Abstract
The amyloidoses constitute a large group of diseases caused by an alteration in the conformation and metabolism of several globular proteins which, under particular conditions, deposit in tissues as insoluble fibrillar aggregates. To date, at least 24 different proteins have been recognized as causative agents of amyloid diseases. Despite a high heterogeneity in amino acid sequence, three‐dimensional structure, and biological function, all amyloidogenic proteins share a reduced folding stability, a strong propensity to acquire more than one conformation, and the capacity to form almost indistinguishable amyloid fibrils. In some cases, the generation of an aggregation‐prone state can be triggered or enhanced by the occurrence of mutations, a proteolytic cleavage, or a seeding process. The interaction between the amyloidogenic precursor, some common components of amyloid deposits, and the extra‐cellular environment also plays a role in fibrillogenesis and in particular in the organ tropism of amyloid deposition. The process of amyloid fibril formation exerts a cytotoxic effect, resulting in tissue damage and organ dysfunction. Prefibrillar aggregates are thought to have an active part in this process. Due to the pathogenic complexity of amyloid diseases, the integration of several therapeutic interventions involving different critical levels of the amyloidogenic cascade is envisaged.
The amyloidoses constitute a large group of diseases which display a highly heterogeneous clinical presentation, but have a homogeneous molecular and histological profile. They are characterized by the presence of amyloid fibrils showing a cross β supersecondary structure in one or more tissues. Hence, proteins aggregating as amyloid fibrils play a central role in the pathogenic mechanisms of the disease.
Biochemical characteristics of amyloidogenic proteins
The isolation of the protein components of natural amyloid fibrils and their chemical characterization represents an indispensable investigative tool for the development of any pathogenic theory. To date, at least 24 different proteins have been identified as causative agents of amyloid diseases Citation1. All these proteins may generate amyloid fibrils which are histologically almost indistinguishable, although a certain degree of variability is disclosed by the accurate evaluation of the number of protofilaments assembled within the mature fibre, ranging from three protofilaments in the fibres deriving from apolipoprotein A–I (apoA–I) Citation2, to six for fibres of different chemical composition Citation3. The organization of amyloid fibres may be regarded as a primordial structure, being also achieved by proteins not associated with amyloid diseases, and acquires the significance of a generic structure enabling aggregated proteins to organize themselves. The stability of the fibre derives from an indefinite succession of hydrogen bonds between the backbones of the polypeptide chains and the contribution of side chains, which may promote or inhibit aggregation.
The variability and conformational plasticity which characterize the different amyloidogenic proteins at the prefibrillar functional state account for the significant role of the conversion of these structures into such a repetitive, ordered fibrillar form in the pathogenesis of these diseases.
Key messages
Amyloidogenic proteins are synthesized by various organs and require distinct therapeutic approaches.
Tissue damage can be halted or prevented by a rapid reduction in the amyloidogenic protein concentration.
The complex pathogenic cascade offers the possibility of targeting several critical passages for therapy.
A dynamic view of the pathogenic process
The transition from one conformation to another is a dynamic process that polypeptide chains undergo at least once during their biological life, when, shortly after their synthesis, they fold and acquire a three‐dimensional structure. Typically, this is an intracellular process consisting in the rapid, sequential formation of intramolecular bonds allowing the protein to achieve its definitive form (the so‐called ‘native structure’). The formation of the right intramolecular bonds and acquisition of the native structure has a solid thermodynamic basis and is guided by the achievement of the most stable energetic level. The exposure to various extra‐cellular denaturating stimuli causes the unfolding of the polypeptide chain, an event which is normally followed by the rapid restoration of the native structure. This extra‐cellular unfolding and refolding process causes the exposure of normally hidden hydrophobic residues, thus making the protein prone to autoaggregation. Moreover, a partially unfolded protein may become the target of ubiquitous endopeptidases. Even minor proteolytic cleavage may destabilize the protein, promote its denaturation and, sometimes, prevent the restoration of the native structure.
