Abstract
Dilated cardiomyopathy (DCM) is a myocardial disease characterized by dilatation and impaired systolic function of the left or both ventricles. The etiology of DCM is multifactorial, and many different clinical conditions can lead to the phenotype of DCM. During recent years it has become evident that genetic factors play an important role in the etiology and pathogenesis of idiopathic DCM. The genetics of DCM have been under intensive investigation lately, and thereby the knowledge on the genetic basis of DCM has increased rapidly. The genetic background of the disease seems to be relatively heterogeneous, and the disease‐associated mutations concern mostly single families and only few affected patients. Disease‐associated mutations have been detected e.g. in genes encoding sarcomere, cytoskeletal, and nuclear proteins, as well as proteins involved with regulation of Ca2+ metabolism. The mechanisms, by which mutations eventually result in clinical heart failure, are complex and not yet totally resolved. DCM causes considerable morbidity and mortality. Better knowledge of the genetic background and disease‐causing mechanisms would probably help us in focusing early treatment on right subjects and potentially also developing new treatment modalities and improving cardiac outcome in the affected patients. This review deals with DCM of genetic origin.
Introduction
Dilated cardiomyopathy (DCM) is a primary myocardial disease characterized by dilatation of the left or both ventricles and impaired systolic function Citation1. DCM causes considerable morbidity and mortality, and it is one of the major causes of sudden cardiac death. DCM can manifest itself at any age, but most commonly after middle age. DCM is clinically a very heterogeneous disease, and patients having this disease may be symptomless but may also have severe heart failure. DCM is the most common cardiomyopathy explaining about 60% of all cardiomyopathies Citation2,3. The incidence of dilated cardiomyopathy has been increasing, which is explained by more accurate diagnosis and a higher index of suspicion among clinicians Citation4. The incidence of DCM has been suggested to be 5–8 in 100,000 per year in the United States and also in European populations Citation4–7, and the prevalence approximately 40 in 100,000 in the United States Citation4,Citation8. The annual incidence of dilated cardiomyopathy in the pediatric population has been reported to be 0.57 cases per 100,000 in USA Citation9. In the Finnish pediatric population the incidence was found to be 0.34 per 100,000 per year and the prevalence 2.6 per 100,000 per year Citation10.
The frequencies of familiarity of DCM vary between different studies, but common estimation is that approximately 30%–50% of cases of idiopathic DCM has a genetic origin Citation11–14. Autosomal dominant inheritance is the most common inheritance form Citation12,Citation15,16. Although genetic factors seem to play an important role in the pathogenesis of DCM, hitherto reported mutations explain only a minority of familial DCM. The first familial cardiomegaly was characterized in 1949, but the first DCM‐associated mutation (in the dystrophin gene) was described as late as in 1993 Citation17–19. Since the beginning of 1990s, the genetics of DCM have been investigated more intensively, especially during the last 8 years. To date, DCM‐associated mutations in many different genes with subsequent alterations in protein structure have been reported, but still these mutations explain only a minority of the etiology of DCM () Citation18–49.
Figure 1. A schematic structure of the myocyte. Dilated cardiomyopathy(DCM)‐associated proteins are marked by italic font.
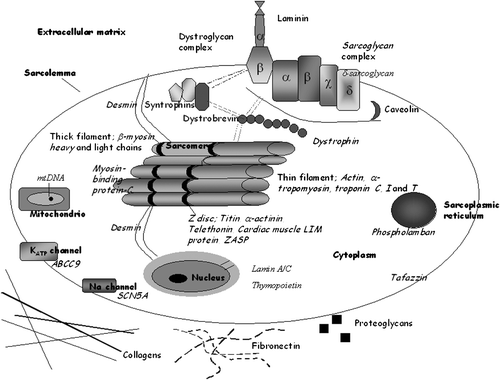
During the last years, research results have highlighted the importance of the lamin A/C gene in familial DCM. The mutations in the lamin A/C gene are one of the most frequently detected mutations in DCM populations Citation20,Citation25,Citation29,Citation32,Citation50–52. The phenotype of DCM patients is very similar in different mutations of the lamin A/C gene. On the other hand, the phenotype seems to differ somewhat from that of a ‘typical’ DCM patient, and therefore we can say that patients with mutations in the lamin A/C gene may form a special subgroup in the DCM population Citation20,Citation25,Citation29,Citation32,Citation50–52.
Although the prognosis of DCM has improved during the last decades, it still causes considerable morbidity and mortality. Identification of mutations in genes associated with this disease adds not only information on the pathogenesis of DCM, but also provides important prognostic information and helps screening other asymptomatic family members.
Monogenetic dilated cardiomyopathy
The prevalence of familial DCM varies between 10% and 50%. The lowest prevalence is reported in studies without systematic screening of family members. If family members are systematically screened, the prevalence of familiarity varies between 20% and 50% Citation12,13,Citation16,Citation44. The mode of inheritance of DCM can be autosomal dominant (∼60%), autosomal recessive (∼16%), X‐linked (∼10%) or mitochondrial (∼8%) Citation53. Also mutations without genetic inheritance can exist (de novo mutations), but these kinds of mutations are quite rare Citation54. The penetrance of DCM is variable, and it is dependent at least on gene, mutation, and age. Also modifying factors can affect the phenotype of a single patient. For example, in the lamin A/C gene the penetrance of the disease is nearly 100% in patients over 40 years.
Although genetic factors play a role in the pathogenesis of DCM, the genetic background of DCM is still largely unknown. Linkage analysis has not been very useful due to the limited size of families and difficulties in the diagnosis of DCM. However, linkage analysis has been applied, and several chromosomal loci for DCM have been found. Specific genes have not yet been identified in all loci, but some clinical characteristics are associated with known loci, e.g. skeletal muscle disease and mitral prolapse. Chromosomal loci reported to be associated with DCM are listed in .
Table I. Chromosomal loci, clinical characteristics and suggested disease‐causing mechanisms associated with DCM.
The most commonly used research method in familial DCM is the candidate gene approach. To date, gene defects have been reported in over 20 genes: cardiac actin, desmin, dystrophin, δ‐sarcoglycan, troponin T and β‐myosin heavy chain, α‐tropomyosin, tafazzin, titin, lamin A/C, metavinculin, myosin‐binding protein‐C, cardiac muscle LIM protein (named according to closely related Lin‐11, Isl‐1 and Mec‐3 genes), phospholamban, α‐actinin, troponin I, troponin C, thymopoietin, SCN5A (sodium channel, voltage gated, type V, alpha polypeptide), ZASP/cypher, and ABCC9 (ATP‐binding cassette, sub‐family C (CFTR/MRP), member 9) genes Citation20,21,Citation23,Citation27,28,Citation30,Citation33–38,Citation40–42,Citation45–49,Citation55,56. These genes can be classified according to their locations or disease‐causing mechanisms. Classification according to disease‐causing mechanisms is presented in .
Figure 2. A schematic figure of the Dilated cardiomyopathy(DCM)‐causing mechanisms.
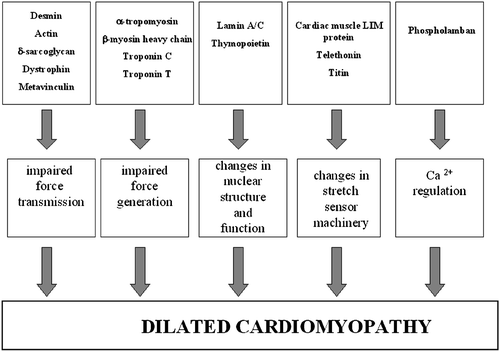
Genes associated with dilated cardiomyopathy
Sarcomere protein and sarcomere‐associated protein genes ( and )
Cardiac actin
The cardiac actin gene (ACTC) on chromosome 15q14 has six exons. Actin is the major component of the thin filament, and it forms a complex with α‐tropomyosin and troponins (troponin C, I and T), essential components of the contraction unit of heart muscle. The cardiac actin gene was the first sarcomere protein gene reported to be associated with DCM. Olson et al. Citation36 found two missense mutations, Arg312His and Glu361Gly, in the ACTC gene in two DCM families. These mutations are located in the region that encodes the immobilized end of the actin filament. This region is near the dystrophin‐binding site, which binds the protein domain to Z bands and intercalated discs Citation44,Citation56,57. These mutations are suggested to affect force transmission to adjacent sarcomeres and myocytes. Gene defects in the cardiac actin gene, which affect force generation, have not been reported since. The mechanisms by which the mutations lead to clinical disease have remained unclear. Overall, the mutations in the cardiac actin gene are thought to be a rare cause of DCM, and therefore it is difficult to describe the typical phenotype of DCM caused by a mutation in the ACTC gene Citation58–60.
α‐Tropomyosin
The α‐tropomyosin gene (TPM1) on chromosome 15q22 has ten exons in the cardiac isoform of the gene. α‐Tropomyosin is a component of the thin filament, and it interacts with troponin T. This interaction is needed in heart muscle contraction. The interaction can be Ca2+‐sensitive or not. Only two DCM‐associated mutations have been reported in the TPM1 gene. Olson et al. Citation37 described two mutations, Glu40Lys and Glu54Lys, in two small DCM families. These two mutations in the TPM1 gene are thought to alter electrostatic interactions between actin and tropomyosin or compromise the structural integrity of tropomyosin Citation37. The family with the Glu40Lys mutation had a variable phenotype. One family member had severe disease and underwent heart transplantation at the age of 10, whereas one family member developed a mild DCM phenotype at the age of 40. The Glu54Lys mutation has been associated with sudden cardiac death at young ages. Other DCM‐associated mutations in the TPM1 gene have not been published yet, and therefore TPM1 gene defects seem to be relatively uncommon in DCM.
β‐Myosin heavy chain
The β‐myosin heavy chain gene (MYH7) on chromosome 14q11 has 38 coding exons. The β‐myosin heavy chain forms part of the thick filament of the sarcomere. The β‐myosin heavy chain protein has two different domains, a heavy meromyosin domain, which consists of two subfragments (S1 and S2) and a light meromyosin (LMM) domain Citation61. The S1 locates in the head domain and has actin and ATP‐binding regions, and it is therefore essential for generation of movement needed in contraction Citation61. The S2 joins the heads at the neck region. The LMM portion forms binding sites for myosin‐binding protein‐C and titin and is essential for maintaining assembly of the filament and stability of the protein Citation61.
