Abstract
Background. Published data support genetic variants, as well as certain infectious agents, as potential risk factors for schizophrenia. Less is known about interactions between the risk factors.
Aim. To evaluate exposure to infectious agents and host genetic variation as joint risk factors.
Methods. We investigated four infectious agents: cytomegalovirus (CMV), herpes simplex viruses 1 and 2 (HSV1, HSV2), and Toxoplasma gondii (TOX). We initially compared exposure using specific serum antibodies, among simplex and multiplex nuclear families (one or more than one affected offspring, respectively). If interactions between infectious agents and host genetic variation are important risk factors for schizophrenia, we reasoned that they would be more prominent among multiplex versus simplex families. We also evaluated the role of variation at chromosome 6p21‐p23 in conjunction with exposure. We used 22 short tandem repeat polymorphisms (STRPs) dispersed across this region.
Results. Though exposure to all four agents was increased among multiplex families versus simplex families, the difference was consistently significant only for CMV (odds of exposure to CMV in multiplex families: 2.47, 95% CI: 1.48–5.33). Transmission disequilibrium tests and case‐control comparisons using STRPs revealed significant linkage/association with D6S2672 among CMV+ schizophrenia patients.
Conclusions. Polymorphisms near D6S2672 could confer risk for schizophrenia in conjunction with CMV exposure.
Introduction
Schizophrenia is a common, severe disorder with a lifetime morbid risk of about 1% Citation1. Inheritance patterns are consistent with a polygenic multifactorial etiology Citation2. Epidemiological studies have indirectly supported an infectious etiology, but the nature and the timing of the infection are uncertain Citation1,Citation3,4. It has also been difficult to obtain consistent evidence for individual infectious agents or for genetic variants Citation5–7. Part of the inconsistency in the genetic and epidemiologic studies of schizophrenia could be explained if interactions between infectious agents and host genetic variation were to play a role in susceptibility, such as the interactions demonstrated for HIV and malaria Citation8,9. To evaluate this hypothesis, we have conducted two lines of research. First, we evaluated exposure to selected infectious agents among patients with schizophrenia and their relatives. Next, we investigated associations with specific genetic variants in conjunction with the exposure data.
The known sharing of environment and genetic variants among members of families provides an opportunity for indirect tests of gene‐environment interaction hypotheses. Such interactions can be complex and are difficult to investigate. We reasoned that if interactions between infectious agents and host genetic variation are important risk factors for schizophrenia, they would be more prominent among families with multiple affected members when compared with families having single affected individuals. We also tested for the interaction between prior exposure to infectious agents and genotypes in the major histocompatibility complex (MHC) region on chromosome 6p21 among schizophrenia patients. Our decision to study the MHC region is logical because it harbors immune‐related genes, including human leukocyte antigen (HLA) genes and has been implicated in schizophrenia susceptibility by linkage and association studies Citation10,11. We targeted short tandem repeat polymorphisms (STRPs) across the entire MHC region as a preliminary step to localize specific regions that may be involved in schizophrenia pathogenesis, infectious disease pathogenesis, or both. The relatively high informativeness of STRPs enhances their utility for family‐based association analyses, though in some cases their relatively high mutation rate could obscure linkage disequilibrium with a disease gene allele Citation12. Nevertheless, similar estimates for linkage disequilibrium (LD) were obtained over a 140‐kb region spanning the von Willebrand factor gene, using bi‐ or multiallelic markers Citation13. The power of STRPs is also comparable to single nucleotide polymorphisms for family‐based association/linkage analyses Citation14.
A panoply of infectious agents has been targeted in schizophrenia research, including those capable of infecting the central nervous system. Of particular interest in this regard are neurotropic members of the human herpesviruses because these agents are capable of establishing a latent infection within the central nervous system. Another agent of interest is the apicomplexa parasite Toxoplasma gondii, because the cyst forms of this organism can also establish a prolonged infection within the central nervous system Citation3,Citation15. We investigated four organisms that have been implicated in schizophrenia genesis: cytomegalovirus (CMV), herpes simplex virus 1 (HSV1), herpes simplex virus 2 (HSV2) and Toxoplasma gondii (TOX) Citation16–18. These agents have very different modes of infection and have divergent population prevalence rates Citation19–21. CMV infection typically occurs in childhood, and the virus is passed by saliva. Cross‐generation transfer from children to adults also occurs. HSV1 infection is airborne, and again transmitted via saliva, while HSV2 is typically a sexually transmitted disease. TOX is a zoonotic infection, and the carriers are typically domestic cats.