Amyloidogenic proteins show a high propensity for autoaggregation during these fluctuations between fully unfolded, locally unfolded, and folded state. Amyloid proteins may be divided into four categories: 1) proteins with an intrinsic propensity for conformational fluctuations, which further increases with age (transthyretin, with its ability to form amyloid deposits in elderly people even in its wild‐type form, is one example of this category of amyloid forming proteins); 2) proteins which, in spite of their susceptibility to partial denaturation and autoaggregation, do not normally form amyloid deposits due to their low serum concentration, but can generate deposits once a persistently high serum concentration is achieved, such as β2‐microglobulin in patients undergoing long‐term dialysis; 3) proteins in which the conversion to fibrils requires the presence of mutations (this is the case with human lysozyme, which does not produce amyloid fibrils in its wild‐type form nor the Thr70Asn variant, but is the causative agent of an autosomal dominant hereditary disease transmitted when one of the variants Phe56Thr, Phe57Ile, Trp64Arg, or Arg67His is present Citation4); and 4) proteins which do not generate amyloid fibrils until a proteolytic cleavage takes place, releasing a polypeptidic fragment with high propensity for fibrillar aggregation, such as apoA–I.
Mutations and the molecular mechanism of amyloid formation
Advancements in our knowledge of the mechanisms of amyloidogenic protein aggregation result from comparative studies of normal proteins and their pathologic mutated counterparts. Autosomal dominant hereditary amyloidoses, as well as immunoglobulin light chain amyloidosis (AL amyloidosis), provide examples of this type.
Amyloidogenic immunoglobulin light chains
Only a small percentage of immunoglobulin light chains are able to form amyloid fibrils in vivo. AL amyloidosis occurs only in 12%–15% of patients with myeloma. It is therefore presumed that only some immunoglobulin light chains display structural features related to amyloidogenicity. This hypothesis, although not new, is based on the well known observation that λ light chains are more frequently amyloidogenic than κ light chains and that, among λ light chains, those derived from some genes such as Vλ3r (λIII family) Citation5 and Vλ6a have a strong propensity to form amyloid fibrils. Comparative analyses of tertiary structure and folding/unfolding dynamics of immunoglobulin light chains are complicated by the intrinsic structural variability of these proteins and the difficulty in identifying which mutations determine the amyloidogenic state. Nevertheless, it is through the comparison of recombinant immunoglobulin light chains with specific mutations identified in AL patients that the pathogenic relevance of the folding/unfolding process was first hypothesized Citation6. On the other hand, crystallographic studies focused on normal and amyloidogenic immunoglobulin light chains have not offered additional insights about the pathogenesis of the disease, but they did demonstrate that a pathologic light chain can acquire a three‐dimensional structure indistinguishable from that of a non‐amyloidogenic light chain.
Amyloidogenic proteins in hereditary amyloidosis
Studies on immunoglobulin light chains have found extensive confirmation by observations obtained with several globular proteins that acquire an amyloidogenic state as a consequence of genetic mutation. Biochemical and biophysical attention has been focused on transthyretin and lysozyme. Transthyretin is a homotetrameric protein with a predominant β‐sheet secondary structure, whereas lysozyme consists of a single polypeptidic chain with a prominent α‐helix organization. To date, more than 100 transthyretin mutations have been reported: some are not associated with amyloidosis, and a few variants protect against the development of the disease. Of the five reported lysozyme variants, only one, Thr70Asn, which is the most frequent mutation in the population, fortunately, is not related to amyloidosis Citation7. Once again, the major contribution to the pathogenetic interpretation originated from studies on the folding/unfolding dynamics of pathologic variants.
Amyloidosis as a prototype of conformational diseases
The generic definition of folding/unfolding dynamics requires a particular predisposition of amyloidogenic globular proteins to acquire more than one conformation during their extra‐cellular biological life. This process is permitted and is highly enhanced with regard to the wild‐type form, because even point mutations can cause the loss of intramolecular bonds which play an important role in maintaining the stability of the globular structure. For proteins with a complex quaternary structure, such as transthyretin, destabilization translates into a weaker cohesion between monomers, whereas for proteins consisting of single polypeptides, like lysozyme, it interferes with the intramolecular bonds responsible for their tertiary structure.
