pressure to maintain the barrier, an altered function of genes involved in permeability and wound closure could lead to disease, and it is perhaps here that MYO9B plays a role in the disease process, if its function has similar effects to MYLK.
Increased barrier is not just confined to CD: is there an overlap in disease mechanisms?
An impaired barrier function has been suggested for multiple disorders, e.g. IBD, asthma, type I diabetes and psoriasis Citation70–72. In all these disorders an increased reaction of the immune system is seen in response to known or unknown pathogens. This could be due to an impaired epithelial cell barrier in the intestine or in one of the other organs lined with epithelial cells Citation70, Citation72–74. These disorders can also co‐occur in families, which might be due to overlapping susceptibility genes, such as MYO9B. Although the same gene might be involved in various disorders, the causal variant may not necessarily be the same.
Gluten peptides evoke an immune response
HLA presents gluten peptides to the T cells
The first gene repeatedly shown to contribute to the genetics of CD was the HLA‐DQ gene. The HLA‐DQ2.5 molecule (built up from the DQA1*0501 and DQB1*0201 variants) predisposes to CD, because of its binding properties for gluten peptides. The HLA‐DQ2.2 (built up from the DQA1*0201 and DQB1*0202 variants) predisposes to CD only when it is expressed together with the DQ2.5. This is due to the DQB1*0202 that can in combination with the DQA1*0501 of DQ2.5 form a functional molecule with similar binding properties for gluten peptides. Lastly, the HLA‐DQ8 molecule (built up from the DQA1*0301 and DQB1*0302 variants) also gives some predisposition to CD. It has been found that over 90% of CD patients carry the DQ2.5 molecule (alone or in combination with DQ2.2 or DQ8), and most of the remaining patients carry the DQ2.2 or DQ8 Citation75, Citation76. Fewer than 6% of the CD patients carry none of these molecules, but some of them do carry one‐half of the DQ2.5 heterodimer. In Europe a gradient is seen for the DQ types in CD patients: southern European populations have more CD patients who carry none of the risk DQ molecules by themselves, but in whom the risk molecules can form due to combinations of the HLA‐DQA1 and ‐DQB1 genes on the different chromosomes (trans effect) than the northern European populations, in whom the combination of genes on one chromosome already forms the molecules (cis effect) Citation75, Citation76. Although these molecules play a large role in CD, they cannot be the only contributing factors, since around 25% of the normal population also carry the DQ2 molecules without having CD, for example. The HLA‐DQ2.5 and ‐DQ8 molecules are therefore seen as necessary, but not sufficient, to cause CD.
Dose‐response effect
The highest RR of developing CD is seen for persons homozygous for the DQ2.5 molecule (homozygous for the variants of HLA‐DQA1 and ‐DQB1 genes that form the DQ2.5 molecule), compared to those heterozygous for DQ2.5 or DQ8 (heterozygous for the HLA‐DQA1 and ‐DQB1 genes that form these molecules) Citation76. This dose‐effect has also been observed functionally Citation77. HLA‐DQ dimers can be formed by cis‐ and trans‐combinations. The HLA‐DQA1 and HLA‐DQB1 genes can therefore give rise to 0–4 functional DQ2.5 molecules, and to correspondingly increasing levels of HLA‐DQ2.5 peptide complexes that can lead to immune responses when the reaction threshold of the T cells is crossed Citation77, Citation78. A recent study showed that HLA‐DQ2.5 homozygosity is more than doubled (from 20.7% to 44.1%) in RCD type II patients Citation79, and in patients with an EATL, the amount of HLA‐DQ2.5 homozygosity increases to 53.3%. This suggests that HLA‐DQ2.5 homozygosity increases the risk of becoming an RCD II patient or of developing an EATL.
tTG modifies gluten peptides and makes them more immunogenic
The role of the HLA‐DQ complex was only partly understood, since it is not a highly potent gluten peptide binder. This changed when it was shown that tTG has an effect on the binding compatibility of gluten peptides to HLA‐DQ2.5 and ‐DQ8, by selectively deamidating specific glutamines in gluten epitopes. This introduces negatively charged glutamic acids in the epitopes that fit better into the binding pocket of the HLA‐DQ2.5 or ‐DQ8 molecule, and are therefore more capable of stimulating the T cells Citation80–83. The complex formation of tTG and gliadin in untreated CD patients is increased compared to treated CD patients and controls Citation84. These tTG‐gliadin complexes are seen more in the epithelial and subepithelial levels and less in the lamina propria, when compared to controls. A recent paper from Sakly et al. found more tTG expressing cells in the basement membrane and lamina propria, together with an increased staining Citation85. The amount of tTG expressing enterocytes in cases compared to controls was decreased. It has now been shown that tTG can incorporate gliadin into the interstitial matrix components of the lamina propria, leading to an increased availability of gliadin that might act as an extra trigger for the reaction processes seen in CD Citation86. Although the tTG gene is important in the pathogenesis of CD in several ways, no causative role has been found. Firstly, there were no differences in the coding sequence—sequenced at RNA level—detected between CD patients and controls Citation87, and secondly a genetic study did not show any linkage or association with CD Citation88.