Each of these infectious agents establishes latency following primary infection, and this latency persists for many years with cycles of reactivation, resulting in persistently elevated immunoglobulin G (IgG) antibody levels. Either the initial infection or subsequent cycles of reactivation could contribute to schizophrenia risk. Retrospective studies of such infectious agents suffer from being unable to identify the timing of the infectious insult, but benefit from the ability to investigate exposure in the relatively distant past Citation22.
Key messages
Interaction between infectious exposure and genetic variation may contribute to schizophrenia risk.
Polymorphisms near D6S2672 in the human leukocyte antigen (HLA) region on chromosome 6p could confer risk for schizophrenia in conjunction with cytomegalovirus (CMV) exposure.
Methods and materials
Clinical sample
Cases and family members
The cases comprised Caucasian patients who were recruited at Western Psychiatric Institute and Clinic, University of Pittsburgh, and other treatment facilities within a 500‐mile radius of Pittsburgh, located in northeastern USA. Both DNA and serum were collected from these individuals. Information about the patients was gathered from the Diagnostic Interview for Genetic Studies (DIGS) Citation23, their medical records, and relevant informants. Board‐certified psychiatrists established consensus diagnoses using DSM‐IV criteria Citation24. They were blind to the genotyping results. When available, affected siblings of the patients were also interviewed and diagnosed in the same way. Blood samples were obtained from these individuals. Available, consenting parents provided blood samples, but formal structured diagnostic evaluations were not conducted. In our sample, one father received a clinical diagnosis of schizoaffective disorder, bipolar type. Samples were not collected from additional unaffected siblings of the probands.
Community controls
Unscreened, Caucasian population‐based controls were obtained from neonatal cord bloods of live births at Magee‐Women's Hospital, Pittsburgh. No details apart from ethnicity and gender were available. Ethnicity was based on maternal report. Genomic DNA was available from the control samples, but serological analyses could not be conducted.
The study was approved by the University of Pittsburgh Institutional Review Board (IRB). Written informed consent was obtained from all participants, except the neonatal controls, in accordance with IRB guidelines.
Immunoassays for antibody titers of sera of subjects
Serum samples were obtained from the peripheral venous blood of the subjects, and stored at −80°C until use. The levels of IgG antibodies to HSV1, HSV2, TOX, and CMV were tested with solid‐phase enzyme immunoassay Citation25,26. Briefly, wells of microtiter plates coated with target antigens (obtained from KMI Diagnostics Inc., Minneapolis, USA) were reacted with test serum diluted 1:100 in phosphate‐buffered saline solution at pH 7.4 containing 0.1% polysorbate (Tween) 20. Following incubation for 2 hours at 37°C, the plates were washed five times with phosphate‐buffered saline solution containing 0.1% polysorbate 20, and incubated with peroxidase‐labeled anti‐human IgG. Following incubation for 1 hour at 37°C, the plates were washed five times and incubated with 2‐2′‐azino‐bis [3‐ethylbenzthiazoline‐6‐sulfonic acid]‐hydrogen peroxide peroxidase substrate. Following reaction for 30 minutes, the amount of color generated by reaction between the antigen‐bound enzyme and the soluble substrate was quantitated by means of a microplate colorimeter at a wavelength of 450 nm. Assays using different microtiter plates were standardized by the use of curves generated from reference samples run on each assay plate. Antibody titers were quantified as signal/cutoff (S/C) ratios, which were calculated for each sample by dividing the optical density measurements generated in the assay by the optical density of cutoff sera that were provided by the manufacturer of the assays. S/C values of 2.0 or more were taken as abnormal, based on the frequency distribution for S/C values. Immunological analyses were performed only for patients and their parents, as sera were unavailable from the cord blood samples.
