Abstract
Scleroderma or systemic sclerosis (SSc) is a complex disease in which the vasculopathy and the activation of the immune system with production of inflammatory mediators lead to dysregulated fibroblast activation. The resulting excessive deposition of collagens and other extracellular matrix proteins ends in fibrosis and organ dysfunction. The cause is unknown, but environmental factors are thought to play a role by triggering abnormal responses in genetically susceptible hosts. The recent past has witnessed important advances in the definition of mechanisms that underlie the persistent activation in fibroblasts of genes involved in uncontrolled fibrosis, a hallmark of SSc. These include the preferential production of type 2 T cell cytokines in target organs, the presence of autoantibodies with fibroblast‐activating capacities, the production of vasoconstrictive mediators that impact on fibroblast biosynthetic properties, the transforming growth factor‐β‐related metabolic signature, and the presence of altered signaling pathways in fibroblasts. Furthermore, while no animal models recapitulate all the features of SSc, they have been instrumental for assessing the relevance of specific processes to the development of fibrosis. More importantly, some of the research findings are leading to therapies that target altered processes with the potential of changing the prognosis of some dismal aspects of the disease.
| Abbreviations | ||
| AFA | = | antifibroblast antibody |
| AP‐1 | = | activating protein‐1 |
| autoAb | = | autoantibody |
| BAFF | = | B cell‐activating factor of the TNF family |
| BAL | = | bronchoalveolar lavage |
| CBP | = | CREB‐binding protein |
| CC chemokine | = | chemokine with two adjacent cysteins |
| CCN | = | connective tissue growth factor/cystein‐rich 61/nephroblastoma‐overexpressed family members |
| CD | = | cluster of differentiation |
| CMV | = | cytomegalovirus |
| CTGF | = | connective tissue growth factor |
| EC | = | endothelial cell |
| ECM | = | extracellular matrix |
| FAK | = | focal adhesion kinase |
| GVHD | = | graft versus host disease |
| HLA | = | human leukocyte antigens |
| ICAM | = | intercellular adhesion molecule |
| IFN−γ | = | interferon–γ |
| IL | = | interleukin |
| IP‐10 | = | interferon‐induced protein of 10 kDa |
| ITAC | = | interferon gamma‐inducible T cell alpha chemoattractant |
| MCP‐1 | = | monocyte chemotactic protein‐1 |
| MHC | = | major histocompatibility complex |
| MMP | = | matrix metalloproteinase |
| NK | = | natural killer cells |
| PAH | = | pulmonary arterial hypertension |
| PCR | = | polymerase chain reaction |
| PDGF | = | platelet derived growth factor |
| R | = | receptor |
| ROS | = | reactive oxygen species |
| SNP | = | single nucleotide polymorphism |
| SSc | = | systemic sclerosis |
| STAT | = | signal transducer and activator of transcription |
| TcR | = | T cell receptor |
| TGF‐β | = | transforming growth factor‐β |
| TGF‐βIIR | = | transforming growth factor‐β receptor II |
| TNF‐α | = | tumor necrosis factor‐α |
| Tsk‐1 | = | tight skin mouse‐1 |
| VEGF | = | vascular endothelial growth factor |
Introduction
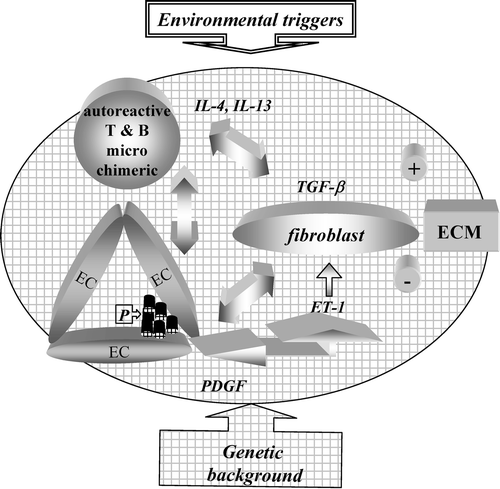
Scleroderma refers to a condition characterized by three distinct aspects that coexist and very likely influence each other during its development: 1) the vasculopathy primarily due to endothelial cell (EC) dysfunction and damage which expresses not only as Raynaud's phenomenon but also as pulmonary arterial hypertension (PAH) and hypertensive renal crisis; 2) the activation of the immune system whose hallmark is the presence of autoantibodies (autoAb) directed against ubiquitous nuclear components. Further, cell‐specific autoAb and activated T cells infiltrating target organs are distinctly present; and 3) fibrosis resulting from fibroblast activation and myofibroblast transdifferentiation resulting in excessive deposition of extracellular matrix (ECM) (). On appropriate but still incompletely defined genetic background, it is thought that environmental events of various nature trigger modifications in the microvasculature and the immune system that then drive phenotypic changes in fibroblasts, which result in an exuberant deposition of ECM exceeding its degradation. Since not only the skin but all organs and tissues may be affected, systemic sclerosis (SSc) is the preferred term for identifying the spectrum of clinical presentations that are encountered in clinical practice. The clinical course of the disease varies, and different subsets have been identified according to localization of skin fibrosis, preferences in internal organ involved, and patterns of autoantibodies (autoAb) present in the serum Citation1–3. Prognosis also varies in different subsets, being relatively benign in SSc with limited cutaneous involvement and much worse in individuals with diffuse cutaneous, renal and/or lung involvement. In these patients quality of life is reduced, and life expectancy is shortened to a few years after diagnosis Citation4. Although recent progress has been made in the management of hypertensive renal crisis and PAH thus improving the prognosis of some patients, no cure is available. Several other clinical conditions are characterized by skin fibrosis usually not accompanied by vasculopathy and the presence of autoAb Citation5. While they should be differentiated from SSc when explained to the suffering patient and a therapeutic approach is sought, they are of great interest since they offer different paradigms for understanding fibrosis development. A novel clinical entity initially characterized by skin thickening in dialyzed individuals and named nephrogenic fibrosing dermopathy has recently been identified Citation6. While the chosen nomenclature stresses the predominant dermal involvement, careful evaluation of affected individuals has permitted identification also of lung, heart, and muscular involvement Citation7, and the term of nephrogenic systemic fibrosis has been proposed for its identification. In this disorder at sites undergoing fibrosis there is an accumulation of CD34+ CD68+ dendritic cells which are distinctly absent in SSc Citation7,8. Additional important information on the mechanisms likely to be at play during the development of SSc is derived from animal models and in vitro studies. None of the existing animal experimental models encompasses the complex pathogenic features of human SSc. Still, in vivo models are instrumental to delineate the key features of the pathogenic processes that drive fibrosis. Furthermore, the in vivo disruption or manipulation of pivotal signaling pathways previously identified with in vitro investigation has been key in exploring how genetic manipulations predispose to fibrosis development Citation9.
An abundant literature has been published in the recent past and has been discussed in a number of excellent papers covering various aspects of SSc and fibrosis in general Citation9–14. I will particularly focus the present review on evidence implicating the activation of the innate and adaptative arms of the immune system resulting in inflammation, fibroblast activation, myofibroblast transdifferentiation, and development of fibrosis within SSc.
Figure 1. Pathogenesis of systemic sclerosis: schematic representation. The arrows depict presumed interactions between cells of the immune system and endothelial cells (EC) and their influence on the capacity of fibroblasts to enhance extracellular matrix (ECM) deposition. The scheme highlights some of the mediators thought to favor a profibrotic fibroblast phenotype. P = platelets.
Key messages
The pathogenesis of systemic sclerosis is extremely complex and related to concomitant abnormalities of endothelial cells, cells of the immune system with dysregulated production of autoantibodies and soluble inflammatory mediators, and fibroblasts.
