Abstract
Ischemia‐reperfusion injury of the myocardium has long been a subject of intense research. Cardiac preconditioning, an associated phenomenon, has also been critically investigated over the past two decades. Although the biochemistry of ischemia‐reperfusion and its association with oxidative metabolism has long been established, recent studies have further revealed a more intricate role of a number of reactive oxygen‐nitrogen species in those processes. Emerging evidence suggests that an elaborate network of enzymes (and other biomolecules) dedicated to the generation, utilization, and diminution of reactive oxygen‐nitrogen species maintains the redox homeostasis in the myocardium, and any perturbation of its status has distinctive effects. It thus appears that while excessive generation of reactive species leads to cellular injury, their regulated generation may cause transient and reversible modifications of cellular proteins leading the transmission of intracellular signals with specific effects. Taken together, generation of reactive oxygen‐nitrogen species in the myocardium plays a nodal role in mediating both ischemic injury and cardioprotection.
| Abbreviations | ||
| GSH | = | glutathione |
| GSSG | = | glutathione disulfide |
| Akt | = | protein kinase B |
| PI | = | phosphatidylinositol |
| MAPK | = | mitogen‐activated protein kinase |
| iNOS | = | inducible nitric oxide synthase |
| GPCR | = | G protein couple receptor |
| Ang | = | angiotensin |
| IGF | = | insulin‐like growth factor |
| EGF | = | epidermal growth factor |
| HO | = | heme oxygenase |
| Nrf | = | NF‐E2‐related factor‐2 |
| ECH | = | epithelioid cell histiocytoma |
Cardiac ischemia—the leading cause of heart failure, acute myocardial infarction, and arrhythmias—has long been a major contributor towards infirmity in the industrialized societies and thus is a subject of intense research Citation1. Reperfusion injury, an essential consequence of the restoration of blood supply to the ischemic myocardium, has also been a subject of intense interest Citation2. Advantageously, following the seminal observation in the mid‐80s that several cycles of brief ischemia followed by reperfusion renders the heart more resistant towards reperfusion injury had led to the concept of ‘ischemic preconditioning’ and the feasibility of ‘cardioprotection’ Citation3–6. However, in spite of the molecular, pharmacological, and biochemical characterization of a plethora of ‘pro‐survival’ and ‘pro‐death’ molecules and their modulators, ‘cardioprotection’, especially in the clinical setup, still remains exasperatingly elusive Citation6,7. Such disillusionment can at least be partially attributed to the consideration of cell signaling and gene regulatory pathways in isolation, rather than in the context of the organismal and cellular milieu. Nevertheless, cardiac biologists over the years have accumulated a wealth of information regarding the role of reactive oxygen species and redox metabolism in cardiac injury as well as protection. However, how the respective signaling pathways ultimately integrate into either pro‐survival or pro‐death phenomenon is yet to be fully understood Citation8,9. The present review is aimed towards the compilation of a number of emerging concepts on redox‐metabolism in the ischemic myocardium, especially in the context of preconditioning.
Key messages
Reactive oxygen‐nitrogen species play a nodal role in ischemia‐reperfusion injury as well as preconditioning of the myocardium.
Regulated generation of certain reactive species may initiate specific cell signaling events with distinct consequences.
Based on the type, amplitude, and duration of the reactive species generated, cardiac myocytes may opt for death or survival pathways.
Reactive oxygen/nitrogen species (ROS/RNS) as modulators of cellular functions
During the past several decades, our understanding of the roles of free radicals in cellular and organismal biology has painstakingly evolved. Although once it was believed that generation of free radicals/reactive oxygen species (ROS) are detrimental to the living organisms, subsequent studies have established that production of ROS to a limited extent may lead to distinctive cellular events like cell proliferation and differentiation Citation10–13. Free radicals are characterized by the presence of one or more unpaired electrons in the outermost orbital of their constituent atoms and thus are extremely reactive (and labile) as compared to their normal counterparts with paired electrons. Furthermore, certain compounds, namely hydrogen peroxide, are not free radicals per se, but in the presence of transition metals it generates highly active hydroxyl radicals (OH·) and thus is a ROS in specific biological contexts (14,15). ROS are generated in various cell types either under normal physiological conditions to mediate certain regulatory functions or under the influence of certain pathological stimuli with deleterious consequences Citation16. As an example, superoxide free radical (O2·−) is produced by nicotinamide adenine dinucleotide phosphate (NADPH)‐oxidase complex in phagocytic cells for the purpose of killing invading microbes Citation17. Exogenous factors like cytokines and growth factors, environmental toxicants like tobacco smoke, ultra‐violet radiation, etc., and intracellular signal mediators like sphingolipids may also cause intracellular ROS generation, thereby influencing cellular events Citation18–20. In addition to ROS, a number of nitrogen‐containing free radicals have also emerged during the past decade as the key mediators of various pathophysiological events. Since the monumental discovery that nitric oxide (NO·) can act as an intracellular signal transducer, numerous studies have established it as a modulator of disparate cellular events such as dilatation of blood vessels, neurotransmission, inflammatory response, and regulation of gene expression Citation21,22. Also, nitric oxide can interact with other oxidants like superoxide radical (O2·−), hydrogen peroxide (H2O2), or transition metal centers resulting in the generation of a number of highly reactive entities, collectively known as reactive nitrogen species (RNS) Citation23. Taken together, due to such close interrelationship between reactive oxygen and nitrogen species, their role(s) in biology are often considered in conjunction with one another.
Intracellular generation of ROS/RNS
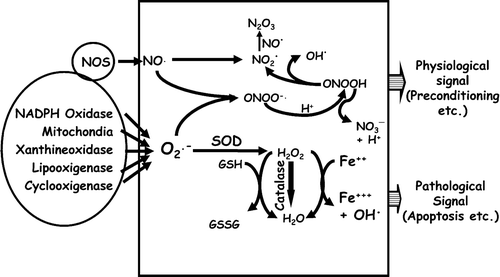
Mitochondria and peroxisomes are the two major intracellular sources of ROS generation. The mitochondrial electron transport chain (complex I to IV) is a highly structured assembly of redox proteins dedicated to the generation of a proton gradient (ΔpH) and a membrane potential (ΔΨm) across the inner mitochondrial membrane resulting in ATP synthesis. When these two values are sufficiently high, electron transport is inhibited by a feedback mechanism, leading to an accumulation of electrons which then leak out and concomitantly bind to oxygen, thereby generating O2·−. Amongst four electron transport complexes, complex I and III are considered to be the primary producers of O2·−Citation24–26. Although O2·− is highly reactive, it is instantaneously converted into hydrogen peroxide by superoxide dismutase (Mn‐SOD in the mitochondria and Cu/Zn‐SOD in the cytosol). Thereafter, H2O2 is converted to oxygen and water by catalase. However, under certain circumstances, especially due to excess generation, O2·− may escape this attenuation process thereby producing other ROS causing oxidative injuries Citation27. Like ROS, a number of RNS can also be generated by various intracellular pathways. As an example, NO· reacts with O2·− by a diffusion‐controlled reaction generating peroxynitrite (ONOO−) which is of potential importance in certain cell types such as macrophages and endothelial cells which simultaneously produce O2·− and NO· Citation28. Peroxynitrite can cause oxidation and nitration of proteins (at thiols, methionine, and tyrosine) as well as lipid peroxidation Citation29. Also upon protonation, peroxynitrite produces ONOOH, which decays either via production of NO2· (nitrogen dioxide radical) and OH·, or NO3− and H+Citation23. Potential protective roles of nitrite and nitrate anions (and nitric oxide) in ischemia‐reperfusion setup have been documented Citation30. Also, excessive generation of NO2· may lead to the formation of N2O3, a potent nitrosating agent Citation31. Generation of other reactive nitrogen species such as nitroxyl radical (HNO), especially in the context of ischemic brain, has also been described Citation32,33. As described in the following sections, and summarized in , many of these ROS/RNS have been implicated in cell signaling, especially when generated at a lower doses, while their excessive generation often leads to the oxidative modifications of cellular proteins (such as formation of carbonyls, oxidation of methionines/cysteines and cross‐linking of polypeptides) with deleterious consequences Citation34–37.
Figure 1. Generation and effects of intracellular ROS/RNS: Superoxide (O2⋅−) and nitric oxide (NO·) generated from various intracellular sources are identified by oval‐shaped boxes. Those two radicals can be converted into various other ROS/RNS by mutually interactive pathways as described in the text. Depending on type, combination, amplitude, and duration of those radicals the respective signals are finally integrated into an output‐like preconditioning effect (physiological) or apoptosis (pathological).