Familiality and infectious exposure
To assess the distribution of infectious exposure within and among families, we performed three analyses involving generalized linear models. All of these analyses were conducted using the logic of generalized estimating equations, as implemented in SAS Proc GENMOD, to correct for within‐family correlations. Within‐family correlations were estimated using the exchangeable or fixed‐correlation options, whichever was appropriate. Fit of the models was adequate.
We first assessed whether infectious exposure (+/−) was a function of the age of the individuals in the study. Exposure was assumed to be binomially distributed and the link function was a logit. We were then interested in the relationship of exposure status among family members. We computed these dependencies in various ways. One ad hoc method was used to regress out the effect of age using the logit model described above, and then to evaluate the Pearson correlation of the standardized residuals. This approach accounts for the dependence on age, when present. Finally, we contrasted the distribution of exposure between simplex and multiplex families by fitting a model in which exposure (+/−) was the outcome, age was a predictor and family (simplex/multiplex) was another predictor. This last analysis was performed on entire families and also by using only children (the generation in which schizophrenia is manifest). Of interest in this analysis is whether the simplex/multiplex levels account for a significant portion of the variability in exposure, after taking the effect of age into account. (It should be noted that generation and age are confounded, so that including generation in the models would not, and did not, account for a substantive amount of the variation.)
DNA markers and genotyping
Selection of markers
Twenty‐two STRP were selected from the Genome database (http://www.gdb.org) and the Psoriasis Genetics Laboratory (PGL) Department of Dermatology, University of Michigan (http://www.psoriasis.umich.edu/hla2000/index.html), mindful of local linkage disequilibrium patterns Citation27. They spanned the HLA region and flanking sequences extending across 21.5 Mb. According to the consensus map at Sanger Center (http://www.sanger.ac.uk/HGP/Chr6/MHC.shtml), the average distance between 22 markers was 1022.19 kb (range: 19 kb to 7865 kb). It was 293.3 kb (106 kb to 673 kb) between markers in the HLA region (see for list of markers).
Table I. Family‐based association analyses of subgroups of probands defined by seropositivity to four infectious agents.
Genomic DNA samples were amplified using a standard polymerase chain reaction (PCR) protocol (primer sequences listed in Supplementary Table 2 online). The amplified products were electrophoresed using an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems, New Jersey, USA) and the identified products were analyzed using GeneScan software. Data were imported into the Genotyper 3.7 program for allele calls. One CEPH individual (#1347‐02) was used as a reference for all assays (PE Applied Biosystems, Foster City, CA). The genotypes for this person were obtained from the websites of the Center for Medical Genetics, Marshfield Medical Research Foundation (http://www.marshfieldclinic.org/research/genetics/) or the PGL. Two investigators blind to the clinical status of samples read all genotypes independently. Markers with disagreement rates exceeding 1.0% were discarded and replaced by adjacent markers, and the final error rate was less than 0.5%.
Statistical analysis
Some probands were not used for transmission disequilibrium test (TDT) analyses, as both parents were unavailable, and only one sibling from multiplex families was used for TDT and case‐control analysis. We conducted TDT analysis using the Monte Carlo Extended TDT (MCETDT) to test for allele‐wise transmission distortion Citation28. To integrate case‐control differences in STRP distribution across all markers and thereby avoiding a multitude of hypothesis tests, we used the Bayesian Adaptive Regression Splines (BARS) Citation29. The BARS test fitted a spline to a set of summary statistics, one for each STRP, by using a free‐knot basis Citation29; the summary statistic, Nei's Gst Citation30 scaled by its variance, measures the differentiation between cases and controls at each of the STRP. (Nei's Gst is a common measure of population differentiation, in this situation cases and controls, and it naturally accommodates multiallelic loci.) BARS determined if the spline had a significant mode by computing the 95% confidence interval (CI) for the mode; if the CI did not span the whole region, then the null hypothesis was rejected.
Results
Our samples consisted of Caucasian patients with schizophrenia or schizoaffective disorder, their parents and affected siblings, where available. Some of our analyses separated families by the number of affected offspring, specifically into simplex (one affected child) and multiplex (more than one affected child) (see Supplementary Table 1 online).