Several DCM‐causing mutations have been detected in the MYH7 gene. Kamisago et al. Citation25 found two mutations, Ser542Pro and Phe764Leu, in the MYH7 gene in two different DCM families. The Ser542Pro mutation was associated with sudden death and heart transplantation at young ages, while the Phe764Leu mutation manifested at birth or at the age of 2 months. These mutations are thought to disrupt interactions between myosin and actin or diminish the efficiency of contraction, respectively Citation30. Daehmlow et al. Citation23 reported two mutations, Ala223Thr and Ser642Leu, in two sporadic DCM patients. The Ala223Thr mutation is thought to affect thermostability and protein folding, and the Ser642Leu mutation in turn conformational structure of protein near the actin‐binding site Citation23. In our own studies we detected two novel mutations in this same gene Citation62. One of the mutations, Arg1053Gln, caused mainly hypertrophic cardiomyopathy (HCM), and only one patient had a phenotype resembling DCM. The Arg1500Trp mutation was found only in one patient with classical DCM. Villard et al. Citation63 screened a large number of familial and sporadic DCM patients and detected eight DCM‐associated mutations. In their study population mutations in the MYH7 gene explained approximately 10% of the cases. In other populations, however, mutation frequencies of MYH7 gene have not been so high.
Troponin T, I and C
The troponin T gene (TNNT2) on chromosome 1q32 has 17 exons. The troponin T forms the troponin complex with troponin C and I, and they are all part of the thin filament. Troponin T has interaction sites with tropomyosin and other troponins. Several DCM‐causing mutations have been reported in this gene. Kamisago et al. Citation30 found one deletion, ΔLys210, in two DCM families. Later Hanson et al. Citation28 detected this same deletion in another DCM family. This deletion reduces ionic interactions between troponin C and troponin T and decreases the activation of calcium‐stimulated actomyosin‐ATPase. These changes are thought to lead to a decrease in power stroke Citation30. Morimoto et al. Citation62 explored the effect of the ΔLys210 mutation under physiological conditions in cardiac muscle fibers of the rabbit. The ΔLys210 mutation was found to decrease Ca2+‐sensitivity of force generation in the sarcomere, which could be one of the primary mechanisms for the pathogenesis of DCM. Robinson et al. Citation64 compared the changes in thin filament function caused by the mutations in DCM and HCM. The DCM‐associated mutation, ΔLys210, decreases ATPase activity, which in turn speeds filament sliding, and Ca2+‐activation becomes non‐cooperative Citation64. One single mutation, Arg141Trp in the troponin T gene, has been found in one DCM family Citation35. The mutation Arg141Trp is located within the tropomyosin‐binding domain of cardiac troponin T and alters the charge of the residue. Mogensen and co‐workers reported about three new DCM‐associated mutations (Arg113Trp, Arg205Leu, and Asp270Asn) in troponin T Citation46. Stefanelli et al. Citation65 found one novel mutation, Ala172Ser, which caused a severe phenotype with sudden cardiac death and low ejection fraction. The previously described ΔLys210 deletion is associated with sudden cardiac death, heart failure, and conduction system disease.
The troponin I gene (TNNI3) on chromosome 19q13 has eight exons. Troponin I is one part of the troponin complex. The role of troponin I is to control heart muscle contraction by inhibiting actomyosin ATPase and revealing active sites of tropomyosin. Murphy et al. Citation42 found the first recessive DCM‐associated mutation, Ala2Val, in this gene. This mutation was shown to impair the interaction between troponin I and troponin T, leading to diminished myocardial contraction. The phenotype of the Ala2Val mutation seemed to be severe.
The troponin C gene (TNNC1) on chromosome 3p21 has six exons. Troponin C forms the last component of the troponin complex. Its function is to take up and bind calcium‐ion to the troponin complex leading to structural changes, which allow the interaction of actin with myosin. The actin connection with myosin is necessary for heart muscle contraction. Mogensen et al. Citation46 reported one DCM‐associated mutation in the troponin C gene with a severe clinical disease.
Myosin‐binding protein‐C
The myosin‐binding protein‐C gene (MYBPC‐3) on chromosome 11p11 has 34 exons. The protein of the MYBPC‐3 gene has different regions and one cardiac‐specific region. There are at least two interacting domains with other proteins e.g. myosin and titin Citation66. Only one DCM‐associated mutation, Asp948Thr, in one sporadic DCM patient has been reported for this gene Citation23. The specific effect of this mutation is not known, and because only one patient with the mutation has been reported, the clinical phenotype remains unclear.
Titin and telethonin
The titin gene (TTN) on chromosome 2q24 has 362 exons. The main components of the sarcomeric cytoskeleton, which provides a scaffold for the sarcomere, are titin, α‐actinin, and myomesin Citation67. Titin, also called connectin, is a giant protein, which spans from the Z line to the M line in the sarcomere. Titin has several functions including the control of thin filament assembly, force transmission, maintenance of resting tension, and elasticity of the muscle Citation68–70. Titin also binds myosin filaments into the Z discs and interacts with myosin‐binding protein‐C, α‐actinin, and telethonin Citation68,Citation70. Gerull et al. Citation27 found two DCM‐associated mutations, a 2‐bp insertion and Trp930Arg, in the TTN gene. The 2‐bp insertion is predicted to truncate the protein, and the Trp930Arg mutation affects the Z disc/I band transition zone Citation27. Itoh‐Satoh et al. Citation68 reported four DCM‐associated mutations in the titin gene. Two of these mutations, Val54Met and Ala743Val, are located in the Z line region of titin. These mutations are thought to decrease binding affinities of titin to Z line proteins. Two other mutations, Glu4053ter and Ser4465Asp, are located in the cardiac‐specific N2‐B domain of titin. The Glu4053ter mutation is thought to encode truncated nonfunctional protein. The main clinical findings in patients with titin mutations are DCM with arrhythmias or conduction system abnormalities and need for implantable cardioverter‐defibrillator (ICD). Recently, Gerull et al. Citation71 reported a novel heterozygous 1‐bp deletion mutation (c.62890delG) in TTN that cosegregated with DCM in a large Australian pedigree. The TTN deletion caused a frameshift, thereby generating a truncated A band titin due to a premature stop codon and the addition of ten novel amino acid residues. In this case the penetrance was incomplete and expressivity was variable Citation71.
The telethonin gene (TCAP) on chromosome 17q12 probably has two exons. Telethonin is actually a substrate of titin. Interaction of titin with telethonin at the Z disc is necessary for proper function of the sarcomere. Telethonin has a role in the assembly of the sarcomere by providing spatially defined binding sites for other sarcomeric proteins. After activation by phosphorylation and calcium/calmodulin binding, titin phosphorylates the C‐terminal domain of telethonin Citation72. Knöll et al. Citation33 recently reported one mutation, Arg87Gln, in the TCAP gene in one DCM patient. The mutated telethonin gene leads to a defect in the interaction between cardiac muscle LIM protein and telethonin. Hayashi et al. Citation73 described one TCAP mutation, E132Q, in a DCM patient. This mutation is suggested to impair the interaction of TCAP with titin, muscle LIM protein, and calsarcin‐1.
The clinical disease in patients with telethonin mutations seems to be quite severe. Sudden cardiac death and early onset of the disease were clinical findings in one family with E132Q mutation Citation73.
Cytoskeletal protein genes ( and )
Desmin
The desmin gene (DES) on chromosome 2q35 has nine exons. Desmin belongs to the intermediate filaments and has typical structural components of intermediate filaments; a central rod, head, and tail domains. Desmin encircles the Z discs and radiates out to connect adjacent myofibrils. It also forms a link between actin and the dystrophin‐sarcoglycan complex Citation74. Desmin attaches and stabilizes the sarcomere, connects adjacent myofibrils together and Z bands to plasma membrane and nuclear envelope. In the desmin gene, one DCM‐associated mutation, Ile451Met, has been reported Citation34,Citation75. The Ile451Met mutation is located in a region of the gene encoding the tail domain of the protein, the specific function of which is unknown. It has been speculated that DCM‐causing mechanism in the desmin gene is related to ineffective force transmission Citation34,Citation76.
Clinically, the Ile451Met mutation has been shown to be associated with a classical type of DCM and sudden death. Gene defects in the desmin gene have also been shown to cause skeletal muscle disease combined with cardiomyopathy and desmin‐related myopathy, which is caused by abnormal accumulation of desmin in skeletal muscle Citation77–79. Generally, mutations in the desmin gene are thought to be rare causes for DCM, at most accounting for about 2% of the cases Citation60,Citation75.
Dystrophin
The dystrophin gene (DMD) on chromosome Xp21 has 79 exons. Dystrophin is a large cytoskeletal protein, which is part of the dystrophin‐associated glycoprotein complex (DAG). The DAG complex includes dystroglycan, sarcoglycan, syntrophin complex and caveolin. The DAG complex forms a transmembrane link between the extracellular matrix and the intracellular cytoskeleton Citation80. Dystrophin interacts with actin and thereby links the sarcomere to the extracellular space. Dystrophin is thought to play a role in force transduction, membrane stability, and intracellular organization. Badorff et al. Citation81 showed that an enterovirus infection can lead to cleavage of dystrophin. This leads to disruption of the cytoskeleton resulting in myocyte dysfunction, decreased force transmission, and increased cell permeability.
Dystrophin mutations are better known to cause skeletal muscle disease with elevated levels of creatine kinase, such as Duchenne and Becker muscular dystrophies, than dilated cardiomyopathy Citation82,83. In a few cases, however, gene defects in the dystrophin gene have been shown to be associated with isolated cardiomyopathy without skeletal muscle disease. This disease is called X‐linked dilated cardiomyopathy (XLCM) Citation84. XLCM often presents in young male patients with heart failure and no signs of skeletal muscle disease. XLCM mutations in the dystrophin gene can be divided into two groups according to localization, 5′end region mutations and midrod domain region mutations Citation85. The majority of the DCM‐associated mutations are located in the 5′end region and are malignant in nature Citation18,19,Citation39, Citation86–89. Patients with malignant disease have suffered from sudden cardiac death and heart transplantation. Several mutations, mainly deletions, have been reported in the midrod domain of the protein, and usually these mutations cause a more benign disease Citation85,Citation87,88,Citation90,91.