Redox equilibrium and cell fate
During evolution, while aerobic organisms acquired the ability to harness energy by reducing atmospheric oxygen, a plethora of other redox‐sensitive molecules carrying out diverse cellular functions has also emerged. Whenever ROS/RNS is generated in living cells, it reacts with lipids, proteins, carbohydrates, and nucleic acids, and the outcome depends upon the cell type and the nature, intracellular localization, amplitude, and the life span of the reactive species Citation38–43. Furthermore, since redox homeostasis is an essentiality of normal cellular functions, nature has also evolved an extensive network of antioxidant defense systems attenuating various ROS/RNS. Antioxidant enzymes like superoxide dismutase (converts O2·− into H2O2), catalase (converts H2O2 into water), glutathione peroxidase (converts H2O2 into water), thioredoxin‐thioredoxin reductase, glutaredoxin‐glutaredoxin reductase, and small molecules like vitamin E and C thus play a major role in maintaining the redox equilibrium Citation44–46. Thus, an intricate network of enzymes (and other biomolecules) dedicated to the generation, utilization, and diminution of ROS/RNS maintains intracellular redox, and any perturbation of redox‐status has distinctive effects Citation43,Citation47–50. Emerging evidences suggest that while general oxidative modifications of proteins have deleterious effects (on both structure and functions), controlled oxidation might have regulatory consequences Citation51–53. Thus, based upon the reactive species and its targets, the effects can be divergent Citation54–58.
ROS/RNS as second messengers
Although the intracellular generation of ROS/RNS and its role in oxidative/nitrosative modifications of proteins have been documented for several decades, their potential role in intracellular signaling became apparent following the discovery of nitric oxide as a transducer of specific signals Citation21. Subsequent years have seen a paradigm shift when other reactive species emerged as the potential mediators of intracellular communications Citation59. Increasing evidence suggests that ROS/RNS can cause transient and reversible modifications (oxidative/nitrosative) of proteins, especially when momentary generated, resulting in loss‐gain of functions, thereby acting as second messengers Citation59,60. Second messengers are generated upon receptor stimulation, are of short lifespan, and act upon specific targets. In that context, while ROS/RNS are also generated upon receptor stimulation and are very transient in nature, their specificity of action (except that of nitric oxide) has often been a subject of intense debate Citation59,Citation61. As an example, while a highly reactive species like OH·− is unlikely to have any specificity, others like O2· and H2O2 are highly diffusible, less reactive, and therefore might act in a specific manner Citation62,63. Generation of ROS/RNS has been attributed to the modulation of a number of signaling/gene regulatory modules including MAP kinase, PI3 kinase/Akt, transcription factors AP‐1 and NFκB Citation64–67. However, only a few direct targets of oxidative/nitrosative modifications have been identified as yet, and those are primarily the cysteine residues present in the catalytic sites of the respective proteins Citation59,Citation68,69. Noticeably, a number of nonsignaling regulatory modules like ryanodine receptor, sarco/endoplasmic Ca2+ pumps (SERCA) have also been identified as the targets of oxidative modifications in the context of cardiovascular biology. Ryanodine receptor contains a large number of cysteine residues, and redox‐modifications of at least some of those modulate its function Citation70,71. Nitric oxide (NO) can also form S‐nitroso derivatives of some of those cysteine residues and thereby play a role in modulating its functions Citation72. Similarly, peroxynitrite inhibits SERCA activity by oxidative modifications thereby affecting calcium transients Citation73.
Thioredoxin/glutaredoxin systems and redox‐homeostasis
Although the side chains of various amino acids are amenable to oxidative and nitrosative modifications, those of cysteine thiols have drawn significant attention Citation74. Many of the cysteine oxidations are reversible and might constitute a highly evolved mechanism of activation‐inactivation of cognate proteins like that by phosphorylation‐dephosphorylation Citation75,76. Oxidative modifications of cysteines also play a critical role in pathobiology in general and cardiovascular biology in particular Citation77. Emerging evidence suggests that a group of enzymes and low molecular weight peptides, namely thioredoxin and glutaredoxin oxidoreductases, plays a key role in restoring protein thiols and thereby maintaining redox homeostasis. Although these proteins are functionally equivalent to free radical scavengers, their role(s) are much wider in the context of metabolism, signaling, and gene regulatory events Citation78–80. Thioredoxin is a general disulphide reductase with a low redox potential and thus capable of acting upon a wide range of oxidized proteins including thioredoxin peroxidase, which is a ROS scavenger. In mammals, there are two thioredoxins, namely Trx1, found in cytosol and the nucleus, while Trx2 is mitochondrial Citation81,82. Trx1 can also be secreted out and act as a chemokine. Heart failure patients show elevated levels of plasma thioredoxin as an adaptive response towards increased oxidative stress Citation83. Thioredoxin also provides protection against oxidative injuries caused by adriamycin and reperfusion Citation84. Protective effects of thioredoxin are mediated via its interaction with a diverse family of regulatory molecules like apoptotic signal regulating kinase 1 (ASK‐1), vitamin D3‐upregulated protein‐1 (VDUP‐1), gene regulatory proteins NFkB and AP‐1. Noticeably, thioredoxin can attenuate ASK‐1 function both in a redox‐dependent and ‐independent manner Citation85,86. VDUP‐1 exerts its pro‐apoptotic effect by the suppression of thioredoxin activity Citation87,88. Thioredoxin has also been attributed to the increased expression of antioxidant genes like Mn‐SOD and heme oxygenase‐1 Citation89. Our laboratory has demonstrated that in ex vivo working heart, the thioredoxin level decreases upon ischemia, while preconditioning upregulates its expression Citation90,91. Treatment with thioredoxin inhibitor cis‐diammine‐dichloroplatinum abolishes cardioprotection by preconditioning. Transgenic mouse hearts overexpressing thioredoxin‐1 shows significant improvement in postischemic ventricular recovery and reduced myocardial infarct size, further supporting its nodal role in cardioprotection Citation90,91.
Like thioredoxin, the glutaredoxin system comprising glutaredoxin, glutathione, and glutathione reductase also plays a key role in maintaining intracellular redox equilibrium. In mammals, there are two glutaredoxin genes, namely Grx 1 and Grx 2. In human, while Grx1 is cytosolic and is involved in the reduction of various enzymes, Grx2 has two splice variants localized in the nucleus and the mitochondrion Citation92. The mitochondrial electron transport chain is a major source of ROS that leads to a reduced GSH/GSSG ratio and glutathionylation of various proteins including the NADH‐binding pockets of complex I (thereby further increasing the ROS generation). Grx 2 plays a major role in regenerating key mitochondrial proteins by deglutathionylation Citation93. In agreement, ablation of Grx 2 by RNAi results in increased susceptibility towards cell death by inducers of ROS like doxorubicin Citation94. In a recent study, decrease in glutaredoxin (and reduced glutathione), in conjunction with increased oxidant levels, has been documented in cardiac interfibrillar (but not in subsarcolemmal) mitochondria of old rats Citation95. Also, in cardiac myoblast cell line H9c2, 17 beta‐estradiol prevents oxidative stress‐induced apoptosis by glutathione/glutaredoxin‐dependent redox modulation of Akt activity Citation96.
Ischemia‐reperfusion injury is caused by the generation of ROS/RNS
It has long been documented that restoration of blood flow into the ischemic myocardium leads to ventricular arrhythmias, contractile dysfunction, myocardial stunning, and myocyte death Citation97–101. Subsequent studies established a cause‐and‐effect relationship between oxidative stress and ischemia‐reperfusion injury Citation102–104. The advent of 1) more sensitive assays for ROS/RNS generation, both in tissues and in cultured cells Citation105,106; 2) molecular cloning, overexpression, and targeted deletion of genes of various antioxidant enzymes Citation62,Citation107–109; and 3) availability of various redox‐responsive markers Citation26,Citation71 have unequivocally established the deleterious role of ROS/RNS in ischemia‐reperfusion injury. Simultaneously, increasing evidence such as generation of free radicals in conjunction with the accumulation of oxidized proteins in the mitochondria had established it as the primary source of ROS generation during ischemic injury Citation110–116. During ischemia, diminished oxygen supply inhibits the TCA cycle and oxidative phosphorylation while increasing the free fatty acid level, NADH/NAD+ ratio, and glycolysis, thereby leading to acidosis with deleterious consequences Citation117. Decrease in respiration also leads to reduced electron transport in the mitochondria, causing one electron leakage followed by the reduction of O2·− to O2·−. Under normal circumstances, O2−· would be acted upon by superoxide dismutase, converting it into H2O2. However, in the reperfused myocardium, due to acidosis and reducing environment, ferric and ferrous ions are released from metalloproteins which in turn catalyse (Fenton reaction) the generation of highly reactive hydroxyl radical from O2·− and H2O2Citation118. Under ischemic conditions, enzymes like xanthine oxidase also reduce hydrogen peroxide into hydroxyl radical Citation119, while upon reperfusion, due to concurrent generation of NO and superoxide ions, highly reactive peroxynitrite is formed Citation120. Also during reperfusion, increased intracellular H+ stimulate the Na+/H+ exchanger, resulting in increased intracellular Na+ which in turn stimulate Na+/Ca2+ exchanger, ultimately causing an increase in intracellular (and intramitochondrial) Ca2+ with detrimental effects like mitochondrial swelling, opening of mitochondrial permeability transition pores (PTP), and apoptosis Citation121,122. Since intracellular Ca2+ regulates a plethora of cellular enzymes like kinases, phosphatases, endonucleases, and proteases, an elevation in Ca2+ level adversely affects other cellular functions as well Citation123.