The hallmark of systemic sclerosis is the persistent and dysregulated activation of genes encoding for collagens and other components of the extracellular matrix. Genome‐wide analysis of affected tissues points to a dominant alteration of the transforming growth factor‐β pathway.
While the cause of systemic sclerosis remains unknown, very likely different environmental agents of chemical or infectious origin act as triggers of the disease on complex genetic predisposing background. Heterogeneous disease presentations may indeed reflect different interactions between triggers and gene patterns.
Possible causative agents
While no definitive proof exists for causative agents, epidemiological evidence coupled to experimental work points to the involvement of viral infections and environmental agents as possible triggers of SSc. Sequence homologies between retroviral proteins and the topoisomerase‐1 antigen target of the anti‐Scl‐70 autoAb have been identified, and expression of retroviral proteins in fibroblasts results in the acquisition of an SSc‐like phenotype Citation15. A stronger case has been put forward linking human cytomegalovirus (CMV) infection and SSc development Citation16. First, the high prevalence of anti‐CMV IgA antibodies capable of inducing endothelial cell apoptosis has been associated with the presence of anti‐Scl‐70 autoAb. Second, by screening a random peptide library, Italian colleagues have found in the sera of SSc individuals Ab recognizing simultaneously the CMV UL‐94 protein and NAG‐2, a protein expressed on the cell surface of human EC and fibroblasts. This is an interesting example of molecular mimicry which may cause loss of tolerance resulting in cross‐reactive antibodies (as elaborated later on) that have the potential for inducing EC apoptosis and activating fibroblasts Citation17,18. Silica, metal dust, organic solvents, pesticides, hair dyes, and industrial fumes have been identified among environmental agents implicated in the development of SSc Citation19,20. In this respect the observation is of interest that in multigravidae, but not in nulligravidae, female mice the exposure to vinylchloride initiates a process resulting in fibrosis. This finding has the merit of putting together microchimerism, thought to be initiated or amplified during pregnancy (see later on), and exposure to an environmental agent (vinylchloride) within a particular genetic background (the mouse strain used) as determinants involved in fibrosis development, none of which separately is sufficient to initiate the disease Citation21.
Genetic background
SSc is not inherited as a simple Mendelian trait, and concordance between monozygotic twins is low. However, clusters of familial cases are well recognized as well as the presence of autoAb in the relatives of SSc patients. These observations have set the stage for linking genetic background to SSc development. A cogent case has been provided when, in the native American Choctaw tribe with high SSc prevalence, a single nucleotide polymorphism (SNP) of fibrillin‐1, a large protein with a role in the structure of ECM and in the availability of bioactive transforming growth factor‐β (TGF‐β), has been strongly associated with the occurrence of SSc Citation22. Further, fibrillin‐1 appears to be the target of autoAb Citation23. Several SNPs with potential to influence gene expression levels have been identified in genes coding for a vast number of cytokines or their receptors, considered of major importance for fibrosis development including, but not only, TGF‐β, connective tissue growth factor (CTGF), monocyte chemotactic protein‐1 (MCP‐1) Citation24. Further, dinucleotide repeats that may increase COL1A2 gene transcription have been identified within regulatory regions of collagen genes Citation25. Finally, significant gene associations have been found between some human leukocyte antigen (HLA) alleles and the presence of anticentromere or anti‐Scl‐70 autoAb, which is associated to distinct clinical subsets. Since HLA genes are key players in controlling the immune response, these findings provide a link between discrete clinical subsets and autoimmunity Citation26.
Vascular disease and endothelial cells
Vascular dysfunction, a very early event in SSc development, is initially due to the unbalance between vasodilation and vasoconstriction controlled by mediators of neural and endothelial origin. Both excessive production of vasoconstrictor endothelins and decreased production of vasodilator nitric oxide (NO) have been documented in SSc. This unbalance leads to tissue hypoxia resulting in ischemia‐reperfusion events that submit the tissues to oxidative stress Citation27,28. This alteration may precede vascular injury or be the consequence of precedent pathological events that are then amplified with increased microvascular permeability and enhanced leukocyte recruitment leading to endothelial cell injury and thrombosis, exposure of platelets to subendothelial structures with consequent activation and release of potent mediators such as platelet‐derived growth factor (PDGF) Citation29 (). PDGF participates in the recruitment of fibroblasts and pericytes and their transdifferentiation into myofibroblasts thus contributing to fibroproliferative events that ultimately result in and amplify tissue damage. Additionally, angiogenesis appears to be impaired in SSc, an event possibly linked with altered production or response to vascular endothelial growth factor (VEGF), decreased differentiation from mesenchymal progenitor cells into endothelial cell precursors, and altered endothelial cell precursor function Citation30,31. Of interest, the USD‐200 chicken animal model of systemic fibrosis shows prominent early microvascular alterations that accompany fibrosis development. AutoAb directed against endothelial cells (EC) capable of inducing EC apoptosis have been described in USD‐200 chickens, further tightening the links between autoimmunity and vasculopathy leading to fibrosis development Citation32.
Fibroblasts and fibrosis
ECM is continuously renewed through balanced synthesis and degradation. Fibroblasts are capable of both synthesis and degradation, mostly achieved via specific enzymes of which matrix metalloproteinases (MMP) are of major importance. In SSc and other fibrotic conditions excess of synthesis over degradation is thought to take place Citation33. It is not yet firmly established whether SSc fibroblasts are set to higher deposition of ECM because of intrinsic changes and/or under the influence of inflammatory mediators or other stimuli. In many respects, fibroblasts in early active SSc resemble those found during the repair phase of tissue injury Citation34. The failure in SSc of mechanisms responsible for shutting down their activated state is a fundamental question that remains to be answered. Collagens, of which type I and III are major ECM components in the skin, are triple‐helix structures secreted as procollagens which subsequently undergo maturation. The transcription of collagen genes is regulated by elements with repressor and enhancer functions that are differently recruited under basal and inflammatory conditions Citation35. MMP participate in ECM degradation, their production is transcriptionally regulated, and require stepwise activation from inactive precursors (proMMP) Citation36. Their activity depends further on interactions with extracellular matrix components and binding to endogenous inhibitors such as tissue inhibitor of metalloproteinases (TIMP) Citation37. MMP‐1, or interstitial collagenase, has the distinctive capacity (shared with MMP‐8, MMP‐13, and MMP‐14) to cleave the triple‐helix of type I collagen allowing the chains to unwind and become susceptible to further degradation by the gelatinases MMP‐2 and MMP‐9 Citation38. TGF‐β upregulates type I and III collagens and TIMP‐1 while downregulating MMP‐1 gene transcription Citation9–12. In contrast, the proinflammatory cytokines (IL‐1, TNF‐α) induce MMP production. Among the many hallmark characteristics that distinguish SSc fibroblasts from normal fibroblasts, are: higher collagen synthesis Citation39, higher TGF‐β, connective tissue growth factor (CTGF), IL‐6, endothelin‐1, plasminogen activator inhibitor‐1, TIMP‐1 