The level of stability in tertiary and quaternary structures of a protein can be measured in energetic terms, and the entity of reduction in stability—expressed in kcal per mole of protein—is known for each amyloidogenic mutation. Biological examples illustrating the role of thermodynamic stability of the three dimensional structure are available for lysozyme and transthyretin. The folding stability of the non‐amyloidogenic Thr70Asn lysozyme variant is between that of the wild‐type lysozyme and its amyloidogenic variants Citation8. The Thr119Met variant of transthyretin, which protects against amyloid formation, has a stabilizing effect which is superior to that of the wild‐type protein and is able to counterbalance the reduction in stability of the amyloidogenic transthyretin variant. Thus in heterozygote subjects manifesting the Thr119Met transthyretin in association with an amyloidogenic variant (e.g. Val30Met), the potentially amyloidogenic transthyretin does not cause disease Citation9.
The reduced intrinsic stability of the native state of the protein is necessary, but not sufficient, to confer a propensity to form amyloid. Amyloid fibril formation requires the concomitant presence of other features directly involving the protein or its physico‐chemical nanoenvironment.
Pathogenetically important elements concerning the primary structure of amyloid fibrils include the distribution of amino acid residues with a global positive or negative charge, the propensity of the polypeptide chain to assume a predominant α or β conformation, and the representation of hydrophobic residues. Experimental data obtained from studies on short peptides have emphasized that the characteristics promoting fibrillar aggregation include the neutralization of charges of opposite sign, the abundance of residues favouring the β‐sheet secondary structure, and the hydrophobicity of side chains. On the other hand, the presence of some key residues like proline, interrupting the β‐strand conformation, and the proximity of self‐repulsing amino acid residues having the same charge, inhibit fibrillar aggregation. These single amino acid residues which inhibit aggregation are called ‘structural gatekeepers’ Citation10. Several algorithms capable of predicting the aggregation propensity of amyloidogenic peptides (such as Aβ peptides of Alzheimer's disease) have been developed, and their results are in complete agreement with experimental measurements Citation11. The possibility of estimating the aggregation propensity of globular proteins is more complex than for short peptides. Nevertheless, there are several projects which are attempting to deal with this question Citation12.
The extensive and frequent fluctuations between a compact and folded conformation and a partially unfolded structure require that the protein assume more than one molecular form. The use of the term ‘conformational disease’ appropriately describes this aspect of diseases associated with protein aggregation and was first introduced in 1997 by Carrell and Lomas Citation13 to define a non‐amyloid protein aggregation disease such as that caused by intracellular aggregation of the pathologic α1‐antitrypsin variants. Another meaningful term is ‘chameleon protein’ which illustrates the conformational multiplicity acquirable by an amyloidogenic protein and was introduced in the same year by Max Perutz Citation14.
Proteomics of amyloid deposits: common components and tissue tropism mechanisms
The biochemical and biophysical characterization of amyloid proteins has provided the basis for the interpretation of events culminating in the formation of amyloid fibrils, but does not explain the organ selectivity of this process, which varies according to the different amyloidogenic proteins and, to a certain extent, to the different mutations of the same protein.
A less recent acquisition is that amyloid fibrils extracted from tissues contain some common components, which are expressed in all amyloid deposits, irrespective of tissue origin or amyloid type. One of these ubiquitous molecules is serum amyloid P component (SAP), a glycoprotein with a specific binding site for the common conformation of amyloid, located on the B surface of the pentameric form of the protein. The common conformational motif of amyloid which is recognized at high affinity by SAP has not been identified, but it is presumed to be present in all types of amyloid fibrils. SAP plays an important role in the process of amyloid deposition in vivo, due to its ability to protect fibrils from several proteases, thus enabling amyloid deposits to escape from proteolytic reabsorption Citation15.