Adaptive and innate immune system in CD
For a long time it was thought that only the adaptive immunity plays a role in CD. It is now clear that both the adaptive and the innate immune responses are important in CD and that some gluten peptides seem to be involved in either one of these responses (reviewed by Jabri et al.) Citation89. This is also reflected by several gluten peptides having distinct pathological mechanisms in CD.
The adaptive immune response involves CD4+ T cells that are activated by cells, like antigen presenting cells, which present the gluten peptides on their HLA‐DQ2.5 or ‐DQ8 molecule. An example of a gluten peptide that stimulates the adaptive immune response via CD4+ T cells of most adult CD patients is the α‐gliadin 56–75 peptide Citation52, Citation89–91. The priming of the CD4+ T cells in CD is somewhat different from a normal CD4+ T cell reaction, since interleukin‐12 (IL‐12) and signal transducer and activator of transcription 4 (STAT‐4), which are normally active in this T cell reaction, do not seem to be involved in the process in CD. The exact role of these CD4+ T cells is still not well defined since it cannot explain all the changes in the intestine since other disorders associated with CD4+ T cells do not show the increase in IELs and the malignant transformation of these IELs into EATL that occurs in some CD patients. Jabri et al. proposed a role for CD4+ T cells in arming IELs Citation89. IFN, which is produced by the activated αβ CD4+ T cells, creates a more immunogenic environment and could make the enterocytes and the IEL more sensitive. This could provide one of the links between the adaptive and the innate immune responses.
Some of the genes involved in the activation of the adaptive immune response have been genetically tested for their role in CD, but no positive results were found except for the HLA‐DQ2.5 and ‐DQ8 variants (reviewed in Diosdado et al.) Citation92. CTLA‐4, which is a co‐stimulatory molecule of the T cells, seems to have a minor involvement in CD although its role is still under debate Citation21–27. Generally, genes involved in the regulation of T cell activation may have a minor effect on disease susceptibility. CTLA‐4 and PTPN22 are two genes involved in the risk to several autoimmune disorders, although especially the role of the latter in CD is not yet clear Citation21–27, Citation93, Citation94.
The role of the innate immune system in CD is becoming more and more widely recognized. The peptide that has been studied most in relation to stimulation of the innate response is the α‐gliadin p31–49, which induces an immune response in the antigen‐presenting cells (APCs) and epithelial cells but not in the CD4+ T cells Citation89. This was shown by the increased production of IL‐15 due to gluten‐induced epithelial stress, leading to enterocytes with an upregulated MHC class I polypeptide‐related chain A (MICA), a change of the cytotoxic CD8+ T cells into lymphokine‐activated cells and the expression of the NKG2D receptor on the IELs Citation95. This reduces the threshold for T cell receptor activation and mediates the direct killing of epithelial cells.
Although it has long been thought that MICA might play a causal role in CD, there is no genetic evidence yet. It is, however, extremely difficult to study the possible genetic effect of MICA in CD. MICA is located in the HLA region and in high linkage disequilibrium with the HLA‐DQ, meaning that certain MICA variants and certain HLA‐DQ molecules segregate together more often than would be expected by chance Citation96. To study the effect of MICA independently of HLA‐DQ, we would need a large HLA‐DQ2.5‐positive case group, as well as a large HLA‐DQ2.5‐positive control group to see if a certain MICA variant is more often present in the CD patients' group compared to the control group, independently of the shared HLA‐DQ2.5 background. A sufficiently large control group is more difficult to obtain since only 25% of controls carry the HLA‐DQ2.5 molecule.
The IL‐15 produced by APCs and enterocytes is also capable of stimulating IELs Citation97, Citation98 and the T cells of the adaptive immune system, and is therefore one of the links between adaptive and innate immune system.
Discussion
To date, CD is the best understood HLA‐related disorder. The HLA‐DQ2.5 and ‐DQ8 molecules are able to present the environmental factor gluten to the T cells, especially when tTG has modified these gluten proteins in order to make them fit better into the binding pocket of the HLA‐DQ molecules. The immune response that is then elicited is involved in the changes observed in the small intestine of CD patients. tTG plays several roles in CD, like modifying the gluten peptides, cross‐linking gliadin with interstitial matrix proteins, and regulating RhoA activation. This last role is probably also performed by MYO9B, with as tTG has an influence on the intestinal barrier formed by the enterocytes in the intestine. This intestinal barrier is impaired in CD patients and loses some of its ability to regulate both the passage of gluten peptides and other molecules, and the protection against pathogens. Recent observations as reviewed in Jabri et al. have shown that not only the adaptive immune response is involved in CD, but also the innate immune response, and that the different gluten peptides can have their own effect on either of these immune responses Citation89.