and MCP‐1 production Citation40–44, resistance to IFN‐γ Citation45 and to inhibitory signals delivered by T cell contact Citation45,46, altered Smad signaling Citation47–49, altered TGF‐β receptor signaling Citation50,51, constitutive phosphorylation of focal adhesion kinase (FAK), altered responses to endothelin Citation40, and constitutive thrombospondin expression which amplifies TGF‐β signaling Citation52. Genome‐wide analysis of affected skin revealed that over a thousand genes distinguish SSc from normal tissues, with prominent alterations of the TGF‐β and other connective tissue genes Citation53,54. Of interest however, fibroblasts imperfectly maintain all these abnormalities when kept in culture. Cytokines play a major role in regulating ECM deposition by fibroblasts Citation55. Among profibrotic cytokines, TGF‐β plays a pivotal role favoring fibroblast proliferation, fibroblast chemotaxis, matrix production, granulation tissue formation, and collagen contraction. Major advances have been made in recent years in understanding TGF‐β signaling in fibroblasts and its implication in fibrosis development Citation13,Citation35,Citation56–59. Indeed, TGF‐β is nowadays seen as the central mediator for the development of fibrosis in SSc and many other conditions. Three different genes encode the TGF‐β isoforms found in mammals, including TGF‐β1, TGF‐β2, and TGF‐β3. TGF‐βs are generated as propeptides, processed intracellularly, and secreted as biologically inactive precursors with latency‐associated peptide (LAP). The complex LAP‐TGF‐β can bind to latency TGF‐β binding protein (LTBP), which is referred to as ‘large’ latent TGF‐β. The unique ability of LAP to confer latency prevents binding of secreted TGF‐β to ubiquitously expressed receptors and assures an extracellular reservoir of TGF‐β that can be activated on demand. A complex and multifactor regulation controls the conversion of latent TGF‐β to active TGF‐β. Extracellular matrix proteins, including thrombospondin 1 and integrin avβ6, mediate TGF‐β activation in physiological conditions. In addition, various proteases, including plasmin, thrombin, cathepsin D, and plasma transglutaminase, have also been implicated as TGF‐β activators. TGF‐β binds to transmembrane serine/threonine kinases. Type I and type II receptors are glycoproteins coexpressed by most cell types. Initiation of signal requires binding to the TGF‐β receptor type II, a constitutively active serine/threonine kinase, resulting in the recruitment and phosphorylation of TGF‐β receptor type I. The formation of this heterodimeric complex results in the activation of a downstream signaling cascade. The active participation of the TGF‐β type I receptor in SSc fibroblast phenotype has recently been underscored Citation60. Among profibrotic cytokines, CTGF, a member of the CCN family of matricellular proteins, is produced in response to TGF‐β and mediates at least some of the TGF‐β effects Citation59,Citation61. Additional profibrotic cytokines are PDGF, epithelial growth factor (EGF), and basic fibroblastic growth factor (bFGF). In addition, a role for intracellular IL‐1α has been sought in the context of SSc, and the presence of aberrant nuclear IL‐1α has been associated with high IL‐6, PDGF, and procollagen levels Citation62. TNF‐α is a cytokine that has attracted considerable interest, since agents capable of blocking its biological activities are available. TNF‐α inhibits collagen production in a dose‐dependent manner by inhibiting the transcription of type I and type III procollagen mRNA Citation63,64. In addition, molecular studies have shown that TNF‐α inhibits TGF‐β signaling in human fibroblasts by a variety of additional mechanisms (AP‐1 activation, downregulation of TGF‐βRII, and CBP/p300 sequestration Citation65). However, TNF blockade prevents fibrosis development in in vivo models of lung fibrosis induced by bleomycin inhalation Citation66. The discrepancy between findings obtained in vitro (in which TNF decreases collagen production) and in vivo (in which TNF blockade prevents fibrosis development), may be related to the importance of the inflammatory component preceding fibrosis induced by bleomycin that is controlled by TNF blockade. Nonetheless, in view of the possible profibrotic effect, the use of TNF‐blockers in SSc individuals should be restricted to selected cases within controlled investigations.
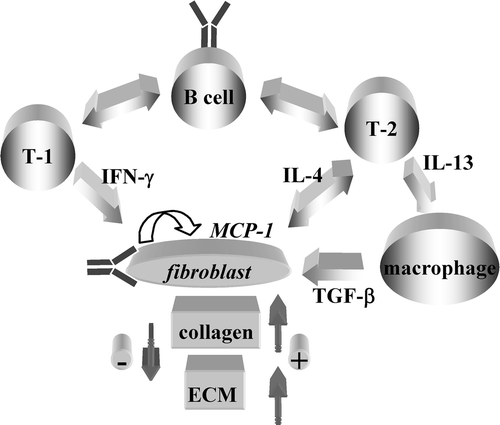
From a polarized T cell point of view, it is interesting to note that IFN‐γ inhibits collagen synthesis Citation67, while IL‐4 and IL‐13 enhance it Citation68–70. Thus, based on the effect on ECM deposition of the prototypic cytokines they produce, Th1‐like T cells have been predicted to decrease while Th2‐like T cells to increase ECM deposition Citation71. The in vivo relevance of IL‐4 for fibrosis development has been documented in several models, including but not exclusively the resistance to developing fibrosis in tight skin mouse‐1 (Tsk‐1) mice with targeted mutation of IL‐4 receptor α‐chain or IL‐4 gene Citation72,73. These models do not assess which cells are responsible for IL‐4 production but identify fibroblasts as cells responsive to IL‐4. Additional data indicate that occupancy of the IL‐13Rα2 by IL‐13 induces in macrophages TGF‐β production that drives enhanced collagen synthesis and fibrosis development Citation74,75 ().
Figure 2. Cells and mediators of the adaptive and natural immune system in relation with fibroblasts and matrix deposition. The arrows depict presumed interactions between cells of the immune system and fibroblasts with potential to modulate extracellular matrix(ECM) deposition. T‐1 = T cells with preferential production of interferon (IFN)‐γ; T‐2 = cells with preferential production of interleukin (IL)‐4; Y = autoAb with fibroblast‐activating capacity.
Fibroblasts and inflammation
Fibroblasts do not only play a key role in ECM deposition and renewal, but by releasing soluble mediators in response to activating stimuli they also participate in shaping and maintaining inflammation. Chemokines are important soluble mediators originally identified for their chemotactic properties Citation76. However, their biological functions are broader, encompassing relevant roles in virus cell entry, angiogenesis, tumor growth, and metastasis formation. Among the chemokines detected by histological or molecular biology methods at sites undergoing fibrosis in humans or mouse models are the CC chemokines RANTES/CCL5, MIP‐1α/CCL3, eotaxin/CCL11, PARC/CCL18, the CXC chemokine IL‐8/CXCL8, ENA‐78/CXCL5, MIP‐2, and the CX3CL1/fractalkine. In addition, the serum levels of CTACK/CCL27, IP‐10/CXCL10, MDC/CCL22, and TARC/CCL17 are increased in SSc. It is not known whether these chemokines play direct pro‐ or antifibrotic roles in addition to recruitment of specific leukocyte subsets. Nonetheless, the proangiogenetic and antiangiogenetic activities of chemokines have been suggested to play an important role in fibrosis Citation77. In bleomycin‐induced lung fibrosis, neutralization of MIP‐2 (murine analogue of IL‐8) attenuates fibrosis Citation78, and systemic administration of IP‐10 and ITAC reduces fibrosis Citation79,80. MCP‐1/CCL2, a CC chemokine that binds to and signals via CCR2, has attracted keen interest since it is abundantly produced by SSc fibroblasts and plays a direct role in collagen and MMP‐1 induction Citation42,Citation81. On the other hand, CCR2 constitutive upregulation in SSc fibroblasts participates in the transdifferentiation process in myofibroblasts Citation82. Of further interest, IL‐4 triggers MCP‐1 production by human fibroblasts, and MCP‐1 may polarize T cells toward the Th2 subset in the mouse and in humans Citation83. In a rodent model of fibrotic versus nonfibrotic pulmonary granulomas, procollagen production is associated with Th2 cells and MCP‐1 production. Furthermore, CCR2−/− mice are resistant to development of lung fibrosis induced by transgenic IL‐13 and bleomycin. Thus, the MCP‐1–CCR2 axis may play a major role in SSc progression, since it can participate in an amplification loop by enhancing collagen production and favoring Th2 cell differentiation with subsequent production of IL‐4, that in turn will favor collagen deposition ().