Ischemic preconditioning, an enigmatic phenomenon
Ischemic preconditioning, a phenomenon where brief repetitive cycles of ischemia‐reperfusion render the myocardium more resistant towards subsequent ischemic insult, was first documented in the mid‐80s Citation3. Thereafter, numerous physiological agonists like norepinephrine, angiotensin II, bradykinin, acetylcholine, cytokines, opioid peptides, and diffusible molecules like adenosine and nitric oxide Citation124–128, pharmacological/chemical agents like peroxynitrite, hydrogen sulfide, and volatile anesthetics have been also identified to have preconditioning effects Citation129–131. However, it is believed that there are subtle differences in the mechanism by which each mediates its cytoprotective effects Citation9,Citation132. A number of signaling kinases (namely PKC, ERK, Src, PI3 kinase‐Akt, p70S6 kinase), gene regulatory proteins (namely AP‐1, NFκB, STAT), mitochondrial permeability transition pores, KATP, Cl−, and the inward rectifier K+ channels are some of the mediators of preconditioning effects Citation124,125,Citation127,Citation133–145. Ischemic preconditioning occurs in two phases: first it is observed within a few minutes of brief ischemia‐reperfusion and stays for several hours (classical or early preconditioning); while the second phase starts after 12–24 hours and stays for 3–4 days (second window of protection or late preconditioning). It is thus believed that while the first phase of preconditioning is primarily achieved through the calibration of biochemical parameters (such as minimizing the usage of ATP), late preconditioning is more of a competent state of the myocardium achieved through a wider alteration of cellular proteome Citation146,147. During the early phase of preconditioning, certain biochemical changes in the intracellular milieu, such as altered pH, oxygen tension, redox equilibrium, etc., are the possible contributors towards preconditioning effects Citation148,149. Preconditioning has also been observed in other tissues like brain and presumably involves conserved pathways rather than a mechanism solely dedicated to cardioprotection Citation150.
Amongst various types of pharmacological preconditioning, that induced by adenosine has been extensively investigated and adenosine receptor (AR) subtypes 1 and 3 are primarily attributed towards this process Citation151,152. While transgenic mice overexpressing A1 AR are more tolerant to ischemic injury, those having deletion of either both or single alleles are refractile towards preconditioning effects and are susceptible to postischemic injury Citation124. Transgenic mice overexpressing A3 AR also show less ATP depletion following ischemia, reduced infarct size following ischemia‐reperfusion, and better postischemic recoveries Citation153,154. Intriguingly, A3 AR gene knockout mice showed improved postischemic recovery as compared to their normal counterparts Citation155. Adenosine receptors are members of the G protein coupled receptor (GPCR) superfamily and, upon stimulation, activate pro‐survival signaling kinases like ERK, p38, PKC, and Akt mediating its beneficial effects Citation156. In addition, rhoA‐phospholipase D1, mitochondrial permeability transition pore, catalase, and superoxide dismutase have been attributed to the preconditioning induced by adenosine Citation152,Citation157,158. Nevertheless, certain discrepancies in the contributions of the downstream mediators have also been reported. For example, while Lasley et al. Citation159 used open chest anesthetized rat and demonstrated the involvement of both ERK and p38 kinase in mediating cardioprotection by A1 AR, Button et al. Citation160 used adult ventricular strips and hypoxia‐reoxygenation protocol and observed that cardioprotection by A1, A2A, and A3 ARs does not require ERK and PI3 kinases. Similarly, while Germack et al. Citation151 used neonatal cardiac myocytes and hypoxia‐reoxygenation protocol demonstrating that A2A AR does not have any role in preventing cell death, Button et al. (adult ventricular strips and hypoxia‐reoxygenation protocol) demonstrated its involvement in preconditioning Citation160. Discrepant observations were also made with reference to other mediators further downstream. While Guo et al. Citation161 used iNOS‐/‐ mouse and demonstrated its essentiality in delayed preconditioning by A1 AR, Lasley et al. Citation159 used a pharmacological inhibitor of iNOS (1400 W) and demonstrated its nonessentiality for the same. Opioids are also involved in cardioprotection during ischemia, and experimental evidence suggests that endogenous opioids released from the myocytes act in an autocrine fashion providing their beneficial effects Citation125. Studies with rabbit and rat heart indicate that amongst three major opioid peptides (endorphins, dynorphins, and enkephalins), enkephalin and the δ1‐receptor are the primary mediators of the protective effects Citation125,Citation162,163. In addition, recent studies have also appreciated the involvement of κ receptor in this process Citation164,165. Like adenosine receptors, opioid receptors also belong to the GPCR superfamily (Gi) and mediate their effect via PKC‐δ, PI3 kinase, tyrosine kinase, ERK, PKA and 12‐lipoxigenase Citation166–170 which modulate their downstream targets like iNOS, sarcolemmal KATP and mitochondrial KATP channels Citation161,Citation164. Two other GPCR agonists, Ang II and norepinephrine, have also been attributed to preconditioning. And depending upon their concentration, both could be beneficial as well as detrimental for the heart Citation171–173. Such disparate behavior is attributed to their ability to initiate concentration‐dependent differential signaling affecting downstream kinases (like p38 MAPK, JNK, and Akt), other regulatory proteins (like IGF‐IR, EGF‐R, and HO‐1), and downstream transcription factors (like NFkB, Nrf2, and AP‐1) Citation173–175. NE‐mediated preconditioning is also mediated by PI3 kinase, PKC, PKA and p38 MAP kinase Citation176–178. Ischemic stimuli also releases bradykinin, primarily from the endothelium, that acts upon the bradykinin receptors (B2) on myocytes and provide preconditioning effects via PKC, PKG, Akt, and nonkinase effectors like iNOS and ATP‐sensitive K+ channel Citation128,Citation141,Citation179–181. Although the physiological and pharmacological stimuli described above provide cardioprotection, it is likely that there are subtle differences in their mechanisms of action Citation9,Citation132. Like in ischemia, reperfusion of the hypoxic myocardium also has deleterious effects (myocyte loss) and has been investigated both in whole heart and in isolated myocytes Citation182,183. Hypoxic injury also involves generation of ROS, reduced glutathione levels, decreased Na+/Ca2+ exchanger activity and calcium overload Citation184,185. Studies with cultured myocytes has also demonstrated that during hypoxia, the generation of ROS from the mitochondrial electron transport chain leads to the activation of p38 MAP kinase and JNK Citation186,187. Finally, nitric oxide, KATP channel, ROS, and gene regulatory protein HIF‐1α have been attributed to the hypoxic preconditioning Citation188,189.
ROS/RNS and ischemic preconditioning: a paradoxical paradigm
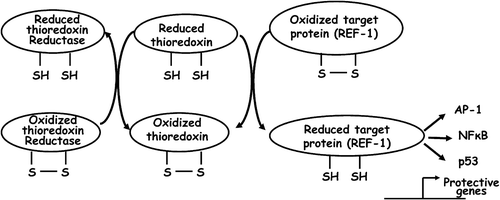
In spite of significant progress in our understanding of the biochemical and molecular events associated with ischemic injury and cardioprotection, the precise mechanism by which cardiac myocytes decide to survive or to die is yet to be deciphered Citation6. Furthermore, studies done over the past decade indicate that the mechanisms of cardioprotection by different pharmacological agents might be divergent, at least partially Citation9. It is thus likely that the vulnerability of the myocardium towards ischemia‐reperfusion insult vis‐à‐vis its attainment of preconditioned status is defined by a distinctive cellular proteome rather than by the presence or absence of a set of ‘protective’ or ‘detrimental’ molecules. In this context, studies done by various laboratories have documented that ROS generation plays a nodal role in mediating both ischemic injury and cardioprotection Citation8. Such paradoxical behavior of ROS can be explained by its disparate regulatory potential. As discussed in the previous sections, reactive oxygen species are well suited as second messengers involved in the activation and/or inactivation of ERK, p38, JNK and PI3 kinases, affecting disparate cellular events Citation190–197. Since intracellular ROS can be generated at multiple sites (like NADPH oxidase, mitochondrial complex I and III) while being simultaneously attenuated by a battery of antioxidant enzymes Citation198, the net ROS threshold in a particular cellular context decides whether it will be deleterious or beneficial Citation172. Emerging evidence also suggests that once specific redox‐responsive signaling pathways are activated, the cognate signal is transmitted to the nucleus initiating discrete gene expression programs Citation172,Citation199,200. In this context, mammalian gene regulatory protein Redox factor‐1 (Ref‐1) has been a paradigm of redox‐sensitive gene regulatory events. Ref‐1 has two distinct functions, while its C‐terminus functions as a DNA repair (oxidative and radiation‐damaged) enzyme, its N‐terminal region contains a redox regulatory domain involved in the reductive modifications of a battery of transcription factors like activator protein‐1 (AP‐1), nuclear factor κB (NFκB), p53, hypoxia inducible factor‐1 (HIF‐1) and thyroid transcription factor‐1 (TTF‐1) () Citation201–204. In response to oxidative stimuli, thioredoxin translocates to the nucleus, interacts with Ref‐1 which in turn transmits the signal by the reduction of specific cysteine residues to its downstream targets Citation205. Two cysteine residues (at positions 65 and 93) of Ref‐1 mediate this process by acting as redox switches (sulfhydryl switches). Protein kinase C can also modulate Ref‐1 activity, further diversifying its regulatory potential Citation206. Ref‐1 thus acts as a coupler of upstream redox events to a plethora of transcription factors and the DNA repair apparatus further downstream Citation207. Although it generally believed that Ref‐1 is a mediator of pro‐survival events, evidence in support of its pro‐apoptotic function also exists Citation208–210. The feasibility of both pro‐ and anti‐apoptotic functions of Ref‐1 can be explained from such dual functions of its targets, namely AP‐1, NFκB, and HIF‐1alpha Citation211–214, while the activation of p53 by Ref‐1 supports its pro‐apoptotic potential Citation215. The diversity of Ref‐1 functions is still being uncovered, and emerging evidence suggests its role in redox‐independent cellular events as well Citation216,217. Taken together, Ref‐1 is a bona fide candidate for playing a nodal role in regulating redox events leading to both ischemic injury and cardioprotection Citation205. Another gene regulatory protein, namely Nrf‐2 (NF‐E2‐related factor 2), has also drawn considerable attention for its role in regulating oxidative events. Nrf‐2 binds the DNA sequence known as antioxidant response element (ARE), commonly found in the regulatory regions of antioxidant and phase II detoxifying enzymes such as glutathione S‐transferase, NAD(P)H, quinone oxidoreductase 1, and heme‐oxygenase‐1. Nrf‐2 is uniquely characterized by it inducibility by a variety of ROS/RNS, cellular lipid oxidation products, and xenobiotics Citation218,219. Under normal conditions, Nrf‐2 is sequestered in the cytoplasm by the cysteine‐rich protein Keap‐1 (Kelch‐like ECH‐associated protein 1). In the presence of ROS/RNS, cysteine residues of Keap‐1 are modified, leading to a conformational change followed by its dissociation, and proteasomal degradation. Nrf‐2 then translocates to the nucleus and activates cognate genes eliciting an antioxidant and detoxification response Citation220. The mechanism of Keap‐1 activation by various stimuli is an active area of research and is less understood in the context of ROS/RNS generated under ischemia‐reperfusion Citation220. Nevertheless, emerging evidence suggests a key role of Nrf‐2 in mediating redox signaling under ischemia‐reperfusion injury as well as cardiac preconditioning Citation221.