Observations from functional studies have shown that the changes in the intestines of CD patients are not just due to damage but also to a deregulation of the proliferation/differentiation ratio of the enterocytes. We have learnt much about the pathological mechanisms in CD over the last 30 years, but still cannot answer all the questions. There are still several black boxes to be discovered, while the players known to be involved in CD might still be hiding some functions that influence the pathology. A combination of studies is needed in order to define how these players act together leading to CD. Genetic studies will reveal many of the small genetic players in CD in the coming years, given the increasing availability of high‐throughput methods. A greater knowledge of pathway analyses and the function of variants in the DNA will also play an important role. Besides this, functional studies will be needed to reveal the role of these genetic variants in CD and to show which genes are only involved in the pathology but not in the genetic susceptibility to CD. These genes may be a consequence, or an enhancer, of the disease process rather than a cause; for example, the expression studies have revealed there are many genes involved in the changes in structure of the intestine, but most of these will not be genetically associated to CD Citation60, Citation99. We should not forget the overlap seen between several autoimmune disorders, both clinically and overlapping linkage regions. The genes found in autoimmune disorders may play a role in multiple disorders (for example, the CTLA‐4 and PTPN22 genes, although they do not play as clear a role in CD as in other disorders) Citation21–27, Citation93, Citation94. The linkage region found in chromosome 6q21 (opposite arm of the HLA region) might also harbor an immune‐related gene since this region has been identified in multiple immune disorders including CD. There are also several disorders where the permeability of the epithelial barrier is impaired and an overlap in disease genes is seen.
The major environmental factor provoking CD is gluten, but other, less important, environmental factors must also play a role, and the search for them has hardly begun. New ideas about pathology will also come from a clinical point of view; for example, clinicians showed years ago that there was an increased intestinal permeability. This is only now being linked to genes involved in CD. Clinicians should also help in defining the criteria for diagnosing a CD patient. For a long time, the diagnosis was made by a biopsy showing Marsh III pathology, but the question as to whether individuals with a Marsh I or Marsh II biopsy are, in fact, also CD patients is arising more and more. Perhaps genetic factors will be used for diagnoses, but so far the only genes playing a large role is are the HLA‐DQA1 and ‐DQB1 and they are used to exclude the possibility of CD. Our main conclusion from the current genetic and functional studies is that we should look for causal genes in the barrier function as well as in the immune system.
Box 1: Challenges in finding genes in complex genetic disorders, with MYO9B as an example
Several factors influence our ability to find the genes involved in CD and to replicate them in multiple populations of CD patients. Since CD is not a monogenic disorder but a complex genetic disorder, multiple genes as well as environmental factors must play a role. The disease‐causing variants will most often be common ones, and each on its own will not be sufficient to cause disease Citation100–103 because they will have a low RR. These variants have to occur frequently enough in the general population for them to co‐occur in the individuals affected by these variants. Not every patient needs to have exactly the same combination of disease‐predisposing variants, so this genetic heterogeneity adds to the difficulty of finding causative genes. In addition, healthy individuals can also harbor several disease‐predisposing gene variants, but not enough or not in the right combination, to cause the disorder. The fact that genetic heterogeneity occurs, that the RR of the variants is low, and that the control population will also harbor these genetic variants, although in a smaller number, means it is necessary to have a large, well defined patient group, as well as a large control group to perform genetic studies.
There are several strategies for searching for disease susceptibility genes, like candidate gene/pathway studies and genome‐wide studies. Both strategies can be studied in families using linkage‐based designs, and in populations using case‐control designs. Candidate gene/pathway studies require biological knowledge about the genes involved, but if they result in an association they often lead to a susceptibility gene being identified. Genome‐wide studies, on the other hand, are hypothesis‐free but when applied to a family‐based linkage design the linkage regions mostly contain ∼50–200 genes. Both strategies have their strengths and weaknesses, and the new possibility of whole‐genome association studies will combine some of the strengths of both studies, since no biological knowledge is required and a positive result will lead to a single, associated gene. These studies will, of course, not avoid all the problems like population stratification, power issues and multiple testing, and a positive replication study will always be needed.
MYO9B as an example
The most recent success in CD research was the finding of MYO9B as a gene causing susceptibility to CD. A family study in the Dutch population found linkage to chromosome 19p13 (MYO9B, previously called the CELIAC4 locus) with an estimated λs of 2.6 Citation104. Subsequently, an association study was performed using microsatellite markers, which resulted in association to a microsatellite marker in intron 1 of MYO9B with CD. Further single nucleotide polymorphism (SNP) typing in and around MYO9B showed association to the linkage disequilibrium (LD) block that covered the 3' site of the gene Citation40. A comprehensive tagging screen using SNPs in the whole MYO9B locus showed no association to other parts of this locus. MYO9B was found in the Dutch population with an estimated RR of 1.7 for heterozygous carriers of the associated variant and an RR of 2.3 for homozygous carriers. The expected population attributable risk of this variant is 23%–25%, meaning that removing this risk factor from the population reduces the risk to CD with 23%–25%. Several questions remain even though the gene responsible for the MYO9B locus has now been identified. First of all, what is the causative variant? The variants found associated to CD are located in a block with high LD and might just be tagging the real causative variant. Perhaps multiple rare variants together cause the association, as seen for the CARD15 gene in IBD Citation105. Secondly, what is the exact RR and does this explain the observed linkage? Most genetic studies suffer from the ‘winner's curse’ and they tend to overestimate the RR Citation43. This could mean that MYO9B does not explain all the linkage and may imply that a second gene should be found even though the comprehensive study in the linkage region provided no evidence for this. If we take the lower boundary of the 95% confidence interval for the odds ratio, the RR would be 1.23, or perhaps even lower in reality. The chance that a replication study will have sufficient power to observe association to CD in a different population is very low. This could explain the negative results from the replication studies of Hunt et al. and Admundsen et al. Citation41, Citation42. The first study had a cohort size that came close to the original Dutch cohort, but the second study was performed in a family setting, which is less prone to population stratification but also less powerful, and a case‐control setting using smaller cohorts than the original study. Both were underpowered if we assume a low RR. The recent observation that MYO9B is also involved in IBD (especially in ulcerative colitis), with an OR of 1.2, could indeed indicate that the studies' power was too low Citation63. Another explanation could be that the original Dutch findings were false‐positive, but we consider the chance of that low given that our result was found in two separate case‐control groups. Since the causative variant has not been uncovered, it is possible that the tag SNPs used were not in complete LD with the causative variant, and there might have been slightly different LD patterns between these three studies. If this proves to be true, then testing the causative variant (once it has been found), in the other two populations might show that MYO9B is associated in multiple CD populations after all.