T cells in SSc
Histological examination of early SSc skin lesions has demonstrated that an inflammatory infiltrate precedes the development of the vasculopathy and fibrosis Citation84. CD4+ T cells, CD8+ T cells, and monocytes are numerically important, but additional cells are B cells, eosinophils, mast cells, and natural killer (NK) cells Citation85–87. Of interest, collagen synthesis determined by in situ localization of procollagen Iα1 appears to be higher in fibroblasts adjacent to inflammatory cells Citation88,89. These findings have led to the hypothesis that inflammatory cells and in particular T cells provide important stimuli that drive collagen synthesis in fibroblasts. While the antigen specificity of T cells infiltrating the skin is not known, the analysis of the T cell receptor (TcR) they express has demonstrated oligoclonality, thus supporting the concept that T cells accumulate in lesional skin in specific response to antigen Citation90. Similar data have been obtained with T cells recovered form bronchoalveolar lavage (BAL) of SSc interstitial lung disease Citation91. T cells infiltrating the skin and other organs in SSc are functionally heterogeneous. Some T cells cloned from SSc skin biopsies are CD4+ producing IFN‐γ or glycosaminoglycan‐stimulating factor when activated by mitogens Citation92, consistent with immunohistochemistry and polymerase chain reaction (PCR)‐based techniques of tissue analysis indicating that T cells infiltrating early active lesions produce IFN‐γ and TNF‐α Citation93. Other authors have described that T cell clones generated from skin biopsies are Th2‐like CD4+ that produce high levels of IL‐4 but no IFN‐γ upon in vitro activation Citation94, and that high IL‐4 but little or no IFN‐γ mRNA expression is detected in infiltrated tissues Citation94,95. In addition, the Th2‐oriented profile of male offspring T cells reactive against maternal MHC antigens has recently been documented in the peripheral blood and skin of women with SSc Citation96. These data are in keeping with the hypothesis that microchimerism may be pathogenetically relevant to fibrosis development, as it could generate a graft versus host disease (GVHD)‐like condition. In bronchoalveolar lavage (BAL) fluid of patients with SSc lung involvement, CD8+ T cells mainly express mRNA for IL‐4 and IL‐5, with or without equal amounts of IFN‐γ mRNA, while in BAL fluids from normal individuals CD8+ T cells preferentially expressed IFN‐γ mRNA with no or little IL‐4 or IL‐5 mRNA Citation97. The DNA array technology used to assess gene expression in lung CD8+ T cells revealed in the majority of examined SSc samples a gene expression pattern consistent with an activated, profibrotic phenotype of CD8+ T cells Citation98. Additional T cell subsets currently being characterized include Treg and Th3 that exert their effector functions at least in part by releasing TGF‐β. The impact of these cells in SSc development has not been evaluated yet. In summary, available data on the characteristics of T cells infiltrating organs affected by SSc are heterogeneous, maybe in relation with different phases of disease evolution; the majority, however, indicate that T cells preferentially produce IL‐4 and therefore belong to the type 2 subset with potential for enhancing collagen deposition by fibroblasts Citation94–98 ().
B cells and autoantibodies (autoAb) in SSc
The presence of autoAb is currently used to identify individuals affected by SSc and overlapping conditions. Indeed, particular autoantibody specificities segregate with specific clinical subsets. These include antibodies directed against ubiquitous antigens such as centromeric proteins B and C, Scl‐70 (topoisomerase‐1), RNA polymerase I and III, Th/T0, PM‐Scl, U1‐RNP, U3‐RNP (fibrillarin). While the titer of Scl‐70 may correlate with clinical severity, and B cell epitopes of Scl‐70 correlate with clinical manifestations Citation99,100, their contribution to pathogenesis remains elusive. Canadian authors have shown that Scl‐70 may be exported to the surface of fibroblasts where it is recognized by autoAb that subsequently may recruit and activate monocytes/macrophages Citation101. It is speculated that recruited monocytes/macrophages may then release profibrotic cytokines that activate fibroblasts. Additional autoAb specificities, not directed against ubiquitous autoantigens, with possible implication in SSc pathogenesis, have recently been identified. These include antibodies directed against MMP‐1 and MMP‐3, more frequently found in diffuse than in limited SSc, capable of inhibiting MMP‐1 and MMP‐3 enzymatic activities thus potentially favoring ECM deposition by inhibiting its normal turnover Citation102.103. Fibrillin‐1 is a matrix protein that forms microfibers whose primary function is architectural. This protein is abnormal in the Tsk‐1 mouse and autoAb to fibrillin‐1 are present in Tsk‐1 sera Citation104. AutoAb antifibrillin‐1 has been detected in the sera of native American SSc patients that correlated significantly with disease expression, but also in sera from Japanese, Caucasian, and African‐American SSc patients Citation23,Citation105. Of foremost importance, antifibrillin‐1 autoAb have been recently shown to induce in dermal fibroblasts phenotypic changes resembling those characteristic of SSc, in particular with the capacity to upregulate collagen transcription in a TGF‐β‐dependent manner Citation106. These findings point to the possibility that autoAb directed against an extracellular protein, a normal constituent of the ECM, could enhance TGF‐β release. In a collaborative work we described the presence in SSc sera, but not in control or sarcoidosis sera, of autoAb that we named AFA (antifibroblast antibodies) capable of binding to the fibroblasts' surface. Upon binding these autoAb are internalized and upregulate intercellular adhesion molecule (ICAM)‐1 expression and IL‐6 production in an IL‐1‐mediated, autocrine loop Citation107. More recently the screening of SSc sera has permitted identification of autoAb that recognize conformation‐dependent epitopes of the PDGF receptor. These autoAb have the remarkable capacity of activating fibroblasts by inducing reactive oxygen species (ROS) and quickly upregulating collagen mRNA transcription Citation108. Finally, as mentioned in a previous paragraph, antibodies selected for binding to a randomly generated peptide have been shown to cross‐recognize a CMV antigen and to bind to endothelial cells and fibroblasts. These cross‐reactive antibodies recognize the CMV UL‐94 protein and the NAG‐2 protein expressed on EC and fibroblasts, and induce the production by fibroblasts of inflammatory mediators in addition to upregulating type I collagen production Citation17,18. Thus, at least three different targets of autoAb have been identified that have shown potential for inducing in normal fibroblasts phenotypic changes with similarities to those characteristic of SSc fibroblasts ().
To understand whether autoAb production could be related to intrinsic abnormalities of the B cell compartment, a number of studies have recently focused on the function and phenotype of B cells in SSc and related animal models. In this respect, of prime importance is the finding of B cell gene expression in SSc skin when analyzed by unbiased gene screening by microarray, indicating thus that B cells are present in tissue seats of fibrosis Citation53. In addition, the frequency of B cells expressing activation markers is increased in SSc, and more interestingly B cells overexpress CD19. CD19 is a critical cell surface signal transduction molecule on B cells that regulates basal signaling thresholds and accelerates signaling through B cell antigen receptor Citation109. Consistent with these findings, in Tsk‐1 mice B cells display enhanced CD19 signaling and a chronically activated phenotype resulting in hypergammaglobulinemia and spontaneous autoantibody production, which are abrogated when CD19 is knocked down Citation110. Among the cytokines that play an important role in B cell homeostasis and activation, BAFF (B cell activating factor of the TNF family) has been reported to be elevated in SSc patients and to correlate positively to the extent of skin fibrosis. In addition, BAFF receptor expression on B cells is increased in SSc patients, and SSc B cells stimulated by BAFF show enhanced IgG and IL‐6 production Citation111. It is important to note that BAFF stimulation enhances CD19 expression and increases the ability of B cell antigen receptor to phosphorylate CD19. Thus, excess BAFF may accelerate B cell hyperactivity via overexpression and phosphorylation of CD19, thereby contributing to B cell abnormalities in SSc.