Proteoglycans represent additional important common components of amyloid deposits and contribute extensively to the carbohydrate composition of amyloid fibres Citation16. Their role is quite complex and seems to act on two different levels: the genesis and the structural stabilization of amyloid fibrils. There is a large amount of information concerning the structure and function of the carbohydrate component of proteoglycans, consisting of glycosaminoglycans with different degrees of sulphation. Proteoglycans could represent the initial structural scaffold which facilitates the adhesion and orientation of the first nuclei of aggregated amyloid protein. In vitro studies clearly demonstrate that heparin and its analogues are able to accelerate the kinetics of fibrillar aggregation of peptides in Alzheimer's disease Citation17, prionic protein Citation18, serum amyloid A (SAA) Citation16, β2‐microglobulin Citation19 and gelsolin Citation20. Despite being a common component of all amyloid deposits, proteoglycans show a certain degree of chemical and structural heterogeneity, depending upon the tissue. Thus, they may play a targeting role in the localization of amyloid deposits in tissues. The fundamental role of a particular glycosaminoglycan (heparan sulphate) in the genesis of amyloid deposits has been elegantly demonstrated in a transgenic experimental model in which the fragmentation of heparan sulphate by heparanase overexpression resulted in resistance against the induction of reactive amyloidosis (AA amyloidosis) Citation21. Other common elements found in amyloid deposits are components of the extra‐cellular matrix, such as laminin, entactin, and collagen IV.
In order to place the biochemical and biophysical evidence within the complex biological scenario, it is necessary to determine in detail the metabolism of proteins expressed in tissues in which amyloid forms, and to detect the interactions between tissue proteins and amyloid fibrils. A simplified model of this problem is the deposition of amyloid fibrils formed by β2‐microglobulin in tissues where collagen type I and III are highly represented. Our group recently demonstrated that the interaction with collagen, though at low affinity (∼100 µM), is effective in generating amyloid fibrils under physical conditions comparable with the biological setting Citation22.
For other types of amyloidosis, tissue proteomics will provide the data required for developing suitable experimental models. At present, efforts mainly focus on cerebral tissue proteomics in the context of amyloid neurodegenerative diseases. These studies indicate that some categories of proteins—particularly the molecular chaperonins, matrix proteins and some energetic mitochondrial metabolism proteins—may play an active role in modulating amyloid deposition. Moreover, proteomic techniques enable the distinction of post‐translational protein modifications within natural amyloid deposits, avoiding complex chromatographic purifications (used in the past), which per se could cause some chemical modifications. We are convinced that the most frequent and important post‐translational modification is represented by proteolytic cleavage.
There has been a lively debate, which is still in progress, on the pathogenic role of proteolytic remodelling of the precursor protein in different types of amyloidosis. In some cases, such as Alzheimer's disease, it is universally accepted that proteolytic cleavage of the precursor protein is indispensable for amyloid formation. In other cases, such as in AL amyloidosis, some investigators are persuaded that proteolysis may occur only when the amyloid has already formed and is deposited in tissues. Despite uncertainties on the timing of the proteolytic event during in vivo fibrillogenesis, the fine determination of proteolytic fragments expressed within natural amyloid fibrils is of paramount importance to develop in vitro models mimicking the biological microenvironment. A paradigmatic case is Alzheimer's disease, in which the presence of the additional amino acid residues at the C‐terminus of Aβ peptides significantly alters the kinetics of fibril aggregation.
Mechanism of tissue damage
The deposition of amyloid fibrils results in cellular injury, tissue damage, and finally organ dysfunction, through mechanisms not completely understood. The simple explanation of a physical, mechanical substitution of parenchymal tissue by amyloid deposits seems to be insufficient. Increasing importance is now attributed to soluble prefibrillar aggregates, which may exert a cytotoxic effect and promote the apoptotic pathway. The cytotoxicity of prefibrillar aggregates has been extensively demonstrated for Aβ Citation23,24 and transthyretin amyloidosis Citation25.