Acknowledgements
We thank Erica van Oort, Jackie Senior and Alexandra Zhernakova for critically reading the manuscript. The authors were supported by grants from the Netherlands Organization for Scientific Research (grant 912‐02‐028), and the Celiac Disease Consortium, an Innovative Cluster approved by the Netherlands Genomics Initiative and partially funded by the Dutch Government (grant BSIK03009).
Conflict of interest
The authors declare no conflict of interest.
References
- Rewers M. Epidemiology of celiac disease: what are the prevalence, incidence, and progression of celiac disease?. Gastroenterology 2005; 128((Suppl 1))S47–51
- Dewar D. H., Ciclitira P. J. Clinical features and diagnosis of celiac disease. Gastroenterology 2005; 128: S19–S24
- Rampertab S. D., Pooran N., Brar P., Singh P., Green P. H. Trends in the presentation of celiac disease. Am J Med 2006; 119: 355.e9–14
- Wahab P. J., Meijer J. W., Mulder C. J. Histologic follow‐up of people with celiac disease on a gluten‐free diet: slow and incomplete recovery. Am J Clin Pathol 2002; 118: 459–63
- Schulzke J. D., Bentzel C. J., Schulzke I., Riecken E. O., Fromm M. Epithelial tight junction structure in the jejunum of children with acute and treated celiac sprue. Pediatr Res 1998; 43((Pt 1))435–41
- Schulzke J. D., Schulzke I., Fromm M., Riecken E. O. Epithelial barrier and ion transport in coeliac sprue: electrical measurements on intestinal aspiration biopsy specimens. Gut 1995; 37: 777–82
- Uil J. J., van Elburg R. M., van Overbeek F. M., Meyer J. W., Mulder C. J., Heymans H. S. Follow‐up of treated coeliac patients: sugar absorption test and intestinal biopsies compared. Eur J Gastroenterol Hepatol 1996; 8: 219–23
- Smecuol E., Sugai E., Niveloni S., Vazquez H., Pedreira S., Mazure R., et al. Permeability, zonulin production, and enteropathy in dermatitis herpetiformis. Clin Gastroenterol Hepatol 2005; 3: 335–41
- Zone J. J. Skin manifestations of celiac disease. Gastroenterology 2005; 128((Suppl 1))S87–91
- Daum S., Cellier C., Mulder C. J. J. Refractory coeliac disease. Best Pract Res Clin Gastroenterol 2005; 19: 413–24
- Green P. H., Jabri B. Celiac disease. Annu Rev Med 2006; 57: 207–21
- Schuppan D., Dieterich W., Ehnis T., Bauer M., Donner P., Volta U., et al. Identification of the Autoantigen of Celiac Disease. Ann NY Acad Sci 1998; 859: 121–6
- Revised criteria for diagnosis of coeliac disease. Report of Working Group of European Society of Paediatric Gastroenterology and Nutrition. Arch Dis Child 1990; 65: 909–11
- Marsh M. Gluten, major histocompatibility complex, and the small intestine. A molecular and immunobiologic approach to the spectrum of gluten sensitivity (‘celiac sprue’). Gastroenterology 1992; 102: 330–54
- Rostami K., Kerckhaert J., Tiemessen R., von Blomberg B. M., Meijer J. W., Mulder C. J. Sensitivity of antiendomysium and antigliadin antibodies in untreated celiac disease: disappointing in clinical practice. Am J Gastroenterol 1999; 94: 888–94
- Dube C., Rostom A., Sy R., Cranney A., Saloojee N., Garritty C., et al. The prevalence of celiac disease in average‐risk and at‐risk Western European populations: a systematic review. Gastroenterology 2005; 128((Suppl 1))S57–67
- Fasano A., Berti I., Gerarduzzi T., Not T., Colletti R. B., Drago S., et al. Prevalence of Celiac Disease in At‐Risk and Not‐At‐Risk Groups in the United States: A Large Multicenter Study. Arch Intern Med 2003; 163: 286–92
- Talley N. J., Valdovinos M., Petterson T. M., Carpenter H. A., Melton L. J 3rd. Epidemiology of celiac sprue: a community‐based study. Am J Gastroenterol 1994; 89: 843–6
- Greco L., Romino R., Coto I., Di Cosmo N., Percopo S., Maglio M., et al. The first large population based twin study of coeliac disease. Gut 2002; 50: 624–8
- Nistico L., Fagnani C., Coto I., Percopo S., Cotichini R., Limongelli M. G., et al. Concordance, disease progression, and heritability of coeliac disease in Italian twins. Gut 2006; 55: 803–8
- King A. L., Moodie S. J., Fraser J. S., Curtis D., Reid E., Dearlove A. M., et al. CTLA‐4/CD28 gene region is associated with genetic susceptibility to coeliac disease in UK families. J Med Genet 2002; 39: 51–4