If confirmed and exploited in animal models, these findings will change our perception of the role of humoral immunity in SSc, since these findings indicate that humoral components of the immune response may be involved in the pathogenesis of SSc to an extent that was previously unthought‐of. At the present time it is, however, unknown whether the presence of these autoAb precedes or accompanies fibrosis development.
From science to the clinics
The identification of endothelin‐1 as a major mediator of vasoconstriction has led to the development of endothelin receptor antagonists of which bosentan has conclusively shown clinical benefit in the management of PAH as well as in the prevention of development of new digital ulcers Citation112. Furthermore, in individuals suffering from PAH secondary to SSc bosentan appears to improve survival Citation113. As said before, fibrosis development is preceded and accompanied by the presence of an inflammatory infiltrate in target organs rich in T cells. While more targeted intervention strategies are needed, cyclophosphamide‐based immunosuppression has shown benefit in a double‐blind, randomized, placebo‐controlled trial assessing its value in interstitial lung diseases associated to early active SSc with stabilization of the forced vital capacity (FVC) in the active, compared to worsening of FVC in the placebo arm Citation114. These results confirm uncontrolled series published in the past. Skin scores aimed at evaluating skin thickening also improved in this study suggesting that immunosuppression has general value in SSc. Along the same lines of thought, ablation of the immune system with very high doses of cyclophosphamide (in conjunction with gamma radiation and antithymocyte globulin in one trial), followed by hematopoietic rescue with the infusion of autologous hematopoietic stem cells, is currently tested in two multicenter randomized trials—one in Europe (ASTIS, www.astistrial.com) the other in the USA (SCOT, www.sclerodermatrial.org). In both trials the control arm consists of monthly pulses of cyclophosphamide. Previous open studies testing this strategy have indicated that the disease may improve, at least transiently, in a high proportion of individuals with highly active disease Citation115; however, the procedure‐related mortality was relatively high. More than 80 individuals have been randomized until now in the ASTIS trial, and no procedure‐related mortality has been recorded yet, by excluding cardiac or pulmonary vascular disease. These trials will tell whether immunoablation followed by reconstitution of the immune system has an advantage over more conventional treatment procedures. They may also offer a proof of the concept that the disease is indeed driven by activation of the immune system since these procedures are not thought to be directly capable of modifying fibroblast metabolism.
Conclusions
A number of fundamental questions remain open in SSc, including: the reason for female preponderance, the preference of type 2 T cell immune response, the connections between microvascular and immunological abnormalities, the nature of the (auto)‐antigen(s) driving the process, their link with the genetic background of the host, the mechanisms leading to the persistence of processes that resemble those initiated for tissue repair, the identification of a hierarchy if any in the events and processes underling the disease, which may be amenable to therapeutic approaches. Nonetheless, recent years have witnessed important advancements in the characterization of molecular and cellular events likely to play a role in SSc. Targeted therapies are become a reality, and indeed we are witnessing revolutionary changes in the approach to the treatment of this disease. Most importantly, cooperative multicentric efforts, such as those conducted by EUSTAR (www.eustar.org), are nowadays favoring the collection of clinical data on large patient populations aiming at better defining clinical presentations and identifying subgroups of patients that could benefit of new therapeutic approaches fostered by lessons derived by novel animal models and in vitro findings Citation116.
Acknowledgements
This work has been financed in part by the Swiss National Science Foundation grant No 310000‐112180/1, by the Association des Sclérodermiques de France, and by the Association Romande des sclérodermiques. I am grateful to my colleagues Dr Camillo Ribi and Gianluca Vanini for their critical reading of this manuscript.
Related Research Data
References
- LeRoy E. C., Black C., Fleischmajer R., Jablonska S., Krieg T., Medsger T. A., Jr., et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol 1988; 15: 202–5
- Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum 1980; 23: 581–90
- Reveille J. D., Solomon D. H. Evidence‐based guidelines for the use of immunologic tests: anticentromere, Scl‐70, and nucleolar antibodies. Arthritis Rheum 2003; 49: 399–412
- Mayes M. D. Scleroderma epidemiology. Rheum Dis Clin North Am 1996; 22: 751–64
- Mori Y., Kahari V. M., Varga J. Scleroderma‐like cutaneous syndromes. Curr Rheumatol Rep 2002; 4: 113–22
- Cowper S. E. Nephrogenic fibrosing dermopathy: the first 6 years. Curr Opin Rheumatol 2003; 15: 785–90
- Jimenez S. A., Artlett C. M., Sandorfi N., Derk C., Latinis K., Sawaya H., et al. Dialysis‐associated systemic fibrosis (nephrogenic fibrosing dermopathy): study of inflammatory cells and transforming growth factor beta1 expression in affected skin. Arthritis Rheum 2004; 50: 2660–6
- Hauser C., Kaya G., Chizzolini C. Nephrogenic fibrosing dermopathy in a renal transplant recipient with tubulointerstitial nephritis and uveitis. Dermatology 2004; 209: 50–2
- Abraham D. J., Varga J. Scleroderma: from cell and molecular mechanisms to disease models. Trends Immunol 2005; 26: 587–95
- Denton C. P., Black C. M. Targeted therapy comes of age in scleroderma. Trends Immunol 2005; 26: 596–602
- Jimenez S. A., Derk C. T. Following the molecular pathways toward an understanding of the pathogenesis of systemic sclerosis. Ann Intern Med 2004; 140: 37–50
- Sakkas L. I., Platsoucas C. D. Is systemic sclerosis an antigen‐driven T cell disease?. Arthritis Rheum 2004; 50: 1721–33
- Pannu J., Trojanowska M. Recent advances in fibroblast signaling and biology in scleroderma. Curr Opin Rheumatol 2004; 16: 739–45
- Sato S., Fujimoto M., Hasegawa M., Takehara K., Tedder T. F. Altered B lymphocyte function induces systemic autoimmunity in systemic sclerosis. Mol Immunol 2004; 41: 1123–33
- Jimenez S. A., Diaz A., Khalili K. Retroviruses and the pathogenesis of systemic sclerosis. Int Rev Immunol 1995; 12: 159–75
- Pandey J. P., LeRoy E. C. Human cytomegalovirus and the vasculopathies of autoimmune diseases (especially scleroderma), allograft rejection, and coronary restenosis. Arthritis Rheum 1998; 41: 10–15