Figure 2. Activation of Ref‐1 by thioredoxin: schematic presentation of interconversion of thioredoxin systems in conjunction with the activation of Ref‐1, followed by that of AP‐1, NFκB and p53.
Summary and conclusion
In the post genome era, our knowledge of various biological phenomena has achieved a new dimension due to the use of integrated approaches such as gene annotation, proteomics, microarray analysis, etc., and it is now expanding at an unprecedented pace Citation9. With the available information, any biological event can thus be considered in a wholesome manner rather than in isolation. In that context, our perception of the role(s) of ROS/RNS in modulating cellular events has also taken a new direction, where it can be either beneficial or detrimental based upon its nature, modes of generation, and its mutual interaction with other cellular constituents in a given context. However, how those ROS/RNS (same or different) can convert a death signal into a survival signal is an emerging theme not only in the context of the biology of ischemia‐reperfusion in specific, but also in the context of the pathophysiology of various diseases in general. It is thus expected that with the identification of more and more redox‐responsive biomolecules and the understanding of their specific roles in respective cellular events, coming years will see further consolidation of our knowledge of the intricacies of cell signaling and gene expression associated with the unique phenomena of ischemia‐reperfusion and cardioprotection.
References
- Nabel E. G. Cardiovascular disease. N Engl J Med 2003; 349: 60–72
- Gross G. J., Auchampach J. A. Reperfusion injury: Does it exist?. J Mol Cell Cardiol 2007; 42: 12–18
- Murry C. E., Jennings R. B., Reimer K. A. Preconditioning with ischemia: a delay of lethal cell injury in ischemic myocardium. Circulation 1986; 74: 1124–36
- Schott R. J., Rohmann S., Braun E. R., Schaper W. Ischemic preconditioning reduces infarct size in swine myocardium. Circ Res 1990; 66: 1133–42
- Das D. K. Cardioprotection with high‐density lipoproteins: fact or fiction?. Circ Res 2003; 92: 258–60
- Bolli R., Becker L., Gross G., Mentzer R., Jr., Balshaw D., Lathrop D. A. Myocardial protection at a crossroads: the need for translation into clinical therapy. Circ Res 2004; 95: 125–34
- Murphy E. Primary and secondary signaling pathways in early preconditioning that converge on the mitochondria to produce cardioprotection. Circ Res 2004; 94: 7–16
- Das D. K., Maulik N. Preconditioning potentiates redox signaling and converts death signal into survival signal. Arch Biochem Biophys 2003; 420: 305–11
- Das D. K., Maulik N. Cardiac genomic response following preconditioning stimulus. Cardiovasc Res 2006; 70: 254–63
- Rice‐Evans C. A., Diplock A. T. Current status of antioxidant therapy. Free Radic Biol Med 1993; 15: 77–96
- Dennery P. A. Role of redox in fetal development and neonatal diseases. Antioxid Redox Signal 2004; 6: 147–53
- Evans M. D., Dizdaroglu M., Cooke M. S. Oxidative DNA damage and disease: induction, repair and significance. Mutat Res 2004; 567: 1–61
- Sauer H., Wartenberg M. Reactive oxygen species as signaling molecules in cardiovascular differentiation of embryonic stem cells and tumor‐induced angiogenesis. Antioxid Redox Signal 2005; 7: 1423–34
- Nordberg J., Arner E. S. Reactive oxygen species, antioxidants, and the mammalian thioredoxin system. Free Radic Biol Med 2001; 31: 1287–312
- Cepinskas G., Rui T., Kvietys P. R. Interaction between reactive oxygen metabolites and nitric oxide in oxidant tolerance. Free Radic Biol Med 2002; 33: 433–40
- Hensley K., Robinson K. A., Gabbita S. P., Salsman S., Floyd R. A. Reactive oxygen species, cell signaling, and cell injury. Free Radic Biol Med 2000; 28: 1456–62
- Moreland J. G., Davis A. P., Bailey G., Nauseef W. M., Lamb F. S. Anion channels including CLC‐3 are required for normal neutrophil oxidative function, phagocytosis, and transendothelial migration. J Biol Chem 2006; 281: 12277–88
- Meng T. C., Buckley D. A., Galic S., Tiganis T., Tonks N. K. Regulation of insulin signaling through reversible oxidation of the protein‐tyrosine phosphatases TC45 and PTP1B. J Biol Chem 2004; 279: 37716–25
- Gensch E., Gallup M., Sucher A., Li D., Gebremichael A., Lemjabbar H., et al. Tobacco smoke control of mucin production in lung cells requires oxygen radicals AP‐1 and JNK. J Biol Chem 2004; 279: 39085–93
- Won J. S., Singh I. Sphingolipid signaling and redox regulation. Free Radic Biol Med 2006; 40: 1875–88
- Moncada S., Palmer R. M., Higgs E. A. The discovery of nitric oxide as the endogenous nitrovasodilator. Hypertension 1988; 12: 365–72
- Hare J. M., Stamler J. S. NO/redox disequilibrium in the failing heart and cardiovascular system. J Clin Invest 2005; 115: 509–17
- Augusto O., Bonini M. G., Amanso A. M., Linares E., Santos C. C., De Menezes S. L. Nitrogen dioxide and carbonate radical anion: two emerging radicals in biology. Free Radic Biol Med 2002; 32: 841–59
- Chen Q., Vazquez E. J., Moghaddas S., Hoppel C. L., Lesnefsky E. J. Production of reactive oxygen species by mitochondria: central role of complex III. J Biol Chem 2003; 278: 36027–31
- Kudin A. P., Bimpong‐Buta N. Y., Vielhaber S., Elger C. E., Kunz W. S. Characterization of superoxide‐producing sites in isolated brain mitochondria. J Biol Chem 2004; 279: 4127–35
- Chen Q., Lesnefsky E. J. Depletion of cardiolipin and cytochrome c during ischemia increases hydrogen peroxide production from the electron transport chain. Free Radic Biol Med 2006; 40: 976–82
- Martens G. A., Cai Y., Hinke S., Stange G., Van de Casteele M., Pipeleers D. Glucose suppresses superoxide generation in metabolically responsive pancreatic beta cells. J Biol Chem 2005; 280: 20389–96
- Klotz L. O., Schroeder P., Sies H. Peroxynitrite signaling: receptor tyrosine kinases and activation of stress‐responsive pathways. Free Radic Biol Med 2002; 33: 737–43
- Sabetkar M., Low S. Y., Naseem K. M., Bruckdorfer K. R. The nitration of proteins in platelets: significance in platelet function. Free Radic Biol Med 2002; 33: 728–36
- Vavrinkova H., Tutterova M., Stopka P., Divisova J., Kazdova L., Drahota Z. The effect of captopril on nitric oxide formation and on generation of radical forms of mitochondrial respiratory chain compounds in ischemic rat heart. Physiol Res 2001; 50: 481–9
- Herold S., Rock G. Mechanistic studies of S‐nitrosothiol formation by NO*/O2 and by NO*/methemoglobin. Arch Biochem Biophys 2005; 436: 386–96
- Donzelli S., Espey M. G., Thomas D. D., Mancardi D., Tocchetti C. G., Ridnour L. A., et al. Discriminating formation of HNO from other reactive nitrogen oxide species. Free Radic Biol Med 2006; 40: 1056–66
- Hewett S. J., Espey M. G., Uliasz T. F., Wink D. A. Neurotoxicity of nitroxyl: insights into HNO and NO biochemical imbalance. Free Radic Biol Med 2005; 39: 1478–88
- Whittier J. E., Xiong Y., Rechsteiner M. C., Squier T. C. Hsp90 enhances degradation of oxidized calmodulin by the 20 S proteasome. J Biol Chem 2004; 279: 46135–42
- Nystrom T. Role of oxidative carbonylation in protein quality control and senescence. EMBO J 2005; 24: 1311–17
- Connelly P. W., Draganov D., Maguire G. F. Paraoxonase‐1 does not reduce or modify oxidation of phospholipids by peroxynitrite. Free Radic Biol Med 2005; 38: 164–74
- Divald A., Powell S. R. Proteasome mediates removal of proteins oxidized during myocardial ischemia. Free Radic Biol Med 2006; 40: 156–64