- Mora B., Bonamico M., Indovina P., Megiorni F., Ferri M., Carbone M. C., et al. CTLA‐4 +49 A/G dimorphism in Italian patients with celiac disease. Hum Immunol 2003; 64: 297–301
- van Belzen M. J., Mulder C. J., Zhernakova A., Pearson P. L., Houwen R. H., Wijmenga C. CTLA4 +49 A/G and CT60 polymorphisms in Dutch coeliac disease patients. Eur J Hum Genet 2004; 12: 782–5
- Clot F., Fulchignoni‐Lataud M. C., Renoux C., Percopo S., Bouguerra F., Babron M. C., et al. Linkage and association study of the CTLA‐4 region in coeliac disease for Italian and Tunisian populations. Tissue Antigens 1999; 54: 527–30
- Djilali‐Saiah I., Schmitz J., Harfouch‐Hammoud E., Mougenot J. F., Bach J. F., Caillat‐Zucman S. CTLA‐4 gene polymorphism is associated with predisposition to coeliac disease. Gut 1998; 43: 187–9
- Holopainen P., Naluai A. T., Moodie S., Percopo S., Coto I., Clot F., et al. Candidate gene region 2q33 in European families with coeliac disease. Tissue Antigens 2004; 63: 212–22
- van Heel D. A., Hunt K., Greco L., Wijmenga C. Genetics in coeliac disease. Best Pract Res Clin Gastroenterol 2005; 19: 323–39
- Falchuk Z. M., Rogentine G. N., Strober W. Predominance of histocompatibility antigen HL‐A8 in patients with gluten‐sensitive enteropathy. J Clin Invest 1972; 51: 1602–5
- Falchuk Z. M., Strober W. HL‐A antigens and adult coeliac disease. Lancet 1972; 2: 1310
- Stokes P. L., Asquith P., Holmes G. K., Mackintosh P., Cooke W. T. Histocompatibility antigens associated with adult coeliac disease. Lancet 1972; 2: 162–4
- Corazza G. R., Tabacchi P., Frisoni M., Prati C., Gasbarrini G. DR and non‐DR Ia allotypes are associated with susceptibility to coeliac disease. Gut 1985; 26: 1210–3
- Keuning J. J., Pena A. S., van Leeuwen A., van Hooff J. P., va Rood J. J. HLA‐DW3 associated with coeliac disease. Lancet 1976; 1: 506–8
- Tosi R., Vismara D., Tanigaki N., Ferrara G. B., Cicimarra F., Buffolano W., et al. Evidence that celiac disease is primarily associated with a DC locus allelic specificity. Clin Immunol Immunopathol 1983; 28: 395–404
- Kagnoff M. F., Harwood J. I., Bugawan T. L., Erlich H. A. Structural analysis of the HLA‐DR, ‐DQ, and ‐DP alleles on the celiac disease‐associated HLA‐DR3 (DRw17) haplotype. Proc Natl Acad Sci U S A 1989; 86: 6274–8
- Rittner C., DeMarchi M., Mollenhauer E., Carbonara A. Coeliac disease and C4A*QO: an association secondary to HLA‐DR3. Tissue Antigens 1984; 23: 130–4
- Sollid L. M., Markussen G., Ek J., Gjerde H., Vartdal F., Thorsby E. Evidence for a primary association of celiac disease to a particular HLA‐DQ alpha/beta heterodimer. J Exp Med 1989; 169: 345–50
- Tosi R., Tanigaki N., Polanco I., De Marchi M., Woodrow J. C., Hetzel P. A. A radioimmunoassay typing study of non‐DQw2‐associated celiac disease. Clin Immunol Immunopathol 1986; 39: 168–72
- Bevan S., Popat S., Braegger C. P., Busch A., O'Donoghue D., Falth‐Magnusson K., et al. Contribution of the MHC region to the familial risk of coeliac disease. J Med Genet 1999; 36: 687–90
- Petronzelli F., Bonamico M., Ferrante P., Grillo R., Mora B., Mariani P., et al. Genetic contribution of the HLA region to the familial clustering of coeliac disease. Ann Hum Genet 1997; 61((Pt 4))307–17
- Monsuur A. J., de Bakker P. I., Alizadeh B. Z., Zhernakova A., Bevova M. R., Strengman E., et al. Myosin IXB variant increases the risk of celiac disease and points toward a primary intestinal barrier defect. Nat Genet 2005; 37: 1341–4
- Amundsen S. S., Monsuur A. J., Wapenaar M. C., Lie B. A., Ek J., Gudjonsdottir A. H., et al. Association Analysis of MYO9B Gene Polymorphisms with Celiac Disease in a Swedish/Norwegian Cohort. Hum Immunol 2006; 67: 341–5
- Hunt K. A., Monsuur A. J., McArdle W., Kumar P. J., Travis S. P., Walters J. R., et al. Lack of association of MYO9B genetic variants with coeliac disease in a British cohort. Gut 2006; 55: 969–72
- Ioannidis J. P., Ntzani E. E., Trikalinos T. A., Contopoulos‐Ioannidis D. G. Replication validity of genetic association studies. Nat Genet 2001; 29: 306–9