- Lunardi C., Dolcino M., Peterlana D., Bason C., Navone R., Tamassia N., et al. Antibodies against human cytomegalovirus in the pathogenesis of systemic sclerosis: a gene array approach. PLoS Med 2006; 3: e2
- Lunardi C., Bason C., Navone R., Millo E., Damonte G., Corrocher R., et al. Systemic sclerosis immunoglobulin G autoantibodies bind the human cytomegalovirus late protein UL94 and induce apoptosis in human endothelial cells. Nat Med 2000; 6: 1183–6
- Diot E., Lesire V., Guilmot J. L., Metzger M. D., Pilore R., Rogier S., et al. Systemic sclerosis and occupational risk factors: a case‐control study. Occup Environ Med 2002; 59: 545–9
- Garabrant D. H., Lacey J. V., Jr., Laing T. J., Gillespie B. W., Mayes M. D., Cooper B. C., et al. Scleroderma and solvent exposure among women. Am J Epidemiol 2003; 157: 493–500
- Christner P. J., Artlett C. M., Conway R. F., Jimenez S. A. Increased numbers of microchimeric cells of fetal origin are associated with dermal fibrosis in mice following injection of vinyl chloride. Arthritis Rheum 2000; 43: 2598–605
- Tan F. K., Wang N., Kuwana M., Chakraborty R., Bona C. A., Milewicz D. M., et al. Association of fibrillin 1 single‐nucleotide polymorphism haplotypes with systemic sclerosis in Choctaw and Japanese populations. Arthritis Rheum 2001; 44: 893–901
- Tan F. K., Arnett F. C., Antohi S., Saito S., Mirarchi A., Spiera H., et al. Autoantibodies to the extracellular matrix microfibrillar protein, fibrillin‐1, in patients with scleroderma and other connective tissue diseases. J Immunol 1999; 163: 1066–72
- Ahmed S. S., Tan F. K. Identification of novel targets in scleroderma: update on population studies, cDNA arrays, SNP analysis, and mutations. Curr Opin Rheumatol 2003; 15: 766–71
- Hata R., Akai J., Kimura A., Ishikawa O., Kuwana M., Shinkai H. Association of functional microsatellites in the human type I collagen alpha2 chain (COL1A2) gene with systemic sclerosis. Biochem Biophys Res Commun 2000; 272: 36–40
- Johnson R. W., Tew M. B., Arnett F. C. The genetics of systemic sclerosis. Curr Rheumatol Rep 2002; 4: 99–107
- Herrick A. L., Matucci Cerinic M. The emerging problem of oxidative stress and the role of antioxidants in systemic sclerosis. Clin Exp Rheumatol 2001; 19: 4–8
- Sambo P., Baroni S. S., Luchetti M., Paroncini P., Dusi S., Orlandini G., et al. Oxidative stress in scleroderma: maintenance of scleroderma fibroblast phenotype by the constitutive up‐regulation of reactive oxygen species generation through the NADPH oxidase complex pathway. Arthritis Rheum 2001; 44: 2653–64
- Silveri F., De Angelis R., Poggi A., Muti S., Bonapace G., Argentati F., et al. Relative roles of endothelial cell damage and platelet activation in primary Raynaud's phenomenon (RP) and RP secondary to systemic sclerosis. Scand J Rheumatol 2001; 30: 290–6
- Distler O., Distler J. H., Scheid A., Acker T., Hirth A., Rethage J., et al. Uncontrolled expression of vascular endothelial growth factor and its receptors leads to insufficient skin angiogenesis in patients with systemic sclerosis. Circ Res 2004; 95: 109–16
- Kahaleh M. B. Raynaud phenomenon and the vascular disease in scleroderma. Curr Opin Rheumatol 2004; 16: 718–22
- Sgonc R., Gruschwitz M. S., Boeck G., Sepp N., Gruber J., Wick G. Endothelial cell apoptosis in systemic sclerosis is induced by antibody‐dependent cell‐mediated cytotoxicity via CD95. Arthritis Rheum 2000; 43: 2550–62
- Trojanowska M., LeRoy E. C., Eckes B., Krieg T. Pathogenesis of fibrosis: type 1 collagen and the skin. J Mol Med 1998; 76: 266–74
- Singer A. J., Clark R. A. Cutaneous wound healing. N Engl J Med 1999; 341: 738–46
- Ghosh A. K. Factors involved in the regulation of type I collagen gene expression: implication in fibrosis. Exp Biol Med (Maywood) 2002; 227: 301–14
- Nagase H., Woessner J. F., Jr. Matrix metalloproteinases. J Biol Chem 1999; 274: 21491–4
- Vincenti M. P. The matrix metalloproteinase (MMP) and tissue inhibitor of metalloproteinase (TIMP) genes. Transcriptional and posttranscriptional regulation, signal transduction and cell‐type‐specific expression. Methods Mol Biol 2001; 151: 121–48
- Shapiro S. D. Matrix metalloproteinase degradation of extracellular matrix: biological consequences. Curr Opin Cell Biol 1998; 10: 602–8
- Shi‐Wen X., Denton C. P., McWhirter A., Bou‐Gharios G., Abraham D. J., du Bois R. M., et al. Scleroderma lung fibroblasts exhibit elevated and dysregulated type I collagen biosynthesis. Arthritis Rheum 1997; 40: 1237–44
- Shi‐Wen X., Denton C. P., Dashwood M. R., Holmes A. M., Bou‐Gharios G., Pearson J. D., et al. Fibroblast matrix gene expression and connective tissue remodeling: role of endothelin‐1. J Invest Dermatol 2001; 116: 417–25
- Eckes B., Hunzelmann N., Ziegler‐Heitbrock H. W., Urbanski A., Luger T., Krieg T., et al. Interleukin‐6 expression by fibroblasts grown in three‐dimensional gel cultures. FEBS Lett 1992; 298: 229–32
- Yamamoto T., Eckes B., Krieg T. High expression and autoinduction of monocyte chemoattractant protein‐1 in scleroderma fibroblasts. Eur J Immunol 2001; 31: 2936–41
- Kessler‐Becker D., Smola S., Krieg T., Eckes B. High plasminogen activator inhibitor type 2 expression is a hallmark of scleroderma fibroblasts in vitro. Exp Dermatol 2004; 13: 708–14
- Igarashi A., Nashiro K., Kikuchi K., Sato S., Ihn H., Grotendorst G. R., et al. Significant correlation between connective tissue growth factor gene expression and skin sclerosis in tissue sections from patients with systemic sclerosis. J Invest Dermatol 1995; 105: 280–4
- Chizzolini C., Rezzonico R., Ribbens C., Burger D., Wollheim F. A., Dayer J. M. Inhibition of type I collagen production by dermal fibroblasts upon contact with activated T cells: different sensitivity to inhibition between systemic sclerosis and control fibroblasts. Arthritis Rheum 1998; 41: 2039–47
- Chizzolini C., Parel Y., De Luca C., Tyndall A., Akesson A., Scheja A., et al. Systemic sclerosis Th2 cells inhibit collagen production by dermal fibroblasts via membrane‐associated tumor necrosis factor alpha. Arthritis Rheum 2003; 48: 2593–604
- Varga J. Scleroderma and Smads: dysfunctional Smad family dynamics culminating in fibrosis. Arthritis Rheum 2002; 46: 1703–13
- Mori Y., Chen S. J., Varga J. Expression and regulation of intracellular SMAD signaling in scleroderma skin fibroblasts. Arthritis Rheum 2003; 48: 1964–78
- Asano Y., Ihn H., Yamane K., Kubo M., Tamaki K. Impaired Smad7‐Smurf‐mediated negative regulation of TGF‐beta signaling in scleroderma fibroblasts. J Clin Invest 2004; 113: 253–64
- Mori Y., Ishida W., Bhattacharyya S., Li Y., Platanias L. C., Varga J. Selective inhibition of activin receptor‐like kinase 5 signaling blocks profibrotic transforming growth factor beta responses in skin fibroblasts. Arthritis Rheum 2004; 50: 4008–21
- Pannu J., Gore‐Hyer E., Yamanaka M., Smith E. A., Rubinchik S., Dong J. Y., et al. An increased transforming growth factor beta receptor type I:type II ratio contributes to elevated collagen protein synthesis that is resistant to inhibition via a kinase‐deficient transforming growth factor beta receptor type II in scleroderma. Arthritis Rheum 2004; 50: 1566–77