Metavinculin
The metavinculin gene (VCL) on chromosome 10q22 has 22 exons. Metavinculin is an isoform of vinculin, encoded by an additional insert of genome (68 amino acids) in the C‐terminal end. Metavinculin belongs to the group of membrane‐associated proteins, which are located in the intercalated discs, costameres and the T tubule system Citation92,93. They are thought to have a role in the anchoring of thin filaments, force transmission, and stability of the cellular membrane, but their specific function in the heart is unknown Citation38,Citation66,Citation94. Maeda et al. Citation95 demonstrated an association between metavinculin deficiency and dilated cardiomyopathy due to a defect in alternative mRNA splicing. Olson et al. Citation38 have found one missense mutation, Arg975Trp and one 3‐bp deletion (Leu954del), in the metavinculin gene in two DCM patients. These authors suggested that the mechanism probably causing DCM was the disruption of force transmission at the thin filament‐intercalated disc interface. Later, Vasile et al. Citation96 detected the same mutation in a patient with hypertrophic cardiomyopathy. This finding highlights the effect of remodeling in the pathophysiology of cardiomyopathies.
The clinical findings in patients with metavinculin mutations are variable. Part of the mutation carriers had well preserved left ventricular diameter and ejection fraction, but one patient presented with rapid progression of heart failure and needed heart transplantation after some years of diagnosis Citation38.
δ‐Sarcoglycan
The δ‐sarcoglycan gene (SGCD) on chromosome 5q33 has eight exons. δ‐Sarcoglycan has three domains: intracellular, transmembrane, and extracellular domains Citation97. Tsubata et al. Citation56 detected two DCM‐causing mutations, Ser151Ala and ΔLys238, in the SGCD gene in single DCM cases. Both mutations are thought to alter the secondary structure of the protein, and the DCM‐causing mechanism may be a defect in force transmission. The DCM‐associated mutations are located in the extracellular domain. Gene defects in the SGCD gene can also lead to limb girdle muscular dystrophy, which is sometimes associated with DCM Citation98. In animal studies, Nigro et al. Citation99 showed that a deletion of the first exon caused a complete absence of the δ‐sarcoglycan protein in the Syrian hamster. This deletion was associated with skeletal muscle disease, ventricular hypertrophy and dilatation, and premature death. In our own study we found one mutation, Arg71Thr, in a small family with DCM, which was, however, associated with a relatively mild phenotype Citation31.
Cardiac muscle LIM protein
The cardiac muscle LIM protein gene (CLP) on chromosome 11p15 has four exons. The CLP protein has two LIM domains, and these domains are highly conserved Citation66. The CLP protein is located in the Z disc, and interacts with α‐actinin and telethonin, and telethonin in turn interacts with titin Citation33. CLP together with telethonin is the key component of the cardiomyocyte stretch sensor machinery Citation33. CLP is also localized in the nucleus, and its function there is to promote myogenesis Citation100. Knöll et al. Citation33 found one DCM‐associated mutation, Trp4Arg, in the CLP gene in ten European patients. The Trp4Arg mutation results in a defect in interaction with telethonin interfering with the cardiac mechanical stretch sensor machinery Citation33. Clinically the Tpr4Arg mutation causes typical DCM with low ejection fraction, but only slightly dilated left ventricle. Mohapatra et al. Citation41 found this same mutation in an European family, but the mutation did not cosegregate with DCM. Therefore, it is possible that the substitution of tryptophan to arginine (W4R) mutation is a modifier mutation rather than a disease‐causing mutation. Mohapatra et al. Citation41 detected another DCM‐associated mutation, Lys69Arg, in the CLP gene. The Lys69Arg mutation abolishes the interaction of telethonin with CLP and α‐actinin and changes the localization of CLP. It is associated with early onset of DCM.
α‐Actinin
The α‐actinin gene (ACTN2) on chromosome 1q42 has 21 exons. α‐Actinin is localized in the Z disc, where it helps to anchor myofibrillar actin filaments. It consists of an N‐terminal actin binding domain, a central rod domain, and a C‐terminal domain, and it functions as a homodimer to cross‐link actin filaments. The rod domain determines the distance between cross‐linked actin filaments and also serves as an interaction site for several cytoskeletal and signaling proteins Citation101. Recently, Mohapatra et al. Citation41 detected one mutation, Gln9Arg, in the α‐actinin gene in one DCM patient with severe disease. The Gln9Arg mutation was predicted to alter the secondary structure of the protein. DCM‐associated mutations in this gene are rare, and therefore clinical characteristics of these gene mutations are difficult to summarize.
Cypher/ZASP
The cypher gene on chromosome 10q22 has 16 exons. It is a cardiac and skeletal muscle‐specific Z line protein, which is expressed in cytoplasm. The proteins in the N‐terminal end interact with each other in cytoskeletal assembly or with other proteins involved in targeting and clustering of membrane proteins. The cypher/ZASP plays a role in bridging the sarcomere to the cytoskeletal network. The interaction site with α‐actinin‐2 has been found Citation48. Vatta et al. reported five mutations (Ser196Leu, Ile352Met, Asp117Asn, Lys136Met, and Thr213Ile) in this gene Citation48. Two of the mutations were found in familial cases, and three were sporadic ones. The authors suggest that the mutated gene could affect the stability of actin cytoskeletal network Citation48. The cypher/ZASP knockout mouse developed congenital myopathy and dilated cardiomyopathy Citation102. Later, Arimura et al. Citation103 found an Asp626Asn mutation in the Japanese population. This mutation was shown to increase affinity of the LIM domain for protein kinase C.
Clinical characteristics for these mutations were dilated cardiomyopathy with severe left ventricular hypertrophy. Some patients had also conduction system disease, but no signs of skeletal muscle disease were found.
Nuclear protein genes ( and )
Lamin A/C gene
The lamin A/C gene (LMNA) on chromosome 1p1 contains 12 exons. Lamin A and C have an identical sequence for the first 566 amino acids in exons 1–10, but differ in the 3′ end of the gene. The nuclear envelope is a double‐layered structure (outer and inner layer) located between the nucleus and cytoplasm. The outer layer of the envelope is directly connected to the endoplasmic reticulum, and ribosomes are attached to it. The nuclear lamina, which is composed of lamins A, B and C, is attached to the inner layer of the nuclear envelope. Lamins connect the nuclear envelope, nuclear matrix and chromatins together. Lamin A/C is also thought to have a role in cell dividing, nuclear growth, and anchorage of nuclear envelope proteins Citation104–107. Lamins are major structural components of the lamina network underlying and supporting mechanically the nuclear envelope Citation107. The lamin A/C protein has three different domains, an N‐terminal head, a coiled‐coil rod, a C‐terminal tail, and several different binding domains, for example lamin B, chromatin, emerin, and lamin‐associated protein binding domains Citation106,Citation108.
Defects in the lamin A/C gene have been shown to be responsible for different diseases e.g. Emery‐Dreifuss muscular dystrophy, limb girdle muscular dystrophy type 1B, Dunnigan‐type familial partial lipodystrophy, Charcot‐Marie‐Tooth disease, mandibuloacral dysplasia, Hutchinson‐Gilford progeria, and dilated cardiomyopathy with conduction abnormalities or mild skeletal muscle disease Citation20,Citation22,Citation25,Citation109–114. Mutations in the lamin A/C gene seem to be the most frequent causes of familial DCM reported so far. During the past 2 years several independent research groups have reported different or similar DCM‐associated mutations in this gene.
It has been speculated that gene defects in the lamin A/C gene could disrupt nuclear function or decompose nuclear structure Citation66. Arbustini et al. Citation20 showed that nuclear membrane damage is associated with gene defects in the lamin A/C gene. It is believed that nuclear membrane damage can lead to disruption of the cell causing myocyte death and tissue damage Citation20,Citation108. Verga et al. Citation52 showed that lamin A/C mutations can cause loss of integrity of myocyte nuclei with blebs of the nuclear membrane, herniations and delamination of the nuclear lamina, and nuclear pore clustering. The authors suggest that gene defects in the lamin A/C gene are associated with loss of protein expression in the selective compartment of noncycling myocyte nuclei. Recently, Nikolova et al. Citation115 reported lamin A/C‐deficient mice, which developed rapidly progressive DCM. Lamin A/C deficiency caused detachment of desmin at the nuclear‐cytoskeletal interface, and lamin A/C deficiency is expected to impair binding between lamin B and desmin Citation115. All known DCM‐associated mutations seem to cause abnormalities in either nuclear structure or function.
It has been estimated that approximately 5% of the DCM population carry the lamin A/C mutation Citation116,117. In our own studies we found that 9% of patients with heart transplantation carried some mutation in the lamin A/C gene Citation54. The phenotype caused by mutations in the lamin A/C gene forms its own subgroup of DCM. Typical clinical findings are progressive conduction system abnormalities, atrial fibrillation, need for permanent pacemakers or automatic implantable cardioverter‐defibrillator (AICDs), ventricular tachycardias, need for heart transplantation and elevated risk for sudden cardiac death Citation32,Citation43,Citation116. These findings can exist with or without skeletal muscle disease. Malignant arrhythmias and need for heart transplantation and ICDs are more common in lamin A/C mutation patients than in other DCM patients Citation32,Citation43,Citation116. Survival is also worse in lamin A/C mutation carriers.
The penetrance of the disease is age‐dependent in the cases of lamin A/C mutation. Almost all patients over 40 years have some kind of sign of the disease, but younger patients might only have first‐degree atrioventricular block. Dilatation of the left ventricle is a later manifestation of the disease, and the left ventricle overall can be only slightly dilated. To our understanding the current diagnostic criteria are not necessarily suitable for these patients.