Analysis of exposure to infectious agents among simplex and multiplex families
We used a sample of 350 individuals from simplex families (123 affected children and 227 parents) and 97 individuals from multiplex families (80 affected children and 17 parents). Results from regression analyses revealed expected patterns of mother‐father, mother‐child, father‐child and child‐child correlation for CMV, HSV1, HSV2, and TOX antibodies (data not shown). Notably, other measures of concordance/agreement yielded the same conclusions, even though they did not account for the effects of age. As expected, families with more than one affected individual were larger on average than families with only one affected individual; however, rates of infectious exposure were not significantly associated with family size or socioeconomic status. To assess the distribution of viral exposure among the simplex versus the multiplex families, we first determined whether viral exposure was a function of age of the individuals. Consistent with published data Citation19, viral exposure increased with age for two of the agents: CMV (slope = 0.0374, SE 0.0078, z = 4.81, P = 1.5×10−6) and HSV1 (slope = 0.0439, SE 0.007, z = 6.24, P = 4.4×10−10), while it was unrelated to age for TOX (slope = 0.0145, SE 0.0103, z = 1.41, P = 0.16) and HSV2 (slope = 0.0074, SE 0.01, z = 0.74, P = 0.46). We then tested whether exposure differs between simplex and multiplex families, while accounting for any (potential) impact of age (). Exposure to three viruses, namely CMV (z = 2.16, P = 0.031), HSV1 (z = 2.19, P = 0.028) and HSV2 (z = 2.81, P = 0.005), showed higher rates in multiplex than in simplex families after accounting for effects of age (TOX, z = 1.21, P = 0.226). Odds of exposure for multiplex families versus those for simplex families were as follows: CMV, 1.96 (CI = 1.15–3.33); HSV1, 1.86 (CI = 1.12–3.10); HSV2, 2.84 (CI = 1.43–5.66); TOX, 1.63 (CI = 0.76–3.50). Correction for multiple testing at this level for four tests would reduce the number of significant differences from three to one (HSV2).
Table II. Antibody status among members of simplex and multiplex families.
Results of the analyses of exposure rates in simplex versus multiplex families are not always simple to interpret; witness the contrast for HSV1 despite the quite similar overall rates (, upper panel). In fact, both parents and their children show elevated rates of HSV1 exposure in the multiplex families; however, due to quite different marginal frequencies the combination of the two subgroups produces the similar exposure rates in the overall table (an example of Simpson's paradox). To explore these results more fully, we also contrasted the rates of exposure for simplex versus multiplex families for affected offspring only (, lower panel). These analyses show that while the rates of viral exposure are always larger in the multiplex families, these differences are most pronounced for CMV exposure (CMV: z = 2.20, P = 0.028; HSV2: z = 1.16, P = 0.246; HSV1: z = 1.06, P = 0.289; TOX: z = 0.02, P = 0.987). The estimated odds of exposure for CMV in multiplex families are 2.47 times higher (CI = 1.48–5.33) than in simplex families. For the other contrasts, the odds ratios and confidence intervals were as follows: HSV1, 1.52 (CI = 0.78–2.98); HSV2, 2.11 (CI = 0.73–6.06); and TOX 1.02 (CI = 0.34–3.08).
Overall, the pattern of CMV exposure is consistently elevated for parents and their affected children from multiplex families relative to simplex families. The same cannot be said for the other infectious agents, but there is evidence that multiplex families tend to have a greater viral burden than simplex families for HSV1 and possibly the other agents as well.
Immunogenetic analyses
The results of the immunological analyses demonstrate an association between CMV exposure and greater familiality of schizophrenia. To investigate whether this familiality could be attributed to genetic variation, we investigated selected genetic markers in the HLA region. We selected genes in this region because the HLA region is often critically involved in immune response. Case trios consisted of cases and both parents, or one case from affected sib pairs with DNA from at least one parent. DNA samples from 184 cases, 292 parents, and 181 controls were used (Tables and ).
Table III. Case‐control association analyses of subgroups of probands defined by seropositivity to cytomegalovirus (CMV).