- Mimura Y., Ihn H., Jinnin M., Asano Y., Yamane K., Tamaki K. Constitutive thrombospondin‐1 overexpression contributes to autocrine transforming growth factor‐beta signaling in cultured scleroderma fibroblasts. Am J Pathol 2005; 166: 1451–63
- Whitfield M. L., Finlay D. R., Murray J. I., Troyanskaya O. G., Chi J. T., Pergamenschikov A., et al. Systemic and cell type‐specific gene expression patterns in scleroderma skin. Proc Natl Acad Sci U S A 2003; 100: 12319–24
- Gardner H., Shearstone J. R., Bandaru R., Crowell T., Lynes M., Trojanowska M., et al. Gene profiling of scleroderma skin reveals robust signatures of disease that are imperfectly reflected in the transcript profiles of explanted fibroblasts. Arthritis Rheum 2006; 54: 1961–73
- Uitto J., Kouba D. Cytokine modulation of extracellular matrix gene expression: relevance to fibrotic skin diseases. J Dermatol Sci 2000; 24(Suppl 1)S60–9
- Denton C. P., Abraham D. J. Transforming growth factor‐beta and connective tissue growth factor: key cytokines in scleroderma pathogenesis. Curr Opin Rheumatol 2001; 13: 505–11
- Ihn H. Pathogenesis of fibrosis: role of TGF‐beta and CTGF. Curr Opin Rheumatol 2002; 14: 681–5
- Schiller M., Javelaud D., Mauviel A. TGF‐beta‐induced SMAD signaling and gene regulation: consequences for extracellular matrix remodeling and wound healing. J Dermatol Sci 2004; 35: 83–92
- Leask A., Abraham D. J. TGF‐beta signaling and the fibrotic response. FASEB J 2004; 18: 816–27
- Chen Y., Shi‐wen X., Eastwood M., Black C. M., Denton C. P., Leask A., et al. Contribution of activin receptor‐like kinase 5 (transforming growth factor beta receptor type I) signaling to the fibrotic phenotype of scleroderma fibroblasts. Arthritis Rheum 2006; 54: 1309–16
- Leask A., Denton C. P., Abraham D. J. Insights into the molecular mechanism of chronic fibrosis: the role of connective tissue growth factor in scleroderma. J Invest Dermatol 2004; 122: 1–6
- Kawaguchi Y., Hara M., Wright T. M. Endogenous IL‐1alpha from systemic sclerosis fibroblasts induces IL‐6 and PDGF‐A. J Clin Invest 1999; 103: 1253–60
- Mauviel A., Heino J., Kahari V. M., Hartmann D. J., Loyau G., Pujol J. P., et al. Comparative effects of interleukin‐1 and tumor necrosis factor‐alpha on collagen production and corresponding procollagen mRNA levels in human dermal fibroblasts. J Invest Dermatol 1991; 96: 243–9
- Solis‐Herruzo J. A., Brenner D. A., Chojkier M. Tumor necrosis factor alpha inhibits collagen gene transcription and collagen synthesis in cultured human fibroblasts. J Biol Chem 1988; 263: 5841–5
- Verrecchia F., Mauviel A. TGF‐beta and TNF‐alpha: antagonistic cytokines controlling type I collagen gene expression. Cell Signal 2004; 16: 873–80
- Piguet P. F., Collart M. A., Grau G. E., Kapanci Y., Vassalli P. Tumor necrosis factor/cachectin plays a key role in bleomycin‐induced pneumopathy and fibrosis. J Exp Med 1989; 170: 655–63
- Rosenbloom J., Feldman G., Freundlich B., Jimenez S. A. Inhibition of excessive scleroderma fibroblast collagen production by recombinant gamma‐interferon. Association with a coordinate decrease in types I and III procollagen messenger RNA levels. Arthritis Rheum 1986; 29: 851–6
- Postlethwaite A. E., Holness M. A., Katai H., Raghow R. Human fibroblasts synthesize elevated levels of extracellular matrix proteins in response to interleukin 4. J Clin Invest 1992; 90: 1479–85
- Serpier H., Gillery P., Salmon‐Ehr V., Garnotel R., Georges N., Kalis B., et al. Antagonistic effects of interferon‐gamma and interleukin‐4 on fibroblast cultures. J Invest Dermatol 1997; 109: 158–62
- Oriente A., Fedarko N. S., Pacocha S. E., Huang S. K., Lichtenstein L. M., Essayan D. M. Interleukin‐13 modulates collagen homeostasis in human skin and keloid fibroblasts. J Pharmacol Exp Ther 2000; 292: 988–94
- Wynn T. A. Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol 2004; 4: 583–94
- McGaha T., Saito S., Phelps R. G., Gordon R., Noben‐Trauth N., Paul W. E., et al. Lack of skin fibrosis in tight skin (TSK) mice with targeted mutation in the interleukin‐4R alpha and transforming growth factor‐beta genes. J Invest Dermatol 2001; 116: 136–43
- Kodera T., McGaha T. L., Phelps R., Paul W. E., Bona C. A. Disrupting the IL‐4 gene rescues mice homozygous for the tight‐skin mutation from embryonic death and diminishes TGF‐beta production by fibroblasts. Proc Natl Acad Sci U S A 2002; 99: 3800–5
- Lee C. G., Homer R. J., Zhu Z., Lanone S., Wang X., Koteliansky V., et al. Interleukin‐13 induces tissue fibrosis by selectively stimulating and activating transforming growth factor beta(1). J Exp Med 2001; 194: 809–21
- Fichtner‐Feigl S., Strober W., Kawakami K., Puri R. K., Kitani A. IL‐13 signaling through the IL‐13alpha2 receptor is involved in induction of TGF‐beta1 production and fibrosis. Nat Med 2006; 12: 99–106
- Baggiolini M. Chemokines and leukocyte traffic. Nature 1998; 392: 565–8
- Keane M. P., Belperio J. A., Moore T. A., Moore B. B., Arenberg D. A., Smith R. E., et al. Neutralization of the CXC chemokine, macrophage inflammatory protein‐2, attenuates bleomycin‐induced pulmonary fibrosis. J Immunol 1999; 162: 5511–8
- Keane M. P., Arenberg D. A., Lynch J. P 3rd., Whyte R. I., Iannettoni M. D., Burdick M. D., et al. The CXC chemokines, IL‐8 and IP‐10, regulate angiogenic activity in idiopathic pulmonary fibrosis. J Immunol 1997; 159: 1437–43
- Keane M. P., Belperio J. A., Arenberg D. A., Burdick M. D., Xu Z. J., Xue Y. Y., et al. IFN‐gamma‐inducible protein‐10 attenuates bleomycin‐induced pulmonary fibrosis via inhibition of angiogenesis. J Immunol 1999; 163: 5686–92
- Burdick M. D., Murray L. A., Keane M. P., Xue Y. Y., Zisman D. A., Belperio J. A., et al. CXCL11 attenuates bleomycin‐induced pulmonary fibrosis via inhibition of vascular remodeling. Am J Respir Crit Care Med 2005; 171: 261–8
- Distler O., Pap T., Kowal‐Bielecka O., Meyringer R., Guiducci S., Landthaler M., et al. Overexpression of monocyte chemoattractant protein 1 in systemic sclerosis: role of platelet‐derived growth factor and effects on monocyte chemotaxis and collagen synthesis. Arthritis Rheum 2001; 44: 2665–78
- Carulli M. T., Ong V. H., Ponticos M., Shiwen X., Abraham D. J., Black C. M., et al. Chemokine receptor CCR2 expression by systemic sclerosis fibroblasts: evidence for autocrine regulation of myofibroblast differentiation. Arthritis Rheum 2005; 52: 3772–82
- Distler J. H. W., Jüngel A., Caretto D., Kowal‐Bielecka O., Gay R. E., Michel B. A., et al. MCP‐1 stimulates the release of the profibrotic cytokine IL‐4 by inducing Th2 differentiation in Th0 cells (Abstract). Ann Rheum Dis 2005; 64: SIII110
- Prescott R. J., Freemont A. J., Jones C. J., Hoyland J., Fielding P. Sequential dermal microvascular and perivascular changes in the development of scleroderma. J Pathol 1992; 166: 255–63