Thymopoietin
Thymopoietin, which is also called lamina‐associated polypeptide 2 (LAP2), on chromosome 12, has eight exons. Different isoforms due to alternative splicing are localized in the nuclear membrane and nucleoplasm Citation118,119. Lamina‐associated proteins are one group of lamin‐binding proteins, and thereby their function is to participate in maintaining nuclear integrity and nuclear functions. The only nucleoplasmic isoform is LAP2α, and it interacts with A‐type lamins Citation120. Other domains of the LAP2α are the N‐terminal domain containing LEM‐domain (LAP2‐emerin‐MAN1), chromosome binding domain, and unique C terminus, which has a critical role during nuclear reassembly Citation114. Taylor et al. Citation47 screened the LAP2 gene and detected one putative mutation, Arg690Cys, in one familial DCM pedigree. This mutation altered interaction with LAP2α and lamin A, but the final consequences remain unknown. However, this finding supports the importance of disturbed nuclear functions in the pathogenesis of DCM. Some of the patients with mutation in the thymopoietin gene have severe disease without signs of skeletal muscle disease Citation47.
Ion channel protein genes or genes regulating Ca2+ metabolism
SCN5A
The SCN5A gene on chromosome 3 has 28 exons. This cardiac sodium channel gene contains four homologous domains, each of which has six putative membrane‐spanning regions. The defects in this gene are better known to cause Brugada syndrome and long QT syndrome Citation121. McNair et al. Citation45 detected one mutation (Asp1275Asn) in a family with DCM. The clinical characteristics for this mutation were atrial fibrillation, conduction abnormalities, and early onset of the disease. The disease‐causing mechanism is different compared with other disease‐causing mechanisms in DCM, i.e. altered ion homeostasis Citation45.
ABCC9
The ABCC9 gene on chromosome 12 has 39 exons. This gene is a member of the superfamily of ATP‐binding cassette (ABC) transporters. ABC proteins transport various molecules across extra‐ and intra‐cellular membranes. Bienengraeber et al. Citation49 identified two heterozygous mutations in the ABCC9 gene, which encodes the C‐terminal domain of SUR2A specific to the cardiac splice variant of the regulatory K(ATP) channel subunit. The authors suggest that these defects in the regulatory K(ATP) channel subunit disrupt catalysis‐dependent gating and impair metabolic decoding, establishing a hitherto unrecognized mechanism of channel malfunction in human disease.
Patients with mutations in the ABCC9 gene have severe DCM with ventricular tachycardia as a common feature. The mutations identified resulted in dysfunction of the SUR2A subunit by disruption of the C terminus Citation49.
Phospholamban
The phospholamban gene on chromosome 6q22 has two exons. Phospholamban has an essential role in Ca2+ metabolism by modulating calcium‐ATPase activity. Thereby phospholamban is a regulator of cardiac relaxation. Schmitt et al. Citation40 detected one DCM‐causing mutation, Arg9Cys, in the phospholamban gene in one DCM family. This Arg9Cys mutation is thought to cause DCM by directly disturbing the myocellular Ca2+ metabolism due to constitutive SERCA2a (sarcoplasmic reticulum calcium‐ATPase) inhibition. The authors also developed a transgenic mouse model with this mutation, which appeared to have a similar type of disease as mutation carriers (heart failure with premature death). Family members with the Arg9Cys mutation had severe disease with frequent heart transplantations and premature deaths.
Unclassified group
G4.5/Tafazzins
The G4.5 gene on chromosome Xq28 encodes tafazzins. G4.5 is expressed in cardiac and skeletal muscle. Tafazzins do not have known similarities to other proteins, and therefore tafazzins compose their own group of proteins. There are two important regions: the N terminus part of the gene may serve as a membrane anchor, and the central portion may serve as an exposed loop interacting with other proteins. Mutations in this gene are known to cause a phenotype of DCM, Barth syndrome, noncompaction of left ventricular myocardium, and endocardial fibroelastosis Citation21,Citation24.
Mitochondrial mutations
The mitochondrial genome is a double‐stranded circular molecule. The mitochondrial genome has some specific features compared with genomic DNA. Mitochondria do not have introns and histones, and they lack an effective DNA repair system. The mutation rate is higher in mtDNA than in nuclear DNA, partially due to ineffective DNA repair and abundant free radical formation Citation122. Maternal inheritance is typical for mitochondria, but inheritance according to Mendelian laws can also occur. Mitochondria play an important role in energy generation and thus in contractile function, and disturbances in these functions may lead to DCM in some cases. Gene defects in the mitochondrial genome can cause syndromes like MELAS (mitochondrial encephalomyopathy, lactic acidosis and stroke‐like episodes), Kearns‐Sayre syndrome and MERFF (myoclonus epilepsy with ragged red fibers), which are caused by point mutations and deletions in the mtDNA. Different cardiac abnormalities are accompanied with these syndromes, especially HCM. Few cases of DCM have been detected in patients with MELAS and Kearns‐Sayre syndrome Citation123–129.
Genetic defects in Finnish population of dilated cardiomyopathy
We have examined clinically and screened genetically 52 DCM patients from Kuopio University Hospital region and 38 DCM patients from Helsinki University Hospital region. An additional study group consisted of heart transplantation patients (n = 66) from all over Finland. The genetic analyses were performed during the last 7 years. We screened all coding regions of nine candidate genes (cardiac actin, lamin A/C, troponin C, I, T, α‐tropomyosin, β‐myosin heavy chain, desmin, δ‐sarcoglycan) and a part of the tenth gene (metavinculin) in the above Finnish patient groups with DCM. The screened number of patients for different candidate genes varied between 32 and 156. Nine disease‐associated mutations in three different genes were detected in the analyses. Altogether these mutations accounted for 9.6% of the etiology of the 156 DCM patients. The Ser143Pro mutation in the lamin A/C gene was found to be quite common in Finnish patients with familial DCM. Haplotype analysis strongly suggested that the Ser143Pro mutation is a founder mutation, probably the first founder mutation described for DCM Citation32. Five other LMNA mutations (Ala132Pro, Arg190Trp, T1085 deletion, G1493 deletion, and Arg541Ser) were detected in patients with heart transplantation Citation54. T1085 deletion was found in two unrelated families.
All LMNA mutation carriers have a very similar phenotype. The main characteristic features are progressive conduction defect (66%), early need for a pacemaker, slow atrial fibrillation, sudden cardiac death, and overt heart failure leading to heart transplantation in some cases. All mutation carriers over 40 years had some manifestation of DCM, and only the young mutation carriers under 30 years were free from clinical disease. The penetrance of the disease in our patient population is almost 100% in subjects over 40 years. The LMNA mutations overall, and especially the Ser143Pro mutation, can be classified as a relatively malignant mutation, because several sudden cardiac deaths occurred in the families carrying the mutation. The index patients with the Ser143Pro mutation had a more severe prognosis than noncarriers Citation32. Also, heart transplantations are frequent among LMNA mutation carriers.
Other mutations (Arg1053Gln and Arg1500Trp in the β‐myosin heavy chain gene, and Arg71Thr in the δ‐sarcoglycan gene) seem to be less common in Finnish DCM patients Citation31,Citation62, the prevalence being quite low, approximately 1% in our studies.
Genetic screening in dilated cardiomyopathy and its clinical significance
Dilated cardiomyopathy is known to be heterogeneous in etiology, clinical features, and prognosis, and it seems that genetics of this disease do not make an exception to the rule. So far, over 20 disease‐causing genes have been reported, and most of these mutations explain the disease only in single families or single patients. The disease‐causing mechanisms vary as well, and they are far from resolved yet. There are some mechanisms that have been proposed to cause the clinical disease of DCM. Sarcomere and cytoskeletal protein genes are generally thought to affect force generation or force transmission, and mitochondrial mutations in turn are assumed to affect energy production Citation76. DCM‐associated mutation in the phospholamban gene probably disturbs the myocellular Ca2+ metabolism Citation40. Defects in the cardiac muscle LIM protein and telethonin complex in Z disc have been proposed to affect the cardiomyocyte stretch sensory machinery Citation33. Defects in the nuclear protein gene, such as lamin A/C and thymopoietin, cause disturbances in nuclear structure and function Citation20,Citation43,Citation52.
Although 50% of patients with idiopathic DCM may have a genetic disease, a specific mutation can be diagnosed only in a small proportion of patients and families at the moment. Our view is, however, that in the near future more DCM‐associated gene defects will be reported in the literature, and at the same time the heterogeneity of the disease genetics will increase. For almost all of the gene defects identified so far, the population carrying the mutation is quite small, and therefore an exact description of gene‐specific clinical phenotypes is difficult to give. Furthermore, at the moment we do not have good prognostic factors for specific gene defects. Several different clinical characteristics have been shown to correlate with poor prognosis in idiopathic DCM, but these characteristics are quite common in congestive heart failure in general Citation130–147.
The lamin A/C gene, however, seems to be an exemption in the rule. Mutations in the lamin A/C gene are probably the most frequent gene defects reported to cause DCM. Moreover, lamin A/C mutations are usually associated with a severe phenotype, which also differs somewhat from the classical or typical phenotype of DCM fulfilling the current international diagnostic criteria Citation148. The clinical manifestations of DCM caused by the mutations in the lamin A/C gene are relatively well described. The main characteristic features are slow atrial fibrillation, some degree of atrioventricular conduction system disorder, need for a permanent pacemaker, only slight dilatation of left ventricle, and an elevated risk for sudden cardiac death Citation32,Citation43,Citation148, as already described earlier in this review. Overall, in our experience the generally accepted diagnostic criteria for DCM might not be applicable for all patients with mutations of the lamin A/C gene, and at least with a positive family history a strong suspicion for a lamin A/C mutation should arise also in the presence of mild to moderate alterations in electrocardiogram (ECG) (conduction defects) or echocardiography (mild dilatation or moderate systolic dysfunction).