Transmission disequilibrium test (TDT) using STRPs
Twenty‐two STRPs spanning the HLA region and flanking sequences extending across 21.5 Mb were selected. There was no significant transmission distortion to affected offspring for the entire sample (). However, on the basis of seropositivity, there was significant transmission distortion at D6S2672, a marker in the HLA Class I region, for probands with elevated CMV antibody levels (P = 0.0085). No significant TDT associations were detected in relation to seropositivity for HSV1, HSV2, or TOX (). After correction for multiple testing, of course, even the transmission distortion for D6S2672 would not be significant. Still, these results are congruent with the immunological analyses, which showed that CMV seropositivity correlated significantly with greater familiality of schizophrenia, and could be interpreted as specific gene‐environment interaction for CMV exposure (CMV+) versus no exposure (CMV−). However, they cannot be considered conclusive due to the multiple comparisons. Therefore, we evaluated the data further to see if convergent lines of evidence support this interaction.
Case‐control analyses of CMV+ patients using STRPs
We next compared the patients with unrelated community controls. Because we genotyped 22 STRPs across the region, we sought a test of regional differentiation of the allele distributions for the different samples. Therefore, we applied a test called Bayesian Adaptive Regression Splines (BARS) Citation29. The BARS test fits a spline to the set of summary statistics, one for each STRP, and then tests whether the spline has a significant mode (i.e. populations are differentiated at the tested STRPs). BARS uses a free‐knot basis and estimates the best locations for placement of a minimum number of knots. BARS determines if the spline has a significant mode by computing the confidence interval (CI) for the mode; if the CI spans the whole region, then the null hypothesis of no mode is not rejected. The model is fitted using a reversible‐jump Markov chain Monte Carlo (MCMC) algorithm. For our analyses, we used Nei's Gst, scaled by its variance, as the summary measure. A 95% CI was used, resulting in a 0.05 size of the test. BARS can only be applied to sets of markers without substantial gaps amongst them Citation29, hence we tested two sets of markers: Set 1, D6S1663‐D6S1621; and Set 2, D6S258‐D6S2657. Combining both sets of STRPs produced implausible locations for the modes. Only for Set 2, which included D6S2672 (the STRP demonstrating significant transmission distortion), were the marker distributions significantly differentiated (P<0.05) (). Contrasts of allele distributions for CMV+ patients versus controls () and CMV+ versus CMV− patients were both significant ().
Figure 1. Plot of BARS spline fit of summary statistics for STRP D6S258 to D6S2657. CMV+/CMV−: subgroup of probands with/without elevated CMV antibodies. The Bayesian Adaptive Regression Splines (BARS) test fits a spline to the set of summary statistics, one for each STRP, and then tests whether the spline has a significant mode (i.e. populations are differentiated at the tested STRPs). Contrasts of allele distributions for CMV+ patients versus controls () and CMV+ versus CMV− patients () were both significant for markers D6S258 to D6S2657.
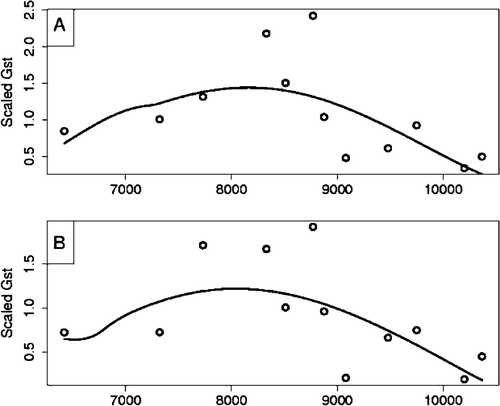
To garner greater insight into which STRPs were substantially different between groups, we tested individual markers comparing all cases with controls, CMV+ cases with controls, and CMV+ cases with CMV− cases using the program CLUMP Citation31. Multiple significant associations were detected when all cases were contrasted with controls, including adjacent markers D6S2672, D6S2676, D6S2694. For CMV+ cases versus controls, significant associations were again seen at D6S2672 and also D6S2694, which is in LD with D6S2672 (D' = 0.577, P = 0.00001). Comparison of CMV+ and CMV− cases yielded a trend for association at D6S2672 (P = 0.0598) and significant association with D6S2676, an adjacent marker (). Therefore, not only are the CMV+ and CMV− samples different, but they are dissimilar to the control population when combined. CMV+/ CMV− analyses also revealed associations at D6S1663 and D6S2657. These associations were not evident using the TDT or the BARS test, indicating that they may be due to population substructure or associations with CMV titer status (see additional details in Supplementary Table 3 online).