- Fleischmajer R., Perlish J. S., Reeves J. R. Cellular infiltrates in scleroderma skin. Arthritis Rheum 1977; 20: 975–84
- Roumm A. D., Whiteside T. L., Medsger T. A., Jr., Rodnan G. P. Lymphocytes in the skin of patients with progressive systemic sclerosis. Quantification, subtyping, and clinical correlations. Arthritis Rheum 1984; 27: 645–53
- Kraling B. M., Maul G. G., Jimenez S. A. Mononuclear cellular infiltrates in clinically involved skin from patients with systemic sclerosis of recent onset predominantly consist of monocytes/macrophages. Pathobiology 1995; 63: 48–56
- Kahari V. M., Sandberg M., Kalimo H., Vuorio T., Vuorio E. Identification of fibroblasts responsible for increased collagen production in localized scleroderma by in situ hybridization. J Invest Dermatol 1988; 90: 664–70
- Scharffetter K., Lankat‐Buttgereit B., Krieg T. Localization of collagen mRNA in normal and scleroderma skin by in‐situ hybridization. Eur J Clin Invest 1988; 18: 9–17
- Sakkas L. I., Xu B., Artlett C. M., Lu S., Jimenez S. A., Platsoucas C. D. Oligoclonal T cell expansion in the skin of patients with systemic sclerosis. J Immunol 2002; 168: 3649–59
- Yurovsky V. V., Wigley F. M., Wise R. A., White B. Skewing of the CD8+ T‐cell repertoire in the lungs of patients with systemic sclerosis. Hum Immunol 1996; 48: 84–97
- Ferrarini M., Steen V., Medsger T. A., Jr., Whiteside T. L. Functional and phenotypic analysis of T lymphocytes cloned from the skin of patients with systemic sclerosis. Clin Exp Immunol 1990; 79: 346–52
- Gruschwitz M. S., Vieth G. Up‐regulation of class II major histocompatibility complex and intercellular adhesion molecule 1 expression on scleroderma fibroblasts and endothelial cells by interferon‐gamma and tumor necrosis factor alpha in the early disease stage. Arthritis Rheum 1997; 40: 540–50
- Mavalia C., Scaletti C., Romagnani P., Carossino A. M., Pignone A., Emmi L., et al. Type 2 helper T‐cell predominance and high CD30 expression in systemic sclerosis. Am J Pathol 1997; 151: 1751–8
- Salmon‐Ehr V., Serpier H., Nawrocki B., Gillery P., Clavel C., Kalis B., et al. Expression of interleukin‐4 in scleroderma skin specimens and scleroderma fibroblast cultures. Potential role in fibrosis. Arch Dermatol 1996; 132: 802–6
- Scaletti C., Vultaggio A., Bonifacio S., Emmi L., Torricelli F., Maggi E., et al. Th2‐oriented profile of male offspring T cells present in women with systemic sclerosis and reactive with maternal major histocompatibility complex antigens. Arthritis Rheum 2002; 46: 445–50
- Atamas S. P., Yurovsky V. V., Wise R., Wigley F. M., Goter Robinson C. J., Henry P., et al. Production of type 2 cytokines by CD8+ lung cells is associated with greater decline in pulmonary function in patients with systemic sclerosis. Arthritis Rheum 1999; 42: 1168–78
- Luzina I. G., Atamas S. P., Wise R., Wigley F. M., Xiao H. Q., White B. Gene expression in bronchoalveolar lavage cells from scleroderma patients. Am J Respir Cell Mol Biol 2002; 26: 549–57
- Rizou C., Ioannidis J. P., Panou‐Pomonis E., Sakarellos‐Daitsiotis M., Sakarellos C., Moutsopoulos H. M., et al. B‐Cell epitope mapping of DNA topoisomerase I defines epitopes strongly associated with pulmonary fibrosis in systemic sclerosis. Am J Respir Cell Mol Biol 2000; 22: 344–51
- Hu P. Q., Fertig N., Medsger T. A., Jr., Wright T. M. Correlation of serum anti‐DNA topoisomerase I antibody levels with disease severity and activity in systemic sclerosis. Arthritis Rheum 2003; 48: 1363–73
- Henault J., Robitaille G., Senecal J. L., Raymond Y. DNA topoisomerase I binding to fibroblasts induces monocyte adhesion and activation in the presence of anti‐topoisomerase I autoantibodies from systemic sclerosis patients. Arthritis Rheum 2006; 54: 963–73
- Sato S., Hayakawa I., Hasegawa M., Fujimoto M., Takehara K. Function blocking autoantibodies against matrix metalloproteinase‐1 in patients with systemic sclerosis. J Invest Dermatol 2003; 120: 542–7
- Nishijima C., Hayakawa I., Matsushita T., Komura K., Hasegawa M., Takehara K., et al. Autoantibody against matrix metalloproteinase‐3 in patients with systemic sclerosis. Clin Exp Immunol 2004; 138: 357–63
- Murai C., Saito S., Kasturi K. N., Bona C. A. Spontaneous occurrence of anti‐fibrillin‐1 autoantibodies in tight‐skin mice. Autoimmunity 1998; 28: 151–5
- Tan F. K., Arnett F. C., Reveille J. D., Ahn C., Antohi S., Sasaki T., et al. Autoantibodies to fibrillin 1 in systemic sclerosis: ethnic differences in antigen recognition and lack of correlation with specific clinical features or HLA alleles. Arthritis Rheum 2000; 43: 2464–71
- Zhou X., Tan F. K., Milewicz D. M., Guo X., Bona C. A., Arnett F. C. Autoantibodies to fibrillin‐1 activate normal human fibroblasts in culture through the TGF‐beta pathway to recapitulate the “scleroderma phenotype”. J Immunol 2005; 175: 4555–60
- Chizzolini C., Raschi E., Rezzonico R., Testoni C., Mallone R., Gabrielli A., et al. Autoantibodies to fibroblasts induce a proadhesive and proinflammatory fibroblast phenotype in patients with systemic sclerosis. Arthritis Rheum 2002; 46: 1602–13
- Baroni S. S., Santillo M., Bevilacqua F., Luchetti M., Spadoni T., Mancini M., et al. Stimulatory autoantibodies to the PDGF receptor in systemic sclerosis. N Engl J Med 2006; 354: 2667–76
- Fujimoto M., Fujimoto Y., Poe J. C., Jansen P. J., Lowell C. A., DeFranco A. L., et al. CD19 regulates Src family protein tyrosine kinase activation in B lymphocytes through processive amplification. Immunity 2000; 13: 47–57
- Sato S., Hasegawa M., Fujimoto M., Tedder T. F., Takehara K. Quantitative genetic variation in CD19 expression correlates with autoimmunity. J Immunol 2000; 165: 6635–43
- Matsushita T., Hasegawa M., Yanaba K., Kodera M., Takehara K., Sato S. Elevated serum BAFF levels in patients with systemic sclerosis: enhanced BAFF signaling in systemic sclerosis B lymphocytes. Arthritis Rheum 2006; 54: 192–201
- Korn J. H., Mayes M., Matucci Cerinic M., Rainisio M., Pope J., Hachulla E., et al. Digital ulcers in systemic sclerosis: prevention by treatment with bosentan, an oral endothelin receptor antagonist. Arthritis Rheum 2004; 50: 3985–93
- Williams M. H., Das C., Handler C. E., Akram M. R., Davar J., Denton C. P., et al. Systemic sclerosis associated pulmonary hypertension: improved survival in the current era. Heart 2006; 92: 926–32
- Tashkin D. P., Elashoff R., Clements P. J., Goldin J., Roth M. D., Furst D. E., et al. Cyclophosphamide versus placebo in scleroderma lung disease. N Engl J Med 2006; 354: 2655–66
- Farge D., Passweg J., van Laar J. M., Marjanovic Z., Besenthal C., Finke J., et al. Autologous stem cell transplantation in the treatment of systemic sclerosis: report from the EBMT/EULAR Registry. Ann Rheum Dis 2004; 63: 974–81
- Tyndall A., Mueller‐Ladner U., Matucci‐Cerinic M. Systemic sclerosis in Europe: first report from the EULAR Scleroderma Trials And Research (EUSTAR) group database. Ann Rheum Dis 2005; 64: 1107