- van Herpen T. W., Goryunova S. V., van der Schoot J., Mitreva M., Salentijn E., Vorst O., et al. Alpha‐gliadin genes from the A, B, and D genomes of wheat contain different sets of celiac disease epitopes. BMC Genomics 2006; 7: 1
- Koning F., Gilissen L., Wijmenga C. Gluten: a two‐edged sword. Immunopathogenesis of celiac disease. Springer Semin Immunopathol 2005; 27: 217–32
- Molberg O., Uhlen A. K., Jensen T., Flaete N. S., Fleckenstein B., Arentz‐Hansen H., et al. Mapping of gluten T‐cell epitopes in the bread wheat ancestors: implications for celiac disease. Gastroenterology 2005; 128: 393–401
- Spaenij‐Dekking L., Kooy‐Winkelaar Y., van Veelen P., Drijfhout J. W., Jonker H., van Soest L., et al. Natural variation in toxicity of wheat: potential for selection of nontoxic varieties for celiac disease patients. Gastroenterology 2005; 129: 797–806
- Spaenij‐Dekking L., Kooy‐Winkelaar Y., Koning F. The Ethiopian cereal tef in celiac disease. N Engl J Med 2005; 353: 1748–9
- Hausch F., Shan L., Santiago N. A., Gray G. M., Khosla C. Intestinal digestive resistance of immunodominant gliadin peptides. Am J Physiol Gastrointest Liver Physiol 2002; 283: G996–G1003
- Vanhoof G., Goossens F., Hendriks L., De Meester I., Hendriks D., Vriend G., et al. Cloning and sequence analysis of the gene encoding human lymphocyte prolyl endopeptidase. Gene 1994; 149: 363–6
- Higaki‐Sato N., Sato K., Esumi Y., Okumura T., Yoshikawa H., Tanaka‐Kuwajima C., et al. Isolation and identification of indigestible pyroglutamyl peptides in an enzymatic hydrolysate of wheat gluten prepared on an industrial scale. J Agric Food Chem 2003; 51: 8–13
- Shan L., Molberg O., Parrot I., Hausch F., Filiz F., Gray G. M., et al. Structural basis for gluten intolerance in celiac sprue. Science 2002; 297: 2275–9
- Matysiak‐Budnik T., Candalh C., Dugave C., Namane A., Cellier C., Cerf‐Bensussan N., et al. Alterations of the intestinal transport and processing of gliadin peptides in celiac disease. Gastroenterology 2003; 125: 696–707
- Diosdado B., Stepniak D. T., Monsuur A. J., Franke L., Wapenaar M. C., Mearin M. L., et al. No genetic association of the human prolyl endopeptidase gene in the Dutch celiac disease population. Am J Physiol Gastrointest Liver Physiol 2005; 289: G495–500
- Monsuur A. J., Stepniak D., Diosdado B., Wapenaar M. C., Mearin M. L., Koning F., et al. Genetic and functional analysis of pyroglutamyl‐peptidase I in coeliac disease. Eur J Gastroenterol Hepatol 2006; 18: 637–44
- Stepniak D., Spaenij‐Dekking L., Mitea C., Moester M., de Ru A., Baak‐Pablo R., et al. Highly efficient gluten degradation with a newly identified prolyl endoprotease: implications for celiac disease. Am J Physiol Gastrointest Liver Physiol 2006; 291: G621–9
- Watson A. J., Chu S., Sieck L., Gerasimenko O., Bullen T., Campbell F., et al. Epithelial barrier function in vivo is sustained despite gaps in epithelial layers. Gastroenterology 2005; 129: 902–12
- Ismail A. S., Hooper L. V. Epithelial cells and their neighbors. IV. Bacterial contributions to intestinal epithelial barrier integrity. Am J Physiol Gastrointest Liver Physiol 2005; 289: G779–84
- Forsberg G., Fahlgren A., Horstedt P., Hammarstrom S., Hernell O., Hammarstrom M. L. Presence of bacteria and innate immunity of intestinal epithelium in childhood celiac disease. Am J Gastroenterol 2004; 99: 894–904
- Diosdado B., Wapenaar M. C., Franke L., Duran K. J., Goerres M. J., Hadithi M., et al. A microarray screen for novel candidate genes in coeliac disease pathogenesis. Gut 2004; 53: 944–51
- Mallant‐Hent R., Mary B., von Blomberg E., Yuksel Z., Wahab P. J., Gundy C., et al. Disappearance of anti‐Saccharomyces cerevisiae antibodies in coeliac disease during a gluten‐free diet. Eur J Gastroenterol Hepatol 2006; 18: 75–8
- Zolotarevsky Y., Hecht G., Koutsouris A., Gonzalez D. E., Quan C., Tom J., et al. A membrane‐permeant peptide that inhibits MLC kinase restores barrier function in in vitro models of intestinal disease. Gastroenterology 2002; 123: 163–72