Discussion
After accounting for confounding effects of age and expected correlation based on relatedness, multiplex families showed increased exposure rate for three of four infectious agents among multiplex families compared with simplex families. The difference was most convincing for CMV exposure, because it was detectable either by using all members of such families or by restricting the comparisons to affected children. Still, the results could be generated by multiple testing. Other possible explanations for these results include greater crowding in multiplex families and medication differences in patients, which could alter serum antibody levels to infectious agents as reported in Leweke et al. Citation16. Our results are consistent with the hypothesis that CMV exposure increases the risk for schizophrenia. However, the lack of exposure data from conventional control samples makes it difficult to evaluate our results in relation to published studies.
We also tested for genetic associations with schizophrenia and viral exposure in family‐based samples. To do so, we selected and genotyped STRPs spanning the HLA region. STRP analyses revealed an association among CMV+ patients at D6S2672, a marker near the boundary between the HLA Class I and Class III regions. This association was specific to CMV+ individuals, as it was not detected among subgroups of patients defined by elevated antibody levels for HSV1, HSV2 and TOX, or among CMV negative patients.
Case‐control comparisons also supported the association between the variation in the HLA region (), especially near D6S2672. In particular, in the contrast of CMV+ patients versus controls, D6S2672 showed sample differentiation as does a nearby STRP; likewise, in the contrast between CMV+ and CMV− patients, the adjacent STRP showed significant association. The results of the case‐control tests for individual STRPs () are not as clear‐cut as the family‐based results (). Significant contrasts were obtained elsewhere in the HLA region for CMV+ patients versus controls or CMV− patients, and, contrary to expectations, some of the overall case‐control contrasts also were significant. In addition, suggestive associations were noted for certain STRP markers among the TOX seropositive groups. These patterns were dissimilar to the CMV‐related associations and were not supported by family‐based analyses (, Supplementary Table 3, online).
The genetic associations detected using the family‐based tests are notable as they are unlikely to be due to factors such as population substructure secondary to ethnic admixture Citation32. Further replicate studies are in progress, including investigations using single nucleotide polymorphisms. The latter will be useful for identifying genes harboring the primary associations. While extensive LD has previously been reported in the HLA region Citation33, recent comprehensive analyses suggest that the levels of recombination in the vicinity of D6S2672 are similar to genome‐wide levels Citation34.
CMV causes lifelong infection, but the majority of infected persons are asymptomatic Citation20. CMV can cause severe neurological conditions such as retinitis and acute inflammatory demyelinating neuropathy among immunosuppressed persons Citation20,Citation35. Since most patients with schizophrenia are not immunosuppressed, other mechanisms need to be invoked. CMV has been implicated in autoimmune diseases such as type 1 diabetes and ‘stiff person syndrome’, both of which are associated with raised antibodies to glutamic acid decarboxylase (GAD). It has been proposed that pathogenesis for these conditions stems from molecular mimicry between CMV peptides and GAD Citation36. A similar mechanism could be at play in schizophrenia, because GAD levels are reduced in postmortem brain samples from patients Citation37. Alternatively, CMV may induce latent infection and apoptosis following perinatal infection Citation38. Subsequent recrudescence in adolescence may precipitate psychopathology and would account for a lag between perinatal infection and the onset of schizophrenia, which typically occurs in adolescence.
A very high prevalence of raised CMV antibody levels (over 80%) have been reported among adults in countries such as India and Korea Citation39,40, yet these countries apparently do not have a proportionately elevated prevalence of schizophrenia compared to the US. It is conceivable that the raised prevalence of CMV is offset by differentiated genetic variation in the HLA or other regions of the genome in these populations. Alternatively, a larger proportion of the etiology of schizophrenia may be attributable to CMV in these locales. These issues are presently being investigated.
In conclusion, our analyses suggest an intriguing interaction between exposure to CMV and a locus in the HLA region in the etiology of schizophrenia. Replicate studies are warranted. The nature of the interaction is uncertain. It could involve genetic variants increasing susceptibility to viral infection, or it could involve more complex mechanisms evoked by genetic variation after infection. Our findings could yet be due to chance or may reflect interactions related to CMV infectivity rather than schizophrenia genesis. If replicated, they would open new vistas of research into gene‐environment interactions and schizophrenia pathogenesis.