In families with two or more patients with DCM, clinical examination, ECG, and echocardiography of the first‐degree relatives seem warranted. In cases of even mild alterations in the above investigations, proper follow‐up should be organized. With clinical features resembling phenotypes typical of lamin A/C mutations, genetic screening for these mutations, whenever possible, should be performed. Genetic screening for lamin A/C mutations is important, because these gene defects are relatively malignant in nature. Mutation carriers should be directed to proper genetic counseling, follow‐up, and treatment including medication, automatic defibrillators, and eventually cardiac transplantation. On the other hand, with genetic screening noncarriers in the family can be liberated from follow‐up. So far, other gene defects associated with DCM seem to be too rare to arrange genetic screening procedures. However, more research is needed to identify novel gene defects in DCM, and on the other hand, to identify patients and families carrying the hitherto reported gene defects. Larger populations with known mutations should be better characterized clinically to allow genotype‐specific phenotyping and prognostic evaluation.
Vatta et al. reported that disruption of integrity of dystrophin may be reversed in patients with cardiomyopathy, provided that patients are treated with left ventricular assist device (LVAD), suggesting that gene defect‐specific treatments may be identified in the future Citation149. On the other hand, it may be that specific polymorphisms, such as angiotensin converting enzyme (ACE) polymorphism and β‐adrenoreceptor polymorphisms as modifier genes may affect the natural course and prognosis in DCM patients with various specific disease‐causing gene defects Citation150,151. McNamara et al. Citation152 reported that higher doses of ACE inhibitors diminished the impact of the ACE D allele, and the benefits of beta‐blockers and high‐dose ACE‐inhibitors appeared maximal for DD patients. Thus in the future, the genotype may also affect the medication chosen for patients with heart failure Citation152,153.
In Finland there is a plan to organize uniform diagnostics, follow‐up, and treatment of all idiopathic DCM patients. According to the plan, we are going to identify one or two cardiologists responsible for cardiomyopathy patients (both DCM and HCM) in bigger hospitals covering the whole country. In clear family cases, first‐degree relatives should be investigated, and blood samples for genetic screening (at least for detection the common founder mutation Ser143Pro) should be taken. The genetic screening in Finland is centralized in the gene laboratory of the Department of Internal Medicine, Kuopio University, the most experienced center for genetic screening of cardiomyopathies.
References
- Richardson P., McKenna W., Bristow M., Maisch B., Mautner B., O'Connell J., et al. Report of the 1995 World Health Organization/International Society and Federation of Cardiology Task Force on the Definition and Classification of cardiomyopathies. Circulation 1996; 93: 841–2
- Abelmann W. H. Doxorubicin cardiomyopathy. Hosp Pract 1982; 17: 17–8
- Valantine H. A., Hunt S. A., Fowler M. B., Billingham M. E., Schroeder J. S. Frequency of familial nature of dilated cardiomyopathy and usefulness of cardiac transplantation in this subset. Am J Cardiol 1989; 63: 959–63
- Codd M. B., Sugrue D. D., Gersh B. J., Melton L. J., 3rd. Epidemiology of idiopathic dilated and hypertrophic cardiomyopathy. A population‐based study in Olmsted County, Minnesota, 1975–1984. Circulation 1989; 80: 564–72
- Williams D., Olsen E. G. Prevalence of overt dilated cardiomyopathy in two regions of England. Br Heart J 1985; 54: 153–55
- Bagger J., Baandrup U., Rasmussen K., Moller M., Vesterlund T. Cardiomyopathy in western Denmark. Br Heart J 1984; 52: 327–31
- Gillum R. Idiopathic dilated cardiomyopathy. Epidemiolgy 1994; 5: 383–5
- Dec G. W., Fuster V. Idiopathic dilated cardiomyopathy. N Engl J Med 1994; 331: 1564–75
- Towbin J., Lowe A., Colan S., Sleeper L., Orav E., Clunie S., et al. Incidence, causes, and outcomes of dilated cardiomyopathy in children. JAMA 2006; 296: 1867–76
- Arola A., Jokinen E., Ruuskanen O., Saraste M., Pesonen E., Kuusela A., et al. Epidemiology of idiopathic cardiomyopathies in children and adolescents. A nationwide study in Finland. Am J Epidemiol 1997; 146: 385–93
- Gregori D., Rocco C., Miocic S., Mestroni L. Estimating th efrequency of familial dilated cardiomyopathy in the presence of misclassification errors. J Appl Stat 2001; 28: 53–62
- Grunig E., Tasman J. A., Kucherer H., Franz W., Kubler W., Katus H. A. Frequency and phenotypes of familial dilated cardiomyopathy. J Am Coll Cardiol 1998; 31: 186–94
- Keeling P. J., Gang Y., Smith G., Seo H., Bent S. E., Murday V., et al. Familial dilated cardiomyopathy in the United Kingdom. Br Heart J 1995; 73: 417–21
- Baig M. K., Goldman J. H., Caforio A. L., Coonar A. S., Keeling P. J., McKenna W. J. Familial dilated cardiomyopathy: cardiac abnormalities are common in asymptomatic relatives and may represent early disease. J Am Coll Cardiol 1998; 31: 195–201
- Mestroni L., Rocco C., Gregori D., Sinagra G., Di Lenarda A., Miocic S., et al. Familial dilated cardiomyopathy: evidence for genetic and phenotypic heterogeneity. Heart Muscle Disease Study Group. J Am Coll Cardiol 1999; 34: 181–90
- Michels V. V., Moll P. P., Miller F. A., Tajik A. J., Chu J. S., Driscoll D. J., et al. The frequency of familial dilated cardiomyopathy in a series of patients with idiopathic dilated cardiomyopathy. N Engl J Med 1992; 326: 77–82
- Evans W. Familial cardiomegaly. Br Heart J 1949; 11: 68–82
- Muntoni F., Cau M., Ganau A., Congiu R., Arvedi G., Mateddu A., et al. Brief report: deletion of the dystrophin muscle‐promoter region associated with X‐linked dilated cardiomyopathy. N Engl J Med 1993; 329: 921–5
- Towbin J. A., Hejtmancik J. F., Brink P., Gelb B., Zhu X. M., Chamberlain J. S., et al. X‐linked dilated cardiomyopathy. Molecular genetic evidence of linkage to the Duchenne muscular dystrophy (dystrophin) gene at the Xp21 locus. Circulation 1993; 87: 1854–65
- Arbustini E., Pilotto A., Repetto A., Grasso M., Negri A., Diegoli M., et al. Autosomal dominant dilated cardiomyopathy with atrioventricular block: a lamin A/C defect‐related disease. J Am Coll Cardiol 2002; 39: 981–90
- Bione S., D'Adamo P., Maestrini E., Gedeon A. K., Bolhuis P. A., Toniolo D. A novel X‐linked gene, G4.5. is responsible for Barth syndrome. Nat Genet 1996; 12: 385–9
- Brodsky G. L., Muntoni F., Miocic S., Sinagra G., Sewry C., Mestroni L. Lamin A/C gene mutation associated with dilated cardiomyopathy with variable skeletal muscle involvement. Circulation 2000; 101: 473–6
- Daehmlow S., Erdmann J., Knueppel T., Gille C., Froemmel C., Hummel M., et al. Novel mutations in sarcomeric protein genes in dilated cardiomyopathy. Biochem Biophys Res Commun 2002; 298: 116
- D'Adamo P., Fassone L., Gedeon A., Janssen E. A., Bione S., Bolhuis P. A., et al. The X‐linked gene G4.5 is responsible for different infantile dilated cardiomyopathies. Am J Hum Genet 1997; 61: 862–7
- Fatkin D., MacRae C., Sasaki T., Wolff M. R., Porcu M., Frenneaux M., et al. Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conduction‐system disease. N Engl J Med 1999; 341: 1715–24
- Genschel J., Schmidt H. H. Mutations in the LMNA gene encoding lamin A/C. Hum Mutat 2000; 16: 451–9
- Gerull B., Gramlich M., Atherton J., McNabb M., Trombitas K., Sasse‐Klaassen S., et al. Mutations of TTN, encoding the giant muscle filament titin, cause familial dilated cardiomyopathy. Nat Genet 2002; 14: 14
- Hanson E. L., Jakobs P. M., Keegan H., Coates K., Bousman S., Dienel N. H., et al. Cardiac troponin T lysine 210 deletion in a family with dilated cardiomyopathy. J Card Fail 2002; 8: 28–32
- Jakobs P. M., Hanson E. L., Crispell K. A., Toy W., Keegan H., Schilling K., et al. Novel lamin A/C mutations in two families with dilated cardiomyopathy and conduction system disease. J Card Fail 2001; 7: 249–56
- Kamisago M., Sharma S. D., DePalma S. R., Solomon S., Sharma P., McDonough B., et al. Mutations in Sarcomere Protein Genes as a Cause of Dilated Cardiomyopathy. N Engl J Med 2000; 343: 1688–96
- Karkkainen S., Miettinen R., Tuomainen P., Karkkainen P., Helio T., Reissell E., et al. A novel mutation, Arg71Thr, in the delta‐sarcoglycan gene is associated with dilated cardiomyopathy. J Mol Med 2003; 81: 795–800
- Kärkkäinen S., Heliö T., Miettinen R., Tuomainen P., Peltola P., Rummukainen J., et al. A novel mutation, Ser143Pro, in the lamin A/C gene is common in finnish patients with familial dilated cardiomyopathy. Eur Heart J 2004; 25: 885–93