- van Bodegraven A. A., Curley C. R., Hunt K. A., Monsuur A. J., Linskens R. K., Onnie C. M., et al. Genetic variation in myosin IXB is associated with ulcerative colitis. Gastroenterology, Published online, Sept 1, 2006
- Post P. L., Bokoch G. M., Mooseker M. S. Human myosin‐IXb is a mechanochemically active motor and a GAP for rho. J Cell Sci 1998; 111((Pt 7))941–50
- Post P. L., Tyska M. J., O'Connell C. B., Johung K., Hayward A., Mooseker M. S. Myosin‐IXb is a single‐headed and processive motor. J Biol Chem 2002; 277: 11679–83
- Matter K., Balda M. S. Signalling to and from tight junctions. Nat Rev Mol Cell Biol 2003; 4: 225–36
- Janiak A., Zemskov E. A., Belkin A. M. Cell surface transglutaminase promotes RhoA activation via integrin clustering and suppression of the Src‐p190RhoGAP signaling pathway. Mol Biol Cell 2006; 17: 1606–19
- Blair S. A., Kane S. V., Clayburgh D. R., Turner J. R. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab Invest 2006; 86: 191–201
- Russo J. M., Florian P., Shen L., Graham W. V., Tretiakova M. S., Gitter A. H., et al. Distinct temporal‐spatial roles for rho kinase and myosin light chain kinase in epithelial purse‐string wound closure. Gastroenterology 2005; 128: 987–1001
- Liu Z., Li N., Neu J. Tight junctions, leaky intestines, and pediatric diseases. Acta Paediatr 2005; 94: 386–93
- Schreiber S. Slipping the barrier: how variants in CARD15 could alter permeability of the intestinal wall and population health. Gut 2006; 55: 308–9
- Schreiber S., Rosenstiel P., Albrecht M., Hampe J., Krawczak M. Genetics of Crohn disease, an archetypal inflammatory barrier disease. Nat Rev Genet 2005; 6: 376–88
- Cookson W. The immunogenetics of asthma and eczema: a new focus on the epithelium. Nat Rev Immunol 2004; 4: 978–88
- Secondulfo M., Iafusco D., Carratu R., deMagistris L., Sapone A., Generoso M., et al. Ultrastructural mucosal alterations and increased intestinal permeability in non‐celiac, type I diabetic patients. Dig Liver Dis 2004; 36: 35–45
- Karell K., Louka A. S., Moodie S. J., Ascher H., Clot F., Greco L., et al. HLA types in celiac disease patients not carrying the DQA1*05‐DQB1*02 (DQ2) heterodimer: results from the European Genetics Cluster on Celiac Disease. Hum Immunol 2003; 64: 469–77
- Margaritte‐Jeannin P., Babron M. C., Bourgey M., Louka A. S., Clot F., Percopo S., et al. HLA‐DQ relative risks for coeliac disease in European populations: a study of the European Genetics Cluster on Coeliac Disease. Tissue Antigens 2004; 63: 562–7
- Vader W., Stepniak D., Kooy Y., Mearin L., Thompson A., van Rood J. J., et al. The HLA‐DQ2 gene dose effect in celiac disease is directly related to the magnitude and breadth of gluten‐specific T cell responses. Proc Natl Acad Sci U S A 2003; 100: 12390–5
- Koning F. Celiac disease: caught between a rock and a hard place. Gastroenterology 2005; 129: 1294–301
- Al‐Toma A., Goerres M. S., Meijer J. W., Pena A. S., Crusius J. B., Mulder C. J. Human leukocyte antigen‐DQ2 homozygosity and the development of refractory celiac disease and enteropathy‐associated T‐cell lymphoma. Clin Gastroenterol Hepatol 2006; 4: 315–9
- Kim C. Y., Quarsten H., Bergseng E., Khosla C., Sollid L. M. Structural basis for HLA‐DQ2‐mediated presentation of gluten epitopes in celiac disease. Proc Natl Acad Sci U S A 2004; 101: 4175–9
- Molberg O., McAdam S. N., Korner R., Quarsten H., Kristiansen C., Madsen L., et al. Tissue transglutaminase selectively modifies gliadin peptides that are recognized by gut‐derived T cells in celiac disease. Nat Med 1998; 4: 713–7
- Qiao S. W., Bergseng E., Molberg O., Jung G., Fleckenstein B., Sollid L. M. Refining the rules of gliadin T cell epitope binding to the disease‐associated DQ2 molecule in celiac disease: importance of proline spacing and glutamine deamidation. J Immunol 2005; 175: 254–61
- Vader L. W., de Ru A., van der Wal Y., Kooy Y. M. C., Benckhuijsen W., Mearin M. L., et al. Specificity of Tissue Transglutaminase Explains Cereal Toxicity in Celiac Disease. J Exp Med 2002; 195: 643–9
- Ciccocioppo R., Di Sabatino A., Ara C., Biagi F., Perilli M., Amicosante G., et al. Gliadin and tissue transglutaminase complexes in normal and coeliac duodenal mucosa. Clin Exp Immunol 2003; 134: 516–24