The role of amyloid fibrils and soluble prefibrillar aggregates in the genesis of cytotoxicity is still debated, as the former contributes to pathogenicity according to histopathogenic observations Citation26, and the latter is regarded as an effector of cytotoxicity from a cellular biochemical and biophysical point of view Citation27. In this context, clinical observation offers a different perspective. Possibly, tissue damage requires the presence of both components: amyloid fibrils and prefibrillar aggregates. There is no sign of disease in the absence of deposits, whereas the elimination of the soluble fibrillar precursor may allow the clearance of signs and symptoms of disease, despite the persistence of unmodified amyloid deposits. This evidence derives from sporadic cases described in the past and particularly from a study conducted by our group in AL amyloidosis, which demonstrated that the reduction of the amyloidogenic free light chain concentration caused by chemotherapy translates into a simultaneous reduction of the serum concentration of the amino‐terminal fragment of pro‐brain natriuretic peptide (NT‐proBNP), a marker of cardiac dysfunction, despite unaltered amyloid deposits in the myocardium as assessed by echocardiography Citation28,29.
An intriguing hypothesis is the creation of a synergic effect between amyloid fibrils and their precursors, exerting their influence on cells situated in proximity of amyloid deposits. A possible explanation might derive from the recent observation Citation30 that an equilibrium is reached, at the growing surface of the fibre, between associating (Kon) and dissociating (Koff) molecules, according to a reversible reaction, with Kon>Koff during the growing phase of the fibre. According to this theory, the Kon–Koff combination at the interface of the growing surface of the fibre would create a high concentration gradient of precursor monomers and oligomers.
Is amyloidosis a protein‐mediated transmissible disease?
AA amyloidosis can be induced in some strains of mice by repeated injections of phlogistic stimuli, such as silver nitrate or casein. After a lag phase of several weeks, a disease very similar to human AA amyloidosis originates. Amyloid fibrils are derived from the acute phase reactant mouse serum AA1. In the 1960s it was reported that the lag phase in the experimental model is dramatically shortened by the administration of an extract of amyloid tissue from another animal. This elusive transmissible element was then called ‘amyloid enhancing factor’ (AEF), and its nature remained obscure for considerable time. There is now general agreement that preformed amyloid fibrils are the essence of the AEF Citation31. In vitro experiments with different types of amyloid polypeptides, in particular with Aβ and islet amyloid polypeptide, demonstrated that seeding a solution of native protein with preformed amyloid fibrils of the same chemical nature abolishes the lag phase, consistently increasing the rate of formation of new amyloid fibrils Citation32,33. Synthetic amyloid fibrils were demonstrated to exert a typical AEF effect in experimental murine AA amyloidosis, acting as a nucleation factor for in vivo newly formed amyloid fibrils Citation34,35. Further studies in the murine model have shown that AA amyloid fibrils are highly infectious even when highly diluted and orally administrated, a mechanism very similar to that of prionoses Citation36. At present, there is no evidence of human transmissibility for any amyloid disease, except for the prion diseases. However, clinical evidence suggests that the formation of amyloid deposits may be enhanced by the presence of fibrillar nuclei. In patients with hereditary transthyretin amyloidosis and initial deposits in the heart receiving a liver transplant, the inexorable progression of amyloid deposits in myocardium takes place, despite the almost complete elimination of circulating mutated transthyretin Citation37 because wild‐type transthyretin accumulates in myocardium Citation38. This observation underscores the necessity of performing timely liver transplantation in patients with familial transthyretin amyloidosis, before the formation of a nidus of amyloid deposits within the myocardium occurs, in order to fully and substantially exploit the benefits of the transplanted organ.