- Knöll R., Hoshijima M., Hoffman H., Person V., Lorenzen‐Schmidt I., Bang M. The cardiac mechanical stretch sensor machinery involves a z disc complex that is defective in a subset of human dilated cardiomyopathy. Cell 2002; 111: 943–55
- Li D., Tapscoft T., Gonzalez O., Burch P. E., Quinones M. A., Zoghbi W. A., et al. Desmin mutation responsible for idiopathic dilated cardiomyopathy. Circulation 1999; 100: 461–4
- Li D., Czernuszewicz G. Z., Gonzalez O., Tapscott T., Karibe A., Durand J. B., et al. Novel cardiac troponin T mutation as a cause of familial dilated cardiomyopathy. Circulation 2001; 104: 2188–93
- Olson T. M., Michels V. V., Thibodeau S. N., Tai Y. S., Keating M. T. Actin mutations in dilated cardiomyopathy, a heritable form of heart failure. Science 1998; 280: 750–2
- Olson T. M., Kishimoto N. Y., Whitby F. G., Michels V. V. Mutations that Alter the Surface Charge of Alpha‐tropomyosin are Associated with Dilated Cardiomyopathy. J Mol Cell Cardiol 2001; 33: 723–32
- Olson T. M., Illenberger S., Kishimoto N. Y., Huttelmaier S., Keating M. T., Jockusch B. M. Metavinculin mutations alter actin interaction in dilated cardiomyopathy. Circulation 2002; 105: 431–7
- Ortiz‐Lopez R., Li H., Su J., Goytia V., Towbin J. A. Evidence for a dystrophin missense mutation as a cause of X‐linked dilated cardiomyopathy. Circulation 1997; 95: 2434–40
- Schmitt J. P., Kamisago M., Asahi M., Li G. H., Ahmad F., Mende U., et al. Dilated cardiomyopathy and heart failure caused by a mutation in phospholamban. Science 2003; 299: 1410–3
- Mohapatra B., Jimenez S., Lin J. H., Bowles K. R., Coveler K. J., Marx J. G., et al. Mutations in the muscle LIM protein and alpha‐actinin‐2 genes in dilated cardiomyopathy and endocardial fibroelastosis. Mol Genet Metab 2003; 80: 207–15
- Murphy R. T., Mogensen J., Shaw A., Kubo T., Hughes S., McKenna W. J. Novel mutation in cardiac troponin I in recessive idiopathic dilated cardiomyopathy. Lancet 2004; 363: 371–2
- Taylor M. R., Fain P. R., Sinagra G., Robinson M. L., Robertson A. D., Carniel E., et al. Natural history of dilated cardiomyopathy due to lamin A/C gene mutations. J Am Coll Cardiol 2003; 41: 771–80
- Mogensen J., Klausen I. C., Pedersen A. K., Egeblad H., Bross P., Kruse T. A., et al. Alpha‐cardiac actin is a novel disease gene in familial hypertrophic cardiomyopathy. J Clin Invest 1999; 103: R39–43
- McNair W., Ku T., Taylor M., Fain P., Dao D., Wolfel E., et al. SCN5A mutation associated with dilated cardiomyopathy, conduction disorder, and arrhythmia. Circulation 2004; 110: 2163–7
- Mogensen J., Murphy R., Shaw T., Bahl A., Redwood C., Watkins H., et al. Severe disease expressing of cardiac troponin C and T mutations in patients with idiopathic dilated cardiomyopathy. J Am Coll Cardiol 2004; 44: 2033–40
- Taylor M., Slavov D., Gajewski A., Vlcek S., Ku L., Fain P., et al. Thymopoietin (lamina‐associated polypeptide 2) gene mutation associated with dilated cardiomyopathy. Hum Mutat 2005; 26: 566–74
- Vatta M., Mohapatra B., Jimenez S., Sanchez X., Faulkner G., Perles Z., et al. Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non‐compaction. J Am Coll Cardiol 2003; 42: 2014–17
- Bienengraeber M., Olson T. M., Selivanov V. A., Kathmann E. C., O'Cochlain F., Gao F., et al. ABCC9 mutations identified in human dilated cardiomyopathy disrupt catalytic K(ATP) channel gating. Nat Genet 2004; 36: 382–7
- Hershberger R. E., Hanson E. L., Jakobs P. M., Keegan H., Coates K., Bousman S., et al. A novel lamin A/C mutation in a family with dilated cardiomyopathy, prominent conduction system disease, and need for permanent pacemaker implantation. Am Heart J 2002; 144: 1081–6
- MacLeod H. M., Culley M. R., Huber J. M., McNally E. M. Lamin A/C truncation in dilated cardiomyopathy with conduction disease. BMC Med Genet 2003; 4: 4
- Verga L., Concardi M., Pilotto A., Bellini O., Pasotti M., Repetto A., et al. Loss of lamin A/C expression revealed by immuno‐electron microscopy in dilated cardiomyopathy with atrioventricular block caused by LMNA gene defects. Virchows Arch 2003; 443: 664–71
- Sinagra G., Di Lenarda A., Brodsky G. L., Taylor M. R., Muntoni F., Pinamonti B., et al. Current perspective new insights into the molecular basis of familial dilated cardiomyopathy. Ital Heart J 2001; 2: 280–6
- Karkkainen S., Reissell E., Helio T., Kaartinen M., Tuomainen P., Toivonen L., et al. Novel mutations in the lamin A/C gene in heart transplant recipients with end stage dilated cardiomyopathy. Heart 2006; 92: 524–526
- Muntoni F., Wilson L., Marrosu G., Marrosu M. G., Cianchetti C., Mestroni L., et al. A mutation in the dystrophin gene selectively affecting dystrophin expression in the heart. J Clin Invest 1995; 96: 693–9
- Tsubata S., Bowles K. R., Vatta M., Zintz C., Titus J., Muhonen L., et al. Mutations in the human delta‐sarcoglycan gene in familial and sporadic dilated cardiomyopathy. J Clin Invest 2000; 106: 655–62
- Komajda M., Charron P., Tesson F. Genetic aspects of heart failure. Eur J Heart Fail 1999; 1: 121–6
- Mayosi B. M., Khogali S., Zhang B., Watkins H. Cardiac and skeletal actin gene mutations are not a common cause of dilated cardiomyopathy. J Med Genet 1999; 36: 796–7
- Takai E., Akita H., Shiga N., Kanazawa K., Yamada S., Terashima M., et al. Mutational analysis of the cardiac actin gene in familial and sporadic dilated cardiomyopathy. Am J Med Genet 1999; 86: 325–7
- Tesson F., Sylvius N., Pilotto A., Dubosq‐Bidot L., Peuchmaurd M., Bouchier C., et al. Epidemiology of desmin and cardiac actin gene mutations in a european population of dilated cardiomyopathy. Eur Heart J 2000; 21: 1872–6
- Blair E., Redwood C., de Jesus Oliveira M., Moolman‐Smook J. C., Brink P., Corfield V. A., et al. Mutations of the light meromyosin domain of the beta‐myosin heavy chain rod in hypertrophic cardiomyopathy. Circ Res 2002; 90: 263–9
- Kärkkäinen S., Heliö T., Jääskeläinen P., Miettinen R., Tuomainen P., Ylitalo K., et al. Two novel mutations in the beta‐myosin heavy chain gene associated with dilated cardiomyopathy. Eur J Heart Fail 2004; 6: 861–8
- Villard E., Duboscq‐Bidot L., Charron P., Benaiche A., Conraads V., Sylvius N., et al. Mutation screening in dilated cardiomyopathy: prominent role of the beta myosin heavy chain gene. Eur Heart J 2005; 26: 794–803
- Robinson P., Mirza M., Knott A., Abdulrazzak H., Willott R., Marston S., et al. Alterations in thin filament regulation induced by a human cardiac troponin T mutant that causes dilated cardiomyopathy are distinct from those induced by troponin T mutants that cause hypertrophic cardiomyopathy. J Biol Chem 2002; 277: 40710–6
- Stefanelli C., Rosenthal A., Borisov A., Ensing G., Russell M. Novel troponin T mutation in familial dilated cardiomyopathy with gender‐dependant severity. Mol Genet Metab 2004; 83: 188–96
- Fatkin D., Graham R. M. Molecular mechanisms of inherited cardiomyopathies. Physiol Rev 2002; 82: 945–80
- Hein S., Kostin S., Heling A., Maeno Y., Schaper J. The role of the cytoskeleton in heart failure. Cardiovasc Res 2000; 45: 273–8
- Itoh‐Satoh M., Hayashi T., Nishi H., Koga Y., Arimura T., Koyanagi T., et al. Titin mutations as the molecular basis for dilated cardiomyopathy. Biochem Biophys Res Commun 2002; 291: 385–93
- Maruyama K. Connectin/titin, giant elastic protein of muscle. FASEB J 1997; 11: 341–5
- Labeit S., Kolmerer B. Titins: giant proteins in charge of muscle ultrastructure and elasticity. Science 1995; 270: 293–6
- Gerull B., Atherton J., Geupel A., Sasse‐Klaassen S., Heuser A., Frenneaux M., et al. Identification of a novel frameshift mutation in the giant muscle filament titin in a large Australian family with dilated cardiomyopathy. J Mol Med 2006; 84: 478–83
- Valle G., Faulkner G., De Antoni A., Pacchioni B., Pallavicini A., Pandolfo D., et al. Telethonin, a novel sarcomeric protein of heart and skeletal muscle. FEBS Lett 1997; 415: 163–8
- Hayashi T., Arimura T., Itoh‐Satoh M., Ueda K., Hohda S., Inagaki N., et al. Tcap gene mutations in hypertrophic cardiomyopathy and dilated cardiomyopathy. J Am Coll Cardiol 2004; 44: 2192–201
- Fuchs E., Cleveland D. W. A structural scaffolding of intermediate filaments in health and disease. Science 1998; 279: 514–9
- Miyamoto Y., Akita H., Shiga N., Takai E., Iwai C., Mizutani K., et al. Frequency and clinical characteristics of dilated cardiomyopathy caused by desmin gene mutation in a Japanese population. Eur Heart J 2001; 22: 2284–9
- Schonberger J., Seidman C. E. Many roads lead to a broken heart: the genetics of dilated cardiomyopathy. Am J Hum Genet 2001; 69: 249–60