- Sakly W., Sriha B., Ghedira I., Bienvenu F., Ayadi A., Sfar M. T., et al. Localization of tissue transglutaminase and N (epsilon)‐(gamma) ‐glutamyl lysine in duodenal cucosa during the development of mucosal atrophy in coeliac disease. Virchows Arch 2005; 446: 613–8
- Dieterich W., Esslinger B., Trapp D., Hahn E., Huff T., Seilmeier W., et al. Cross linking to tissue transglutaminase and collagen favours gliadin toxicity in coeliac disease. Gut 2006; 55: 478–84
- Aldersley M. A., Hamlin P. J., Jones P. F., Markham A. F., Robinson P. A., Howdle P. D. No polymorphism in the tissue transglutaminase gene detected in coeliac disease patients. Scand J Gastroenterol 2000; 35: 61–3
- van Belzen M. J., Mulder C. J., Pearson P. L., Houwen R. H., Wijmenga C. The tissue transglutaminase gene is not a primary factor predisposing to celiac disease. Am J Gastroenterol 2001; 96: 3337–40
- Jabri B., Kasarda D. D., Green P. H. Innate and adaptive immunity: the yin and yang of celiac disease. Immunol Rev 2005; 206: 219–31
- Anderson R. P., Degano P., Godkin A. J., Jewell D. P., Hill A. V. In vivo antigen challenge in celiac disease identifies a single transglutaminase‐modified peptide as the dominant A‐gliadin T‐cell epitope. Nat Med 2000; 6: 337–42
- Arentz‐Hansen H., Korner R., Molberg O., Quarsten H., Vader W., Kooy Y. M., et al. The intestinal T cell response to alpha‐gliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J Exp Med 2000; 191: 603–12
- Diosdado B., Wijmenga C. Molecular mechanisms of the adaptive, innate and regulatory immune responses in the intestinal mucosa of celiac disease patients. Expert Rev Mol Diagn 2005; 5: 681–700
- Bottini N., Musumeci L., Alonso A., Rahmouni S., Nika K., Rostamkhani M., et al. A functional variant of lymphoid tyrosine phosphatase is associated with type I diabetes. Nat Genet 2004; 36: 337–8
- Zhernakova A., Eerligh P., Wijmenga C., Barrera P., Roep B. O., Koeleman B. P. Differential association of the PTPN22 coding variant with autoimmune diseases in a Dutch population. Genes Immun 2005; 6: 459–61
- Hue S., Mention J. J., Monteiro R. C., Zhang S., Cellier C., Schmitz J., et al. A direct role for NKG2D/MICA interaction in villous atrophy during celiac disease. Immunity 2004; 21: 367–77
- van Belzen M. J., Koeleman B. P., Crusius J. B., Meijer J. W., Bardoel A. F., Pearson P. L., et al. Defining the contribution of the HLA region to cis DQ2‐positive coeliac disease patients. Genes Immun 2004; 5: 215–20
- Di Sabatino A., Ciccocioppo R., Cupelli F., Cinque B., Millimaggi D., Clarkson M. M., et al. Epithelium derived interleukin 15 regulates intraepithelial lymphocyte Th1 cytokine production, cytotoxicity, and survival in coeliac disease. Gut 2006; 55: 469–77
- Meresse B., Chen Z., Ciszewski C., Tretiakova M., Bhagat G., Krausz T. N., et al. Coordinated induction by IL15 of a TCR‐independent NKG2D signaling pathway converts CTL into lymphokine‐activated killer cells in celiac disease. Immunity 2004; 21: 357–66
- Juuti‐Uusitalo K., Maki M., Kaukinen K., Collin P., Visakorpi T., Vihinen M., et al. cDNA microarray analysis of gene expression in coeliac disease jejunal biopsy samples. J Autoimmun 2004; 22: 249–65
- Chakravarti A. Population genetics—making sense out of sequence. Nat Genet 1999; 21((Suppl))56–60
- Glazier A. M., Nadeau J. H., Aitman T. J. Finding genes that underlie complex traits. Science 2002; 298: 2345–9
- Risch N. J. Searching for genetic determinants in the new millennium. Nature 2000; 405: 847–56
- Zondervan K. T., Cardon L. R. The complex interplay among factors that influence allelic association. Nat Rev Genet 2004; 5: 89–100
- Van Belzen M. J., Meijer J. W., Sandkuijl L. A., Bardoel A. F., Mulder C. J., Pearson P. L., et al. A major non‐HLA locus in celiac disease maps to chromosome 19. Gastroenterology 2003; 125: 1032–41
- Rioux J. D., Daly M. J., Silverberg M. S., Lindblad K., Steinhart H., Cohen Z., et al. Genetic variation in the 5q31 cytokine gene cluster confers susceptibility to Crohn disease. Nat Genet 2001; 29: 223–8