Conclusions
The attempt to create a schematic synopsis of the molecular mechanisms of amyloidosis inevitably brings to light several paradoxes. One of the controversial aspects concerns the relationship between the structural heterogeneity of amyloidogenic proteins and the clinical heterogeneity of different types of amyloidosis. One enigmatic aspect is that amyloidogenic precursors are supposed to lose a great part of their structural heterogeneity and functional individuality in order to acquire their pathologic role. From a single, largely shared pathogenic agent, one would expect a single disease, whereas it is paradoxical that amyloid deposits are manifested as diverse diseases differing for the organ involved and, within the same organ, for tissues or cellular types affected. This enigma will hopefully be solved when two key steps of the pathologic process are better understood: 1) the connection between the mechanism of amyloid fibril formation and environmental conditions which render the process possible in vivo; and 2) the mechanism of cellular and tissue damage due to amyloid deposition. The most mysterious part of the process is that represented by the conformational heterogeneity, which leads to heterogeneity of a clinical phenotype. This domain sets forth several complex, multifactorial questions, which are difficult to explore; in coming years, basic, clinical and pharmaceutical research will have to face these challenges.
It is possible that the pathogenetic complexity requires the integration of several therapeutic interventions involving different critical levels of the amyloidogenic cascade. For instance, therapeutic strategies aimed at reducing the concentration of the circulating amyloidogenic protein could be associated with the use of molecules inhibiting the interaction between amyloid precursor and tissue factors involved in organ tropism and consequent tissue damage.
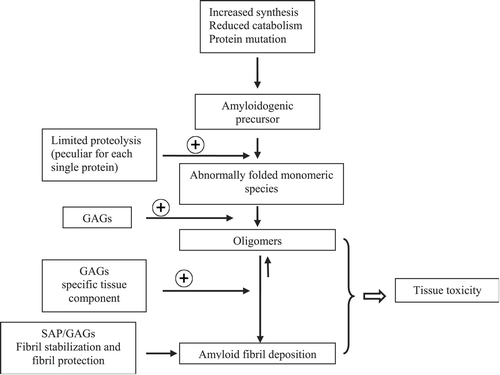
Figure 1. Pathogenic cascade of amyloidosis. The⌖‐symbol indicates a positive effect on the reaction kinetics. Amyloidogenic proteins can be synthesized in excess and persist at a high serum concentration, as in the case with immunoglobulin monoclonal light chains or acute phase protein serum amyloid A protein. Alternatively, amyloid proteins can achieve high serum levels due to reduced catabolism, as with β2‐microglobulin in chronic haemodialysis. In hereditary amyloidoses, the amyloidogenic precursor differs from its normal counterpart due to the presence of a mutation. Some proteins with an intrinsic amyloidogenic predisposition can, at a very low rate, form amyloid deposits, which clinically manifest only in the elderly, such as transthyretin in senile systemic amyloidosis. Some of the amyloidogenic proteins undergo a proteolytic cleavage, which promotes polymerization. Oligomers in equilibrium with fibrils can exert a direct cytotoxic effect. The interaction between amyloidogenic proteins, tissue factors and some common components of amyloid deposits, such as glycosaminoglycans (GAGs) and serum amyloid P component (SAP), induce the formation and persistence of tissue deposits, which contribute to causing functional damage of affected organs.
Acknowledgements
MN, GP, and FL are partly supported by an investigator fellowship from Collegio Ghislieri, Pavia, Italy.