- Dalakas M. C., Park K. Y., Semino‐Mora C., Lee H. S., Sivakumar K., Goldfarb L. G. Desmin myopathy, a skeletal myopathy with cardiomyopathy caused by mutations in the desmin gene. N Engl J Med 2000; 342: 770–80
- Goldfarb L. G., Park K. Y., Cervenakova L., Gorokhova S., Lee H. S., Vasconcelos O., et al. Missense mutations in desmin associated with familial cardiac and skeletal myopathy. Nat Genet 1998; 19: 402–3
- Sjoberg G., Saavedra‐Matiz C. A., Rosen D. R., Wijsman E. M., Borg K., Horowitz S. H., et al. A missense mutation in the desmin rod domain is associated with autosomal dominant distal myopathy, and exerts a dominant negative effect on filament formation. Hum Mol Genet 1999; 8: 2191–8
- Cox G. F., Kunkel L. M. Dystrophies and heart disease. Curr Opin Cardiol 1997; 12: 329–43
- Badorff C., Berkely N., Mehrotra S., Talhouk J. W., Rhoads R. E., Knowlton K. U. Enteroviral protease 2A directly cleaves dystrophin and is inhibited by a dystrophin‐based substrate analogue. J Biol Chem 2000; 275: 11191–7
- Hart K. A., Hodgson S., Walker A., Cole C. G., Johnson L., Dubowitz V., et al. DNA deletions in mild and severe Becker muscular dystrophy. Hum Genet 1987; 75: 281–5
- Koenig M., Hoffman E. P., Bertelson C. J., Monaco A. P., Feener C., Kunkel L. M. Complete cloning of the Duchenne muscular dystrophy (DMD) cDNA and preliminary genomic organization of the DMD gene in normal and affected individuals. Cell 1987; 50: 509–17
- Berko B. A., Swift M. X‐linked dilated cardiomyopathy. N Engl J Med 1987; 316: 1186–91
- Arbustini E., Diegoli M., Morbini P., Dal Bello B., Banchieri N., Pilotto A., et al. Prevalence and characteristics of dystrophin defects in adult male patients with dilated cardiomyopathy. J Am Coll Cardiol 2000; 35: 1760–8
- Milasin J., Muntoni F., Severini G. M., Bartoloni L., Vatta M., Krajinovic M., et al. A point mutation in the 5′ splice site of the dystrophin gene first intron responsible for X‐linked dilated cardiomyopathy. Hum Mol Genet 1996; 5: 73–9
- Feng J., Yan J., Buzin C., Towbin J., Sommer S. Mutations in the dystrophin gene are associated with sporadic dilated cardiomyopathy. Mol Genet Metab 2002; 77: 119
- Feng J., Yan J. Y., Buzin C. H., Sommer S. S., Towbin J. A. Comprehensive mutation scanning of the dystrophin gene in patients with nonsyndromic X‐linked dilated cardiomyopathy. J Am Coll Cardiol 2002; 40: 1120–4
- Bies R. D., Maeda M., Roberds S. L., Holder E., Bohlmeyer T., Young J. B., et al. A 5′ dystrophin duplication mutation causes membrane deficiency of alpha‐dystroglycan in a family with X‐linked cardiomyopathy. J Mol Cell Cardiol 1997; 29: 3175–88
- Franz W. M., Muller M., Muller O. J., Herrmann R., Rothmann T., Cremer M., et al. Association of nonsense mutation of dystrophin gene with disruption of sarcoglycan complex in X‐linked dilated cardiomyopathy. Lancet 2000; 355: 1781–5
- Muntoni F., Di Lenarda A., Porcu M., Sinagra G., Mateddu A., Marrosu G., et al. Dystrophin gene abnormalities in two patients with idiopathic dilated cardiomyopathy. Heart 1997; 78: 608–12
- Kostin S., Scholz D., Shimada T., Maeno Y., Mollnau H., Hein S., et al. The internal and external protein scaffold of the T‐tubular system in cardiomyocytes. Cell Tissue Res 1998; 294: 449–60
- Belkin A. M., Ornatsky O. I., Glukhova M. A., Koteliansky V. E. Immunolocalization of meta‐vinculin in human smooth and cardiac muscles. J Cell Biol 1988; 107: 545–53
- Simpson D. G., Terracio L., Terracio M., Price R. L., Turner D. C., Borg T. K. Modulation of cardiac myocyte phenotype in vitro by the composition and orientation of the extracellular matrix. J Cell Physiol 1994; 161: 89–105
- Maeda M., Holder E., Lowes B., Valent S., Bies R. D. Dilated cardiomyopathy associated with deficiency of the cytoskeletal protein metavinculin. Circulation 1997; 95: 17–20
- Vasile V., Will M., Ommen S., Edwards W., Olson T., Ackerman M. Identification of a metavinculin missense mutation, R975W, associated with both hypertrophic and dilated cardiomyopathy. Mol Genet Metab 2005; 87: 169–74
- Delta‐sarcoglycan (SGCD), Leiden muscular dystrophy pages. Available from: http://www.dmd.nl/sgcd_home.html
- Nigro V., de Sa Moreira E., Piluso G., Vainzof M., Belsito A., Politano L., et al. Autosomal recessive limb‐girdle muscular dystrophy, LGMD2F, is caused by a mutation in the delta‐sarcoglycan gene. Nat Genet 1996; 14: 195–8
- Nigro V., Okazaki Y., Belsito A., Piluso G., Matsuda Y., Politano L., et al. Identification of the Syrian hamster cardiomyopathy gene. Hum Mol Genet 1997; 6: 601–7
- Kong Y., Flick M. J., Kudla A. J., Konieczny S. F. Muscle LIM protein promotes myogenesis by enhancing the activity of MyoD. Mol Cell Biol 1997; 17: 4750–60
- Yoshida K., Nakamura A., Yazaki M., Ikeda S., Takeda S. Insertional mutation by transposable element, L1, in the DMD gene results in X‐linked dilated cardiomyopathy. Hum Mol Genet 1998; 7: 1129–32
- Zhou Q., Ruiz‐Lozano P., Martone M. E., Chen J. Ablation of Cypher, a PDZ‐LIM domain Z‐line protein, causes a severe form of congenital myopathy. J Cell Biol 2001; 155: 605–12
- Arimura T., Hayashi T., Terada H., Lee S‐Y., Zhou Q., Takahashi M., et al. A Cypher/ZASP mutation associated with dilated cardiomyopathy alters the binding affinity to protein kinase. J Biol Chem 2004; 279: 6746–52
- Goldman R. D., Gruenbaum Y., Moir R. D., Shumaker D. K., Spann T. P. Nuclear lamins: building blocks of nuclear architecture. Genes Dev 2002; 16: 533–47
- Franz W. M., Muller O. J., Katus H. A. Cardiomyopathies: from genetics to the prospect of treatment. Lancet 2001; 358: 1627–37
- Stuurman N., Heins S., Aebi U. Nuclear lamins: their structure, assembly, and interactions. J Struct Biol 1998; 122: 42–66
- Mounkes L. C., Burke B., Stewart C. L. The A‐type lamins: nuclear structural proteins as a focus for muscular dystrophy and cardiovascular diseases. Trends Cardiovasc Med 2001; 11: 280–5
- Hutchison C. J., Alvarez‐Reyes M., Vaughan O. A. Lamins in disease: why do ubiquitously expressed nuclear envelope proteins give rise to tissue‐specific disease phenotypes?. J Cell Sci 2001; 114: 9–19
- Bonne G., Di Barletta M. R., Varnous S., Becane H. M., Hammouda E. H., Merlini L., et al. Mutations in the gene encoding lamin A/C cause autosomal dominant Emery‐Dreifuss muscular dystrophy. Nat Genet 1999; 21: 285–8
- Emery A. E. Emery‐Dreifuss syndrome. J Med Genet 1989; 26: 637–41
- van der Kooi A. J., van Meegen M., Ledderhof T. M., McNally E. M., de Visser M., Bolhuis P. A. Genetic localization of a newly recognized autosomal dominant limb‐girdle muscular dystrophy with cardiac involvement (LGMD1B) to chromosome 1q11‐21. Am J Hum Genet 1997; 60: 891–5
- Chaouch M., Allal Y., De Sandre‐Giovannoli A., Vallat J. M., Amer‐el‐Khedoud A., Kassouri N., et al. The phenotypic manifestations of autosomal recessive axonal Charcot‐Marie‐Tooth due to a mutation in Lamin A/C gene. Neuromuscul Disord 2003; 13: 60–7
- Cao H., Hegele R. A. LMNA is mutated in Hutchinson‐Gilford progeria (MIM 176670) but not in Wiedemann‐Rautenstrauch progeroid syndrome (MIM 264090). J Hum Genet 2003; 48: 271–4
- Novelli G., Muchir A., Sangiuolo F., Helbling‐Leclerc A., D'Apice M. R., Massart C., et al. Mandibuloacral dysplasia is caused by a mutation in LMNA‐encoding lamin A/C. Am J Hum Genet 2002; 71: 426–31
- Nikolova V., Leimena C., McMahon A. C., Tan J. C., Chandar S., Jogia D., et al. Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C‐deficient mice. J Clin Invest 2004; 113: 357–69
- van Berlo J. H., deVoogt W. G., van der Kooi A. J., van Tintelen J. P., Bonne G., Yaou R. B., et al. Meta‐analysis of clinical characteristics of 299 carriers of LMNA gene mutations: do lamin A/C mutations portend a high risk of sudden death?. J Mol Med 2005; 83: 79–83
- Meune C., Van Berlo J., Anselme F., Bonne G., Pinto Y. M., Duboc D. Primary prevention of sudden death in patients with lamin A/C gene mutations. N Engl J Med 2006; 354: 209–10
- Foisner R., Gerace L. Integral membrane proteins of the nuclear envelope interact with lamins and chromosomes, and binding is modulated by mitotic phosphorylation. Cell 1993; 73: 1267–79
- Dechat T., Gotzmann J., Stockinger A., Harris C., Talle M., Siekierka J., et al. Detergent‐salt resistance of LAP2alpha in interphase nuclei and phosphorylation‐dependent association with chromosomes early in nuclear assembly implies functions in nuclear structure dynamics. EMBO J 1998; 17: 4887–902
- Vlcek S., Korbei B., Foisner R. Distinct functions of the unique C terminus of LAP2alpha in cell proliferation and nuclear assembly. J Biol Chem 2002; 277: 18898–907
- Bezzina C., Veldkamp M. W., van den Berg M. P., Postma A. V., Rook M. B., Viersma J‐W., et al. A single Na+ channel mutation causing both long‐QT and Brugada syndromes. Circ Res 1999; 85: 1206–13