- Le Moan N., Clement G., Le Maout S., Tacnet F., Toledano M. B. The Saccharomyces cerevisiae proteome of oxidized protein thiols: contrasted functions for the thioredoxin and glutathione pathways. J Biol Chem 2006; 281: 10420–30
- Dimitrov J. D., Ivanovska N. D., Lacroix‐Desmazes S., Doltchinkova V. R., Kaveri S. V., Vassilev T. L. Ferrous ions and reactive oxygen species increase antigen‐binding and anti‐inflammatory activities of immunoglobulin G. J Biol Chem 2006; 281: 439–46
- Nishida M., Tanabe S., Maruyama Y., Mangmool S., Urayama K., Nagamatsu Y., et al. G alpha 12/13‐ and reactive oxygen species‐dependent activation of c‐Jun NH2‐terminal kinase and p38 mitogen‐activated protein kinase by angiotensin receptor stimulation in rat neonatal cardiomyocytes. J Biol Chem 2005; 280: 18434–41
- Heo J., Campbell S. L. Superoxide anion radical modulates the activity of Ras and Ras‐related GTPases by a radical‐based mechanism similar to that of nitric oxide. J Biol Chem 2005; 280: 12438–45
- Schmitt T. L., Hotz‐Wagenblatt A., Klein H., Droge W. Interdependent regulation of insulin receptor kinase activity by ADP and hydrogen peroxide. J Biol Chem 2005; 280: 3795–801
- Howe C. J., Lahair M. M., McCubrey J. A., Franklin R. A. Redox regulation of the calcium/calmodulin‐dependent protein kinases. J Biol Chem 2004; 279: 44573–81
- Katsuoka F., Motohashi H., Ishii T., Aburatani H., Engel J. D., Yamamoto M. Genetic evidence that small maf proteins are essential for the activation of antioxidant response element‐dependent genes. Mol Cell Biol 2005; 25: 8044–51
- Salazar M., Rojo A. I., Velasco D., de Sagarra R. M., Cuadrado A. Glycogen synthase kinase‐3beta inhibits the xenobiotic and antioxidant cell response by direct phosphorylation and nuclear exclusion of the transcription factor Nrf2. J Biol Chem 2006; 281: 14841–51
- Lu H., Dalgard C. L., Mohyeldin A., McFate T., Tait A. S., Verma A. Reversible inactivation of HIF‐1 prolyl hydroxylases allows cell metabolism to control basal HIF‐1. J Biol Chem 2005; 280: 41928–39
- Catalano A., Rodilossi S., Caprari P., Coppola V., Procopio A. 5‐Lipoxygenase regulates senescence‐like growth arrest by promoting ROS‐dependent p53 activation. EMBO J 2005; 24: 170–9
- Colbert C. L., Wu Q., Erbel P. J., Gardner K. H., Deisenhofer J. Mechanism of substrate specificity in Bacillus subtilis ResA, a thioredoxin‐like protein involved in cytochrome c maturation. Proc Natl Acad Sci U S A 2006; 103: 4410–15
- Wu R. F., Terada L. S. Oxidative modification of protein tyrosine phosphatases. Sci STKE 2006; 2006: pl2
- Okamoto A., Iwamoto Y., Maru Y. Oxidative stress‐responsive transcription factor ATF3 potentially mediates diabetic angiopathy. Mol Cell Biol 2006; 26: 1087–97
- Canton M., Neverova I., Menabo R., Van Eyk J., Di Lisa1 F. Evidence of myofibrillar protein oxidation induced by postischemic reperfusion in isolated rat hearts. Am J Physiol Heart Circ Physiol 2004; H870–7
- Chen Y. R., Chen C. L., Zhang L., Green‐Church K. B., Zweier J. L. Superoxide generation from mitochondrial NADH dehydrogenase induces self‐inactivation with specific protein radical formation. J Biol Chem 2005; 280: 37339–48
- Jacob C., Knight I., Winyard P. G. Aspects of the biological redox chemistry of cysteine: from simple redox responses to sophisticated signalling pathways. Biol Chem 2006; 387: 1385–97
- Murdoch C. E., Zhang M., Cave A. C., Shah A. M. NADPH oxidase‐dependent redox signalling in cardiac hypertrophy, remodelling and failure. Cardiovasc Res 2006; 71: 208–15
- Rojas A., Figueroa H., Morales M. A., Re L. Facing up the ROS labyrinth—Where to go?. Curr Vasc Pharmacol 2006; 4: 277–89
- Storz P. Reactive oxygen species‐mediated mitochondria‐to‐nucleus signaling: a key to aging and radical‐caused diseases. Sci STKE 2006; 2006: re3
- Ushio‐Fukai M. Localizing NADPH oxidase‐derived ROS. Sci STKE 2006; 2006: re8
- Kregel K. C., Zhang H. J. An Integrated View of Oxidative Stress in Aging: Basic Mechanisms, Functional Effects and Pathological Considerations. Am J Physiol Regul Integr Comp Physiol 2006; 292: R18–36
- Forman H. J., Fukuto J. M., Torres M. Redox signaling: thiol chemistry defines which reactive oxygen and nitrogen species can act as second messengers. Am J Physiol Cell Physiol 2004; 287: C246–56
- Terada L. S. Specificity in reactive oxidant signaling: think globally, act locally. J Cell Biol 2006; 174: 615–23
- Esposito F., Ammendola R., Faraonio R., Russo T., Cimino F. Redox control of signal transduction, gene expression and cellular senescence. Neurochem Res 2004; 29: 617–28
- Tanaka M., Mokhtari G. K., Terry R. D., Balsam L. B., Lee K. H., Kofidis T., et al. Overexpression of human copper/zinc superoxide dismutase (SOD1) suppresses ischemia‐reperfusion injury and subsequent development of graft coronary artery disease in murine cardiac grafts. Circulation 2004; 110: II200–06
- Bossis G., Melchior F. Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes. Mol Cell 2006; 21: 349–57
- Bubici C., Papa S., Dean K., Franzoso G. Mutual cross‐talk between reactive oxygen species and nuclear factor‐kappa B: molecular basis and biological significance. Oncogene 2006; 25: 6731–48
- Nakashima I., Takeda K., Kawamoto Y., Okuno Y., Kato M., Suzuki H. Redox control of catalytic activities of membrane‐associated protein tyrosine kinases. Arch Biochem Biophys 2005; 434: 3–10
- Han M. J., Kim B. Y., Yoon S. O., Chung A. S. Cell proliferation induced by reactive oxygen species is mediated via mitogen‐activated protein kinase in Chinese hamster lung fibroblast (V79) cells. Mol Cells 2003; 15: 94–101
- Ross B., Mikkelsen R. B., Wardman P. Biological chemistry of reactive oxygen and nitrogen and radiation‐induced signal transduction mechanisms. Oncogene 2003; 22: 5734–54
- O'Brian C. A., Chu F. Post‐translational disulfide modifications in cell signaling—role of inter‐protein, intra‐protein, S‐glutathionyl, and S‐cysteaminyl disulfide modifications in signal transmission. Free Radic Res 2005; 39: 471–80
- Mikkelsen R. B., Wardman P. Biological chemistry of reactive oxygen and nitrogen and radiation‐induced signal transduction mechanisms. Oncogene 2003; 22: 5734–54
- Cheong E., Tumbev V., Abramson J., Salama G., Stoyanovsky D. A. Nitroxyl triggers Ca2+ release from skeletal and cardiac sarcoplasmic reticulum by oxidizing ryanodine receptors. Cell Calcium 2005; 37: 87–96
- Zima A. V., Blatter L. A. Redox regulation of cardiac calcium channels and transporters. Cardiovasc Res 2006; 71: 310–21
- Martinez‐Moreno M., Alvarez‐Barrientos A., Roncal F., Albar J. P., Gavilanes F., Lamas S., et al. Direct interaction between the reductase domain of endothelial nitric oxide synthase and the ryanodine receptor. FEBS Lett 2005; 579: 3159–63
- Cohen R. A., Adachi T. Nitric‐oxide‐induced vasodilatation: regulation by physiologic s‐glutathiolation and pathologic oxidation of the sarcoplasmic endoplasmic reticulum calcium ATPase. Trends Cardiovasc Med 2006; 16: 109–14
- Cross J. V., Templeton D. J. Regulation of signal transduction through protein cysteine oxidation. Antioxid Redox Signal 2006; 8: 1819–27
- Rahman I., Biswas S. K., Jimenez L. A., Torres M., Forman H. J. Glutathione, stress responses, and redox signaling in lung inflammation. Antioxid Redox Signal 2005; 7: 42–59
- Droge W. Oxidative enhancement of insulin receptor signaling: experimental findings and clinical implications. Antioxid Redox Signal 2005; 7: 1071–7
- Ilbert M., Graf P. C., Jakob U. Zinc center as redox switch—new function for an old motif. Antioxid Redox Signal 2006; 8: 835–46