Acknowledgements
Funded in part by grants from the Stanley Medical Research Institute (RHY) and NIH (#MH01489, MH56242 and MH53459 to VLN; MH57881 to BD). We thank Ms Bogdana Krivogorsky for technical assistance and Ms Ann Cusic for data‐entry assistance.
Notes
Supplementary data are available on our website (http://www.pitt.edu/˜nimga/research/mhc/data/).
References
- Jablensky A. The 100‐year epidemiology of schizophrenia. Schizophr Res 1997; 28: 111–25
- Gottesman I. Schizophrenia Genesis: The Origins of Madness. WH Freeman, New York 1991
- Torrey E. F., Yolken R. H. Could schizophrenia be a viral zoonosis transmitted from house cats?. Schizophr Bull 1995; 21: 167–71
- Pearce B. D. Schizophrenia and viral infection during neurodevelopment: a focus on mechanisms. Mol Psychiatry 2001; 6: 634–46
- Jablensky A. Schizophrenia: recent epidemiologic issues. Epidemiol Rev 1995; 17((1))10–20
- Moldin S. O. The maddening hunt for madness genes. Nat Genet 1997; 17: 127–9
- Shirts B. H., Nimgaonkar V. The genes for schizophrenia: finally a breakthrough?. Curr Psychiatry Rep 2004; 6: 303–12
- Hill A. V. The genomics and genetics of human infectious disease susceptibility. Annu Rev Genomics Hum Genet 2001; 2: 373–400
- Joseph J., Behar T. Viral and host genetic factors regulating HIV/CNS disease. J Neurovirol 2004; 10(Suppl)1–6
- Schwab S. G., Knapp M., Mondabon S., Hallmayer J., Borrmann‐Hassenbach M., Albus M., et al. Support for association of schizophrenia with genetic variation in the 6p22.3 gene, dysbindin, in sib‐pair families with linkage and in an additional sample of triad families. Am J Hum Genet 2003; 72: 185–90
- Wright P., Nimgaonkar V. L., Donaldson P. T., Murray R. M. Schizophrenia and HLA: a review. Schizophr Res 2001; 47: 1–12
- Weber J. L., Wong C. Mutation of human short tandem repeats. Hum Mol Genet 1993; 2: 1123–8
- Watkins W. S., Zenger R., O'Brien E., Nyman D., Eriksson A. W., Renlund M., et al. Linkage Disequilibrium Patterns Vary with Chromosomal Location: A Case Study from the von Willebrand Factor Region. Am J Hum Genet 1994; 55: 348–55
- Xiong M., Jin L. Comparison of the power and accuracy of biallelic and microsatellite markers in population‐based gene‐mapping methods. Am J Hum Genet 1999; 64: 629–40
- Yolken R. H., Torrey E. F. Viruses, schizophrenia and bipolar disorder. Clin Microbiol Rev 1995; 8: 131–45
- Leweke F. M., Gerth C. W., Koethe D., Klosterkotter J., Ruslanova I., Krivogorsky B., et al. Antibodies to infectious agents in individuals with recent onset schizophrenia. Eur Arch Psychiatry Clin Neurosci 2004; 254: 4–8
- Yolken R. Viruses and schizophrenia: a focus on herpes simplex virus. Herpes 2004; 11: 83A–88A
- Torrey E. F., Yolken R. H. Toxoplasma gondii and schizophrenia. Emerg Infect Dis 2003; 9: 1375–80
- Smith J. S., Robinson N. J. Age‐specific prevalence of infection with herpes simplex virus types 2 and 1: a global review. J Infect Dis 2002; 186(Suppl 1)S3–28
- Sissons J. G., Carmichael A. J., McKinney N., Sinclair J. H., Wills M. R. Human cytomegalovirus and immunopathology. Springer Semin Immunopathol 2002; 24: 169–85
- Bhopale G. M. Pathogenesis of toxoplasmosis. Comp Immunol Microbiol Infect Dis 2003; 26: 213–22
- Sinclair J., Sissons P. Latency and reactivation of human cytomegalovirus. J Gen Virol 2006; 87(Pt 7)1763–79