- Johns D. R. The other human genome: mitochondrial DNA and disease. Nat Med 1996; 2: 1065–8
- Majamaa‐Voltti K., Peuhkurinen K., Kortelainen M. L., Hassinen I. E., Majamaa K. Cardiac abnormalities in patients with mitochondrial DNA mutation 3243A>G. BMC Cardiovasc Disord 2002; 2: 12
- Mestroni L., Giacca M. Molecular genetics of dilated cardiomyopathy. Curr Opin Cardiol 1997; 12: 303–9
- Marin‐Garcia J., Goldenthal M. J., Ananthakrishnan R., Pierpont M. E., Fricker F. J., Lipshultz S. E., et al. Specific mitochondrial DNA deletions in idiopathic dilated cardiomyopathy. Cardiovasc Res 1996; 31: 306–13
- Anan R., Nakagawa M., Miyata M., Higuchi I., Nakao S., Suehara M., et al. Cardiac involvement in mitochondrial diseases. A study on 17 patients with documented mitochondrial DNA defects. Circulation 1995; 91: 955–61
- Schowengerdt K. O., Jr., Towbin J. A. Genetic basis of inherited cardiomyopathies. Curr Opin Cardiol 1995; 10: 312–21
- Suomalainen A., Paetau A., Leinonen H., Majander A., Peltonen L., Somer H. Inherited idiopathic dilated cardiomyopathy with multiple deletions of mitochondrial DNA. Lancet 1992; 340: 1319–20
- Dimauro S., Davidzon G. Mitochondrial DNA and disease. Ann Med 2005; 37: 222–32
- Manolio T. A., Baughman K. L., Rodeheffer R., Pearson T. A., Bristow J. D., Michels V. V., et al. Prevalence and etiology of idiopathic dilated cardiomyopathy (summary of a National Heart, Lung, and Blood Institute workshop). Am J Cardiol 1992; 69: 1458–66
- Sugrue D. D., Rodeheffer R. J., Codd M. B., Ballard D. J., Fuster V., Gersh B. J. The clinical course of idiopathic dilated cardiomyopathy. A population‐based study. Ann Intern Med 1992; 117: 117–23
- Hofmann T., Meinertz T., Kasper W., Geibel A., Zehender M., Hohnloser S., et al. Mode of death in idiopathic dilated cardiomyopathy: a multivariate analysis of prognostic determinants. Am Heart J 1988; 116: 1455–63
- Schoeller R., Andresen D., Buttner P., Oezcelik K., Vey G., Schroder R. First‐ or second‐degree atrioventricular block as a risk factor in idiopathic dilated cardiomyopathy. Am J Cardiol 1993; 71: 720–6
- Baldasseroni S., De Biase L., Fresco C., Marchionni N., Marini M., Masotti G., et al. Cumulative effect of complete left bundle‐branch block and chronic atrial fibrillation on 1‐year mortality and hospitalization in patients with congestive heart failure. A report from the Italian network on congestive heart failure (in‐CHF database). Eur Heart J 2002; 23: 1692–8
- Goedel‐Meinen L., Hofmann M., Ryba S., Schömig A. Prognostic value of an abnormal signal‐averaged electrocardiogram in patients with nonischemic dilated cardiomyopathy. Am J Cardiol 2001; 87: 809–12
- Mancini M., Eisen H., Kussmaul W., Mull R., Edmunds L., Wilson J. Value of peak exercise oxygen consumption for optimal timing of cardiac transplantation in ambulatory patients with heart failure. Circulation 1991; 83: 778–86
- Junker A., Thayssen P., Nielsen B., Andersen P. E. The hemodynamic and prognostic significance of echo‐Doppler‐proven mitral regurgitation in patients with dilated cardiomyopathy. Cardiology 1993; 83: 14–20
- Stelken A., Younis L., Jennison T., Miller D., Miller L., Shaw L. Prognostiv value of cardiopulmonary exercise testing using percent achieved of predicted peak oxygen uptake for patients with ischemic and dilated cardiomyopathy. J Am Coll Cardiol 1996; 27: 345–52
- Blondheim D. S., Jacobs L. E., Kotler M. N., Costacurta G. A., Parry W. R. Dilated cardiomyopathy with mitral regurgitation: decreased survival despite a low frequency of left ventricular thrombus. Am Heart J 1991; 122: 763–71
- Hung J., Koelling T., Semigran M. J., Dec G. W., Levine R. A., Di Salvo T. G. Usefulness of echocardiographic determined tricuspid regurgitation in predicting event‐free survival in severe heart failure secondary to idiopathic‐dilated cardiomyopathy or to ischemic cardiomyopathy. Am J Cardiol 1998; 82: 1301–3, A10
- Koelling T. M., Aaronson K. D., Cody R. J., Bach D. S., Armstrong W. F. Prognostic significance of mitral regurgitation and tricuspid regurgitation in patients with left ventricular systolic dysfunction. Am Heart J 2002; 144: 524–9
- Gavazzi A., De Maria R., Parolini M., Porcu M. Alcohol abuse and dilated cardiomyopathy in men. Am J Cardiol 2000; 85: 1114–8
- Fauchier L., Babuty D., Poret P., Casset‐Senon D., Autret M. L., Cosnay P., et al. Comparison of long‐term outcome of alcoholic and idiopathic dilated cardiomyopathy. Eur Heart J 2000; 21: 306–14
- Keogh A. M., Baron D. W., Hickie J. B. Prognostic guides in patients with idiopathic or ischemic dilated cardiomyopathy assessed for cardiac transplantation. Am J Cardiol 1990; 65: 903–8
- Klappacher G., Franzen P., Haab D., Mehrabi M., Binder M., Plesch K., et al. Measuring extracellular matrix turnover in the serum of patients with idiopathic or ischemic dilated cardiomyopathy and impact on diagnosis and prognosis. Am J Cardiol 1995; 75: 913–8
- Zannad F., Dousset B., Alla F. Treatment of congestive heart failure: interfering the aldosterone‐cardiac extracellular matrix relationship. Hypertension 2001; 38: 1227–32
- Niebauer J., Volk H. D., Kemp M., Dominguez M., Schumann R. R., Rauchhaus M., et al. Endotoxin and immune activation in chronic heart failure: a prospective cohort study. Lancet 1999; 353: 1838–42
- Mestroni L., Maisch B., McKenna W. J., Schwartz K., Charron P., Rocco C., et al. Guidelines for the study of familial dilated cardiomyopathies. Collaborative Research Group of the European Human and Capital Mobility Project on Familial Dilated Cardiomyopathy. Eur Heart J 1999; 20: 93–102
- Vatta M., Stetson S., Jimenez S., Entman M., Noon G., Bowles N., et al. Molecular normalization of dystrophin in the failing left and right ventricle of patients treated with either pulsatile or continuous flow‐type ventricular assist devices. J Am Coll Cardiol 2004; 43: 811–17
- Andersson B., Sylven C. The DD genotype of the angiotensin‐converting enzyme gene is associated with increased mortality in idiopathic heart failure. J Am Coll Cardiol 1996; 28: 162–7
- Liggett S. B., Wagoner L. E., Craft L. L., Hornung R. W., Hoit B. D., McIntosh T. C., et al. The Ile164 beta2‐adrenergic receptor polymorphism adversely affects the outcome of congestive heart failure. J Clin Invest 1998; 102: 1534–9
- McNamara D., Holubkov R., Postava L., Janosko K., MacGowan G., Mathier M., et al. Pharmacogenetic interactions between angiotensin‐converting enzyme inhibitor therapy and the angiotensin‐converting enzyme deletion polymorphism in patients with congestive heart failure. J Am Coll Cardiol 2004; 44: 2019–26
- O'Toole L., Stewart M., Padfield P., Channer K. Effect of the insertion/deletion polymorphism of the angiotensin‐converting enzyme gene on response to angiotensin‐converting enzyme inhibitors in patients with heart failure. J Cardiovasc Pharmacol 1998; 32: 988–94
- Sebillon P., Bouchier C., Bidot L., Bonne G., Ahamed K., Charron P. Expanding the phenotype of LMNA mutations in dilated cardiomyopathy and functional consequences of these mutations. J Med Genet 2003; 40: 560–7
- Durand J. B., Bachinski L. L., Bieling L. C., Czernuszewicz G. Z., Abchee A. B., Yu Q. T., et al. Localization of a gene responsible for familial dilated cardiomyopathy to chromosome 1q32. Circulation 1995; 92: 3387–9
- Jung M., Poepping I., Perrot A., Ellmer A. E., Wienker T. F., Dietz R., et al. Investigation of a Family with Autosomal Dominant Dilated Cardiomyopathy Defines a Novel Locus on Chromosome 2q14–q22. Am J Hum Genet 1999; 65: 1068–77
- Siu B. L., Niimura H., Osborne J. A., Fatkin D., MacRae C., Solomon S., et al. Familial dilated cardiomyopathy locus maps to chromosome 2q31. Circulation 1999; 99: 1022–6
- Olson T. M., Keating M. T. Mapping a cardiomyopathy locus to chromosome 3p22-p25. J Clin Invest 1996; 97: 528–32
- Sylvius N., Tesson F., Gayet C., Charron P., Benaiche A., Peuchmaurd M., et al. A New Locus for Autosomal Dominant Dilated Cardiomyopathy Identified on Chromosome 6q12–q16. Am J Hum Genet 2001; 68: 241–6
- Messina D. N., Speer M. C., Pericak‐Vance M. A., McNally E. M. Linkage of familial dilated cardiomyopathy with conduction defect and muscular dystrophy to chromosome 6q23. Am J Hum Genet 1997; 61: 909–17
- Krajinovic M., Pinamonti B., Sinagra G., Vatta M., Severini G. M., Milasin J., et al. Linkage of familial dilated cardiomyopathy to chromosome 9. Heart Muscle Disease Study Group. Am J Hum Genet 1995; 57: 846–52
- Bowles K. R., Gajarski R., Porter P., Goytia V., Bachinski L., Roberts R., et al. Gene mapping of familial autosomal dominant dilated cardiomyopathy to chromosome 10q21–23. J Clin Invest 1996; 98: 1355–60