- Nakamura H. Thioredoxin and its related molecules: update 2005. Antioxidant Redox Signal 2005; 7: 823–8
- Holmgren A., Johansson C., Berndt C., Lonn M. E., Hudemann C., Lillig C. H. Thiol redox control via thioredoxin and glutaredoxin systems. Biochem Soc Trans 2005; 33: 1375–7
- Ghezzi P. Oxidoreduction of protein thiols in redox regulation. Biochem Soc Trans 2005; 33: 1378–81
- Turanov A. A., Su D., Gladyshev V. N. Characterization of alternative cytosolic forms and cellular targets of mouse mitochondrial thioredoxin reductase. J Biol Chem 2006; 281: 22953–63
- Nissom P. M., Lo S. L., Lo J. C., Ong P. F., Lim J. W., Ou K., et al. Hcc‐2, a novel mammalian ER thioredoxin that is differentially expressed in hepatocellular carcinoma. FEBS Lett 2006; 580: 2216–26
- Jekell A., Hossain A., Alehagen U., Dahlstrom U., Rosen A. Elevated circulating levels of thioredoxin and stress in chronic heart failure. Eur J Heart Fail 2004; 6: 883–90
- Shioji K., Kishimoto C., Nakamura H., Masutani H., Yuan Z., Oka S., et al. Overexpression of thioredoxin‐1 in transgenic mice attenuates adriamycin‐induced cardiotoxicity. Circulation 2002; 106: 1403–9
- Saitoh M., Nishitoh H., Fujii M., Takeda K., Tobiume K., Sawada Y., et al. Mammalian thioredoxin is a direct inhibitor of apoptosis signal‐regulating kinase (ASK) 1. EMBO J 1998; 17: 2596–606
- Liu Y., Min W. Thioredoxin promotes ASK1 ubiquitination and degradation to inhibit ASK1‐mediated apoptosis in a redox activity‐independent manner. Circ Res 2002; 90: 1259–66
- Wang Y., De Keulenaer G. W., Lee R. T. Vitamin D (3)‐up‐regulated protein‐1 is a stress‐responsive gene that regulates cardiomyocyte viability through interaction with thioredoxin. J Biol Chem 2002; 277: 26496–500
- Butler L. M., Zhou X., Xu W. S., Scher H. I., Rifkind R. A., Marks P. A., et al. The histone deacetylase inhibitor SAHA arrests cancer cell growth, up‐regulates thioredoxin‐binding protein‐2, and down‐regulates thioredoxin. Proc Natl Acad Sci U S A 2002; 99: 11700–05
- Wiesel P., Foster L. C., Pellacani A., Layne M. D., Hsieh C. M., Huggins G. S., et al. Thioredoxin facilitates the induction of heme oxygenase‐1 in response to inflammatory mediators. J Biol Chem 2000; 275: 24840–6
- Turoczi T., Chang V. W., Engelman R. M., Maulik N., Ho Y. S., Das D. K. Thioredoxin redox signaling in the ischemic heart: an insight with transgenic mice overexpressing Trx1. J Mol Cell Cardiol 2003; 35: 695–704
- Das D. K. Thioredoxin regulation of ischemic preconditioning. Antioxid Redox Signal 2004; 6: 405–12
- Lundberg M., Johansson C., Chandra J., Enoksson M., Jacobsson G., Ljung J., et al. Cloning and expression of a novel human glutaredoxin (Grx2) with mitochondrial and nuclear isoforms. J Biol Chem 2001; 276: 26269–75
- Beer S. M., Taylor E. R., Brown S. E., Dahm C. C., Costa N. J., Runswick M. J., et al. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: implications for mitochondrial redox regulation and antioxidant DEFENSE. J Biol Chem 2004; 279: 47939–51
- Lillig C. H., Lonn M. E., Enoksson M., Fernandes A. P., Holmgren A. Short interfering RNA‐mediated silencing of glutaredoxin 2 increases the sensitivity of HeLa cells toward doxorubicin and phenylarsine oxide. Proc Natl Acad Sci U S A 2004; 101: 13227–32
- Suh J. H., Heath S. H., Hagen T. M. Two subpopulations of mitochondria in the aging rat heart display heterogenous levels of oxidative stress. Free Radic Biol Med 2003; 35: 1064–72
- Urata Y., Ihara Y., Murata H., Goto S., Koji T., Yodoi J., et al. 17Beta‐estradiol protects against oxidative stress‐induced cell death through the glutathione/glutaredoxin‐dependent redox regulation of Akt in myocardiac H9c2 cells. J Biol Chem 2006; 281: 13092–102
- Cox J. L., Daniel T. M., Boineau J. P. The electrophysiologic time‐course of acute myocardial ischemia and the effects of early coronary artery reperfusion. Circulation 1973; 48: 971–83
- Lang T. W., Corday E., Gold H., Meerbaum S., Rubins S., Costantini C., et al. Consequences of reperfusion after coronary occlusion. Effects on hemodynamic and regional myocardial metabolic function. Am J Cardiol 1974; 33: 69–81
- Bulkely B. H., Hutchins G. M. Myocardial consequences of coronary artery bypass graft surgery. The paradox of necrosis in areas of revascularization. Circulation 1977; 56: 906–13
- Kusuoka H., Porterfield J. K., Weisman H. F., Weisfeldt M. L., Marban E. Pathophysiology and pathogenesis of stunned myocardium. Depressed Ca2+ activation of contraction as a consequence of reperfusion‐induced cellular calcium overload in ferret hearts. J Clin Invest 1987; 79: 950–61
- Reimer K. A., Lowe J. E., Rasmussen M. M., Jennings R. B. The wave front phenomenon of ischemic cell death. 1. Myocardial infarct size vs duration of coronary occlusion in dogs. Circulation 1977; 56: 786–94
- Darley‐Usmar V. M., Stone D., Smith D. R. Oxygen and reperfusion damage: an overview. Free Radic Res Commun 1989; 7: 247–54
- McCord J. M. Oxygen‐derived free radicals in postischemic tissue injury. N Engl J Med 1985; 312: 159–63
- McCord J. M. Oxygen‐derived radicals: a link between reperfusion injury and inflammation. Fed Proc 1987; 46: 2402–06
- Mason R. P. Using anti‐5,5‐dimethyl‐1‐pyrroline N‐oxide (anti‐DMPO) to detect protein radicals in time and space with immuno‐spin trapping. Free Radic Biol Med 2004; 36: 1214–23
- Liu Y., Zhao H., Li H., Kalyanaraman B., Nicolosi A. C., Gutterman D. D. Mitochondrial sources of H2O2 generation play a key role in flow‐mediated dilation in human coronary resistance arteries. Circ Res 2003; 93: 573–80
- Matsushima S., Ide T., Yamato M., Matsusaka H., Hattori F., Ikeuchi M., et al. Overexpression of mitochondrial peroxiredoxin‐3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation 2006; 113: 1779–86
- Jones S., Hoffmeyer M. R., Sharp B. R., Ho Y. S., Lefer D. J. Role of intracellular antioxidant enzymes after in vivo myocardial ischemia and reperfusion. Am J Physiol Heart Circ Physiol 2003; 284: H277–82
- Asimakis G. K., Lick S., Patterson C. Postischemic recovery of contractile function is impaired in SOD2(+/−) but not SOD1(+/−) mouse hearts. Circulation 2002; 105: 981–6
- Das D. K., George A., Liu X. K., Rao P. S. Detection of hydroxyl radical in the mitochondria of ischemic‐reperfused myocardium by trapping with salicylate. Biochem Biophys Res Commun 1989; 165: 1004–09
- Ruuge E. K., Ledenev A. N., Lakomkin V. L., Konstantinov A. A., Ksenzenko MYu. Free radical metabolites in myocardium during ischemia and reperfusion. Am J Physiol 1991; 261: 81–6
- Cortassa S., Aon M. A., Winslow R. L., O'Rourke B. A mitochondrial oscillator dependent on reactive oxygen species. Biophys J 2004; 87: 2060–73
- Solaini G., Harris D. A. Biochemical dysfunction in heart mitochondria exposed to ischaemia and reperfusion. Biochem J 2005; 390: 377–94
- Aon M. A., Cortassa S., Akar F. G., O'Rourke B. Mitochondrial criticality: a new concept at the turning point of life or death. Biochim Biophys Acta 2006; 1762: 232–40
- Prabu S. K., Anandatheerthavarada H. K., Raza H., Srinivasan S., Spear J. F., Avadhani N. G. Protein kinase A‐mediated phosphorylation modulates cytochrome c oxidase function and augments hypoxia and myocardial ischemia‐related injury. J Biol Chem 2006; 281: 2061–70
- Petrosillo G., Di Venosa N., Ruggiero F. M., Pistolese M., D'Agostino D., Tiravanti E., et al. Mitochondrial dysfunction associated with cardiac ischemia/reperfusion can be attenuated by oxygen tension control. Role of oxygen‐free radicals and cardiolipin. Biochim Biophys Acta 2005; 1710: 78–86