- Nurnberger J. I., Jr., Berrettini W., Simmons‐Alling S., Lawrence D., Brittain H. Blunted ACTH and cortisol response to afternoon tryptophan infusion in euthymic bipolar patients. Psychiatry Res 1990; 31: 57–67
- A.P.A., American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders—DSM‐IV. American Psychiatric Pub, Inc, Arlington, VA 1994
- Ashley R. L., Wu L., Pickering J. W., Tu M. C., Schnorenberg L. Premarket evaluation of a commercial glycoprotein G‐based enzyme immunoassay for herpes simplex virus type‐specific antibodies. J Clin Microbiol 1998; 36: 294–5
- Buka S. L., Tsuang M. T., Torrey E. F., Klebanoff M. A., Bernstein D., Yolken R. H. Maternal infections and subsequent psychosis among offspring. Arch Gen Psychiatry 2001; 58: 1032–7
- Cullen M., Malasky M., Harding A., Carrington M. High‐density map of short tandem repeats across the human major histocompatibility complex. Immunogenetics 2003; 54: 900–10
- Zhao J. H., Curtis D., Sham P. C. Model‐free analysis and permutation tests for allelic associations. Hum Hered 2000; 50: 133–9
- Zhang X., Roeder K., Wallstrom G., Devlin B. Integration of association statistics over genomic regions using Bayesian Adaptive Regression Splines. Hum Genomics 2003; 1: 20–9
- Nei M. Analysis of gene diversity in subdivided populations. Proc Natl Acad Sci U S A 1973; 70: 3321–3
- Sham P. C., Curtis D. Monte Carlo tests for associations between disease and alleles at highly polymorphic loci. Ann Hum Genet 1995; 59(Pt 1)97–105
- Ewens W. J., Spielman R. S. The transmission/disequilibrium test: history, subdivision, and admixture. Am J Hum Genet 1995; 57: 455–64
- Carrington M., Nelson G. W., Martin M. P., Kissner T., Vlahov D., Goedert J. J., et al. HLA and HIV‐1: heterozygote advantage and B*35‐Cw*04 disadvantage [see comments]. Science 1999; 283: 1748–52
- Walsh E. C., Mather K. A., Schaffner S. F., Farwell L., Daly M. J., Patterson N., et al. An integrated haplotype map of the human major histocompatibility complex. Am J Hum Genet 2003; 73: 580–90, Epub 2003 Aug 14
- Khalili‐Shirazi A., Gregson N., Gray I., Rees J., Winer J., Hughes R. Antiganglioside antibodies in Guillain‐Barre syndrome after a recent cytomegalovirus infection. J Neurol Neurosurg Psychiatry 1999; 66: 376–9
- Hiemstra H. S., Schloot N. C., van Veelen P. A., Willemen S. J., Franken K. L., van Rood J. J., et al. Cytomegalovirus in autoimmunity: T cell crossreactivity to viral antigen and autoantigen glutamic acid decarboxylase. Proc Natl Acad Sci U S A 2001; 98: 3988–91
- Akbarian S., Huntsman M. M., Kim J. J., Tafazzoli A., Potkin S. G., Bunney W. E., Jr., et al. GABAA receptor subunit gene expression in human prefrontal cortex: comparison of schizophrenics and controls. Cereb Cortex 1995; 5: 550–60
- DeBiasi R. L., Kleinschmidt‐DeMasters B. K., Richardson‐Burns S., Tyler K. L. Central nervous system apoptosis in human herpes simplex virus and cytomegalovirus encephalitis. J Infect Dis 2002; 186((11))1547–57
- Kothari A., Ramachandran V. G., Gupta P., Singh B., Talwar V. Seroprevalence of cytomegalovirus among voluntary blood donors in Delhi, India. J Health Popul Nutr 2002; 20: 348–51
- Sohn Y. M., Park K. I., Lee C., Han D. G., Lee W. Y. Congenital cytomegalovirus infection in Korean population with very high prevalence of maternal immunity. J Korean Med Sci 1992; 7: 47–51