Related Research Data
References
- Merlini G., Bellotti V. Molecular mechanisms of amyloidosis. N Engl J Med 2003; 349: 583–96
- Relini A., Rolandi R., Bolognesi M., Aboudan M., Merlini G., Bellotti V., et al. Ultrastructural organization of ex vivo amyloid fibrils formed by the apolipoprotein A–I Leu174Ser variant: an atomic force microscopy study. Biochim Biophys Acta 2004; 1690: 33–41
- Serpell L. C., Sunde M., Benson M. D., Tennent G. A., Pepys M. B., Fraser P. E. The protofilament substructure of amyloid fibrils. J Mol Biol 2000; 300: 1033–9
- Merlini G., Bellotti V. Lysozyme: a paradigmatic molecule for the investigation of protein structure, function and misfolding. Clin Chim Acta 2005; 357: 168–72
- Perfetti V., Casarini S., Palladini G., Vignarelli M. C., Klersy C., Diegoli M., et al. Analysis of V lambda–J lambda expression in plasma cells from primary (AL) amyloidosis and normal bone marrow identifies 3r (lambda III) as a new amyloid‐associated germline gene segment. Blood 2002; 100: 948–53
- Wetzel R. Domain stability in immunoglobulin light chain deposition disorders. Adv Protein Chem 1997; 50: 183–242
- Esposito G., Garcia J., Mangione P., Giorgetti S., Corazza A., Viglino P., et al. Structural and folding dynamics properties of T70N variant of human lysozyme. J Biol Chem 2003; 28: 25190–8
- Johnson R. J., Christodoulou J., Dumoulin M., Caddy G. L., Alcocer M. J., Murtagh G. J., et al. Rationalising lysozyme amyloidosis: insights from the structure and solution dynamics of T70N lysozyme. J Mol Biol 2005; 352: 823–36
- Sebastiao M. P., Lamzin V., Saraiva M. J., Damas A. M. Transthyretin stability as a key factor in amyloidogenesis: X‐ray analysis at atomic resolution. J Mol Biol 2001; 306: 733–44
- Chiti F., Calamai M., Taddei N., Stefani M., Ramponi G., Dobson C. M. Studies of the aggregation of mutant proteins in vitro provide insights into the genetics of amyloid diseases. Proc Natl Acad Sci U S A 2002; 99(Suppl 4)16419–26
- Fernandez‐Escamilla A. M., Rousseau F., Schymkowitz J., Serrano L. Prediction of sequence‐dependent and mutational effects on the aggregation of peptides and proteins. Nat Biotechnol 2004; 22: 1302–6
- Chiti F., Dobson C. M. Protein misfolding, functional amyloid, and human disease. Annu Rev Biochem 2006; 75: 333–66
- Carrell R. W., Lomas D. A. Conformational disease. Lancet 1997; 350: 134–8
- Perutz M. F. Amyloid fibrils. Mutations make enzymes polymerize. Nature 1997; 385: 773–5
- Tennent G. A., Lovat L. B., Pepys M. B. Serum amyloid P component prevents proteolysis of the amyloid fibrils of Alzheimer disease and systemic amyloidosis. Proc Natl Acad Sci U S A 1995; 92: 4299–303
- Kisilevsky R. Review: Amyloidogenesis—unquestioned answers and unanswered questions. J Struct Biol 2000; 130: 99–108
- Castillo G. M., Lukito W., Wight T. N., Snow A. D. The sulfate moieties of glycosaminoglycans are critical for the enhancement of beta‐amyloid protein fibril formation. J Neurochem 1999; 72: 1681–7
- Diaz‐Nido J., Wandosell F., Avila J. Glycosaminoglycans and beta‐amyloid, prion and tau peptides in neurodegenerative diseases. Peptides 2002; 23: 1323–32
- Yamamoto S., Yamaguchi I., Hasegawa K., Tsutsumi S., Goto Y., Gejyo F., et al. Glycosaminoglycans enhance the trifluoroethanol‐induced extension of beta(2)‐microglobulin‐related amyloid fibrils at a neutral pH. J Am Soc Nephrol 2004; 15: 126–33
- Suk J. Y., Zhang F., Balch W. E., Linhardt R. J., Kelly J. W. Heparin accelerates gelsolin amyloidogenesis. Biochemistry 2006; 45: 2234–42