- Kubasiak L. A., Hernandez O. M., Bishopric N. H., Webster K. A. Hypoxia and acidosis activate cardiac myocyte death through the Bcl‐2 family protein BNIP3. Proc Natl Acad Sci U S A 2002; 99: 12825–30
- Williams R. E., Zweier J. L., Flaherty J. T. Treatment with deferoxamine during ischemia improves functional and metabolic recovery and reduces reperfusion‐induced oxygen radical generation in rabbit hearts. Circulation 1991; 83: 1006–14
- Kuppusamy P., Zweier J. L. Characterization of free radical generation by xanthine oxidase. Evidence for hydroxyl radical generation. J Biol Chem 1989; 264: 9880–4
- Wang P., Zweier J. L. Measurement of nitric oxide and peroxynitrite generation in the post‐ischemic heart. Evidence for peroxynitrite‐mediated reperfusion injury. J Biol Chem 1996; 271: 29223–30
- Hagihara H., Yoshikawa Y., Ohga Y., Takenaka C., Murata K. Y., Taniguchi S., et al. Na+/Ca2+ exchange inhibition protects the rat heart from ischemia‐reperfusion injury by blocking energy‐wasting processes. Am J Physiol Heart Circ Physiol 2005; 288: H1699–707
- Stromer H., de Groot M. C., Horn M., Faul C., Leupold A., Morgan J. P., et al. Na(+)/H(+) exchange inhibition with HOE642 improves postischemic recovery due to attenuation of Ca(2+) overload and prolonged acidosis on reperfusion. Circulation 2000; 101: 2749–55
- Netticadan T., Temsah R. M., Kawabata K., Dhalla N. S. Ca2+‐overload inhibits the cardiac SR Ca2+‐calmodulin protein kinase activity. Biochem Biophys Res Commun 2002; 293: 727–32
- Lankford A. R., Yang J. N., Rose'Meyer R., French B. A., Matherne G. P., Fredholm B. B., et al. Effect of modulating cardiac A1 adenosine receptor expression on protection with ischemic preconditioning. Am J Physiol Heart Circ Physiol 2006; 290: H1469–73
- Pepe S., van den Brink O. W., Lakatta E. G., Xiao R. P. Cross‐talk of opioid peptide receptor and beta‐adrenergic receptor signalling in the heart. Cardiovasc Res 2004; 63: 414–22
- Das S., Engelman R. M., Maulik N., Das D. K. Angiotensin preconditioning of the heart: evidence for redox signaling. Cell Biochem Biophys 2006; 44: 103–10
- Feng J., Bianchi C., Sandmeyer J. L., Sellke F. W. Bradykinin preconditioning improves the profile of cell survival proteins and limits apoptosis after cardioplegic arrest. Circulation 2005; 112: I190–5
- Krieg T., Qin Q., Philipp S., Alexeyev M. F., Cohen M. V., Downey J. M. Acetylcholine and bradykinin trigger preconditioning in the heart through a pathway that includes Akt and NOS. Am J Physiol Heart Circ Physiol 2004; 287: H2606–11
- Tsutsumi Y. M., Patel H. H., Huang D., Roth D. M. Role of 12‐lipoxygenase in volatile anesthetic‐induced delayed preconditioning in mice. Am J Physiol Heart Circ Physiol 2006; 291: H979–83
- Turan N., Csonka C., Csont T., Giricz Z., Fodor G., Bencsik P., et al. The role of peroxynitrite in chemical preconditioning with 3‐nitropropionic acid in rat hearts. Cardiovasc Res 2006; 70: 384–90
- Pan T., Feng Z. N., Lee S. W., Moore P. K., Bian J. S. Endogenous hydrogen sulfide contributes to the cardioprotection by metabolic inhibition preconditioning in the rat ventricular myocytes. J Mol Cell Cardiol 2006; 40: 119–30
- Liem D. A., te Lintel Hekkert M., Manintveld O. C., Boomsma F., Verdouw P. D., Duncker D. J. Myocardium tolerant to an adenosine‐dependent ischemic preconditioning stimulus can still be protected by stimuli that employ alternative signaling pathways. Am J Physiol Heart Circ Physiol 2005; 288: H1165–72
- Cohen M. V., Yang X. M., Liu G. S., Heusch G., Downey J. M. Acetylcholine, bradykinin, opioids, and phenylephrine, but not adenosine, trigger preconditioning by generating free radicals and opening mitochondrial K(ATP) channels. Circ Res 2001; 89: 273–8
- Oldenburg O., Critz S. D., Cohen M. V., Downey J. M. Acetylcholine‐induced production of reactive oxygen species in adult rabbit ventricular myocytes is dependent on phosphatidylinositol 3‐ and Src‐kinase activation and mitochondrial K(ATP) channel opening. J Mol Cell Cardiol 2003; 35: 653–60
- Yamaura G., Turoczi T., Yamamoto F., Siddqui M. A., Maulik N., Das D. K. STAT signaling in ischemic heart: a role of STAT5A in ischemic preconditioning. Am J Physiol Heart Circ Physiol 2003; 285: H476–82
- Kis A., Yellon D. M., Baxter G. F. Second window of protection following myocardial preconditioning: an essential role for PI3 kinase and p70S6 kinase. J Mol Cell Cardiol 2003; 35: 1063–71
- Zhang J., Ping P., Vondriska T. M., Tang X. L., Wang G. W., Cardwell E. M., et al. Cardioprotection involves activation of NF‐kappa B via PKC‐dependent tyrosine and serine phosphorylation of I kappa B‐alpha. Am J Physiol Heart Circ Physiol 2003; 285: H1753–8
- Chen H., Liu L. L., Ye L. L., McGuckin C., Tamowski S., Scowen P., et al. Targeted inactivation of cystic fibrosis transmembrane conductance regulator chloride channel gene prevents ischemic preconditioning in isolated mouse heart. Circulation 2004; 110: 700–04
- Diaz R. J., Zobel C., Cho H. C., Batthish M., Hinek A., Backx P. H., et al. Selective inhibition of inward rectifier K+ channels (Kir2.1 or Kir2.2) abolishes protection by ischemic preconditioning in rabbit ventricular cardiomyocytes. Circ Res 2004; 95: 325–32
- Tsukamoto O., Asanuma H., Kim J., Minamino T., Takashima S., Ogai A., et al. A role of opening of mitochondrial ATP‐sensitive potassium channels in the infarct size‐limiting effect of ischemic preconditioning via activation of protein kinase C in the canine heart. Biochem Biophys Res Commun 2005; 338: 1460–6
- Krieg T., Philipp S., Cui L., Dostmann W. R., Downey J. M., Cohen M. V. Peptide blockers of PKG inhibit ROS generation by acetylcholine and bradykinin in cardiomyocytes but fail to block protection in the whole heart. Am J Physiol Heart Circ Physiol 2005; 288: H1976–81
- Lecour S., Rochette L., Opie L. Free radicals trigger TNF alpha‐induced cardioprotection. Cardiovasc Res 2005; 65: 239–43
- Ahmad N., Wang Y., Haider K. H., Wang B., Pasha Z., Uzun O., et al. Cardiac protection by mitoKATP channels is dependent on Akt translocation from cytosol to mitochondria during late preconditioning. Am J Physiol Heart Circ Physiol 2006; 290: H2402–48
- Das S., Otani H., Maulik N., Das D. K. Redox regulation of angiotensin II preconditioning of the myocardium requires MAP kinase signaling. J Mol Cell Cardiol 2006; 41: 248–55
- Jones S. P., Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol 2006; 40: 16–23
- Eisen A., Fisman E. Z., Rubenfire M., Freimark D., McKechnie R., Tenenbaum A., et al. Ischemic preconditioning: nearly two decades of research. A comprehensive review. Atherosclerosis 2004; 172: 201–10
- Kim N., Lee Y., Kim H., Joo H., Youm J. B., Park W. S., et al. Potential biomarkers for ischemic heart damage identified in mitochondrial proteins by comparative proteomics. Proteomics 2006; 6: 1237–49
- Nadtochiy S. M., Tompkins A. J., Brookes P. S. Different mechanisms of mitochondrial proton leak in ischaemia/reperfusion injury and preconditioning: implications for pathology and cardioprotection. Biochem J 2006; 395: 611–18
- Heinzel F. R., Luo Y., Li X., Boengler K., Buechert A., Garcia‐Dorado D., et al. Impairment of diazoxide‐induced formation of reactive oxygen species and loss of cardioprotection in connexin 43 deficient mice. Circ Res 2005; 97: 583–6