- Li J. P., Galvis M. L., Gong F., Zhang X., Zcharia E., Metzger S., et al. In vivo fragmentation of heparan sulfate by heparanase overexpression renders mice resistant to amyloid protein A amyloidosis. Proc Natl Acad Sci U S A 2005; 102: 6473–7
- Relini A., Canale C., De Stefano S., Rolandi R., Giorgetti S., Stoppini M., et al. Collagen plays an active role in the aggregation of beta 2‐microglobulin under physio‐pathological conditions of dialysis‐related amyloidosis. J Biol Chem 2006; 24: 16521–9
- Lambert M. P., Barlow A. K., Chromy B. A., Edwards C., Freed R., Liosatos M., et al. Diffusible, nonfibrillar ligands derived from Abeta1‐42 are potent central nervous system neurotoxins. Proc Natl Acad Sci U S A 1998; 95: 6448–53
- Hartley D. M., Walsh D. M., Ye C. P., Diehl T., Vasquez S., Vassilev P. M., et al. Protofibrillar intermediates of amyloid beta‐protein induce acute electrophysiological changes and progressive neurotoxicity in cortical neurons. J Neurosci 1999; 19: 8876–84
- Andersson K., Olofsson A., Nielsen E. H., Svehag S. E., Lundgren E. Only amyloidogenic intermediates of transthyretin induce apoptosis. Biochem Biophys Res Commun 2002; 294: 309–14
- Pepys M. B. Amyloidosis. Annu Rev Med 2006; 57: 223–41
- Cecchi C., Pensalfini A., Baglioni S., Fiorillo C., Caporale R., Formigli L., et al. Differing molecular mechanisms appear to underlie early toxicity of prefibrillar HypF‐N aggregates to different cell types. FEBS J 2006; 273: 2206–22
- Palladini G., Campana C., Klersy C., Balduini A., Vadacca G., Perfetti V., et al. Serum N‐terminal pro‐brain natriuretic peptide is a sensitive marker of myocardial dysfunction in AL amyloidosis. Circulation 2003; 107: 2440–5
- Palladini G., Lavatelli F., Russo P., Perlini S., Perfetti V., Bosoni T., et al. Circulating amyloidogenic free light chains and serum N‐terminal natriuretic peptide type B decrease simultaneously in association with improvement of survival in AL amyloidosis. Blood 2006; 10: 3854–8
- Carulla N., Caddy G. L., Hall D. R., Zurdo J., Gairi M., Feliz M., et al. Molecular recycling within amyloid fibrils. Nature 2005; 436: 554–8
- Niewold T. A., Gruys E., Kisilevsky R., Shirahama T. S. Fibril amyloid enhancing factor (FAEF)‐accelerated amyloidosis in the hamster is not dependent on serine esterase activity and mononuclear phagocytosis. Scand J Immunol 1991; 34: 101–7
- Jarrett J. T., Lansbury P. T., Jr. Seeding ‘one‐dimensional crystallization’ of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie?. Cell 1993; 73: 1055–8
- Kelly J. W. Mechanisms of amyloidogenesis. Nat Struct Biol 2000; 7: 824–6
- Ganowiak K., Hultman P., Engstrom U., Gustavsson A., Westermark P. Fibrils from synthetic amyloid‐related peptides enhance development of experimental AA‐amyloidosis in mice. Biochem Biophys Res Commun 1994; 199: 306–12
- Johan K., Westermark G., Engstrom U., Gustavsson A., Hultman P., Westermark P. Acceleration of amyloid protein A amyloidosis by amyloid‐like synthetic fibrils. Proc Natl Acad Sci U S A 1998; 95: 2558–63
- Lundmark K., Westermark G. T., Nystrom S., Murphy C. L., Solomon A., Westermark P. Transmissibility of systemic amyloidosis by a prion‐like mechanism. Proc Natl Acad Sci U S A 2002; 99: 6979–84
- Olofsson B. O., Backman C., Karp K., Suhr O. B. Progression of cardiomyopathy after liver transplantation in patients with familial amyloidotic polyneuropathy, Portuguese type. Transplantation 2002; 73: 745–51
- Yazaki M., Tokuda T., Nakamura A., Higashikata T., Koyama J., Higuchi K., et al. Cardiac amyloid in patients with familial amyloid polyneuropathy consists of abundant wild‐type transthyretin. Biochem Biophys Res Commun 2000; 274: 702–6