- Lang S. C., Elsasser A., Scheler C., Vetter S., Tiefenbacher C. P., Kubler W., et al. Myocardial preconditioning and remote renal preconditioning—identifying a protective factor using proteomic methods?. Basic Res Cardiol 2006; 101: 149–58
- Germack R., Dickenson J. M. Adenosine triggers preconditioning through MEK/ERK1/2 signalling pathway during hypoxia/reoxygenation in neonatal rat cardiomyocytes. J Mol Cell Cardiol 2005; 39: 429–42
- Hochhauser E., Kaminski O., Shalom H., Leshem D., Shneyvays V., Shainberg A., et al. Role of adenosine receptor activation in antioxidant enzyme regulation during ischemia‐reperfusion in the isolated rat heart. Antioxid Redox Signal 2004; 6: 335–44
- Cross H. R., Murphy E., Black R. G., Auchampach J., Steenbergen C. Overexpression of A(3) adenosine receptors decreases heart rate, preserves energetics, and protects ischemic hearts. Am J Physiol Heart Circ Physiol 2002; 283: H1562–8
- Black R. G., Jr., Guo Y., Ge Z. D., Murphree S. S., Prabhu S. D., Jones W. K., et al. Gene dosage‐dependent effects of cardiac‐specific overexpression of the A3 adenosine receptor. Circ Res 2002; 91: 165–72
- Harrison G. J., Cerniway R. J., Peart J., Berr S. S., Ashton K., Regan S., et al. Effects of A(3) adenosine receptor activation and gene knock‐out in ischemic‐reperfused mouse heart. Cardiovasc Res 2002; 53: 147–55
- Jacobson K. A., Gao Z. G. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov 2006; 5: 247–64
- Mozzicato S., Joshi B. V., Jacobson K. A., Liang B. T. Role of direct RhoA‐phospholipase D1 interaction in mediating adenosine‐induced protection from cardiac ischemia. FASEB J 2004; 18: 406–08
- Hausenloy D., Wynne A., Duchen M., Yellon D. Transient mitochondrial permeability transition pore opening mediates preconditioning‐induced protection. Circulation 2004; 109: 1714–17
- Lasley R. D., Keith B. J., Kristo G., Yoshimura Y., Mentzer R. M., Jr. Delayed adenosine A1 receptor preconditioning in rat myocardium is MAPK dependent but iNOS independent. Am J Physiol Heart Circ Physiol 2005; 289: H785–91
- Button L., Mireylees S. E., Germack R., Dickenson J. M. Phosphatidylinositol 3‐kinase and ERK1/2 are not involved in adenosine A1, A2A or A3 receptor‐mediated preconditioning in rat ventricle strips. Exp Physiol 2005; 90: 747–54
- Guo Y., Stein A. B., Wu W. J., Zhu X., Tan W., Li Q., et al. Late preconditioning induced by NO donors, adenosine A1 receptor agonists, and delta1‐opioid receptor agonists is mediated by iNOS. Am J Physiol Heart Circ Physiol 2005; 289: H2251–7
- Pugsley M. K. The diverse molecular mechanisms responsible for the actions of opioids on the cardiovascular system. Pharmacol Ther 2002; 93: 51–75
- Younes A., Pepe S., Yoshishige D., Caffrey J. L., Lakatta E. G. Ischemic preconditioning increases the bioavailability of cardiac enkephalins. Am J Physiol Heart Circ Physiol 2005; 289: H1652–61
- Patel H. H., Gross E. R., Peart J. N., Hsu A. K., Gross G. J. Sarcolemmal KATP channel triggers delayed ischemic preconditioning in rats. Am J Physiol Heart Circ Physiol 2005; 288: H445–7
- Cao Z., Liu L., Van Winkle D. M. Activation of delta‐ and kappa‐opioid receptors by opioid peptides protects cardiomyocytes via KATP channels. Am J Physiol Heart Circ Physiol 2003; 285: H1032–9
- Peart J. N., Gross G. J. Cardioprotective effects of acute and chronic opioid treatment are mediated via different signaling pathways. Am J Physiol Heart Circ Physiol 2006; 291: H1746–53
- Barrere‐Lemaire S., Combes N., Sportouch‐Dukhan C., Richard S., Nargeot J., Piot C. Morphine mimics the antiapoptotic effect of preconditioning via an Ins(1,4,5) P3 signaling pathway in rat ventricular myocytes. Am J Physiol Heart Circ Physiol 2005; 288: H83–8
- Fryer R. M., Pratt P. F., Hsu A. K., Gross G. J. Differential activation of extracellular signal regulated kinase isoforms in preconditioning and opioid‐induced cardioprotection. J. Pharmacol Exp Ther 2001; 296: 647–54
- Fryer R. M., Wang Y., Hsu A. K., Gross G. J. Essential activation of PKC‐δ in opioid‐initiated cardioprotection. Am J Physiol 2001; 280: H1346–53
- Fryer R. M., Wang Y., Hsu A. K., Nagase H., Gross G. J. Dependence of δ1‐opioid receptor‐induced cardioprotection on a tyrosine kinase‐dependent but not a Src‐dependent pathway. J Pharmacol Exp Ther 2001; 299: 477–82
- Baker K. M., Kumar R. Intracellular Angiotensin II Induces Cell Proliferation Independent of AT1 Receptor. Am J Physiol Cell Physiol 2006; 291: C995–1001
- Kossmehl P., Kurth E., Faramarzi S., Habighorst B., Shakibaei M., Wehland M., et al. Mechanisms of apoptosis after ischemia and reperfusion: role of the renin‐angiotensin system. Apoptosis 2006; 11: 347–58
- Gupta M. K., Neelakantan T. V., Sanghamitra M., Tyagi R. K., Dinda A., Maulik S., et al. An assessment of the role of reactive oxygen species and redox signaling in norepinephrine‐induced apoptosis and hypertrophy of h9c2 cardiac myoblasts. Antioxid Redox Signal 2006; 8: 1081–93
- Eguchi S., Iwasaki H., Ueno H., Frank G. D., Motley E. D., Eguchi K., et al. Intracellular signaling of angiotensin II‐induced p70 S6 kinase phosphorylation at Ser(411) in vascular smooth muscle cells. Possible requirement of epidermal growth factor receptor, Ras, extracellular signal‐regulated kinase, and Akt. J Biol Chem 1999; 274: 36843–51
- Senbonmatsu T., Saito T., Landon E. J., Watanabe O., Price E J. r., Roberts R. L., et al. A novel angiotensin II type 2 receptor signaling pathway: possible role in cardiac hypertrophy. EMBO J 2003; 22: 6471–82
- Karliner J. S., Honbo N., Epstein C. J., Xian M., Lau Y. F., Gray M. O. Neonatal mouse cardiac myocytes exhibit cardioprotection induced by hypoxic and pharmacologic preconditioning and by transgenic overexpression of human Cu/Zn superoxide dismutase. J Mol Cell Cardiol 2000; 32: 1779–86
- Marais E., Genade S., Strijdom H., Moolman J. A., Lochner A. p38 MAPK activation triggers pharmacologically‐induced beta‐adrenergic preconditioning, but not ischaemic preconditioning. J Mol Cell Cardiol 2001; 33: 2157–77
- Robinet A., Hoizey G. Millart H. PI 3‐kinase, protein kinase C, and protein kinase A are involved in the trigger phase of beta1‐adrenergic preconditioning. Cardiovasc Res 2005; 66: 530–42
- Schoemaker R. G., van Heijningen C. L. Bradykinin mediates cardiac preconditioning at a distance. Am J Physiol Heart Circ Physiol 2000; 278: H1571–6
- Parratt J. R., Vegh A. Coronary vascular endothelium‐myocyte interactions in protection of the heart by ischaemic preconditioning. J Physiol Pharmacol 1999; 50: 509–24
- Broadhead M. W., Kharbanda R. K., Peters M. J., MacAllister R. J. KATP channel activation induces ischemic preconditioning of the endothelium in humans in vivo. Circulation 2004; 110: 2077–82
- Sen C. K., Khanna S., Roy S. Perceived hyperoxia: Oxygen‐induced remodeling of the reoxygenated heart. Cardiovasc Res 2006; 71: 280–8
- Mourad O. G. B. I., Johnson J. A. Protein kinase Cϵ interacts with cytochrome c oxidase subunit IV and enhances cytochrome c oxidase activity in neonatal cardiac myocyte Preconditioning. Biochem J 2006; 393: 191–9
- Mansfield K. D., Simon M. C., Keith B. Hypoxic reduction in cellular glutathione levels requires mitochondrial reactive oxygen species. J Appl Physiol 2004; 97: 1358–66
- Eigel B. N., Gursahani H., Hadley R. W. ROS are required for rapid reactivation of Na+/Ca2+ exchanger in hypoxic reoxygenated guinea pig ventricular myocytes. Am J Physiol Heart Circ Physiol 2004; 286: H955–63
- Kulisz A., Chen N., Chandel N. S., Shao Z., Schumacker P. T. Mitochondrial ROS initiate phosphorylation of p38 MAP kinase during hypoxia in cardiomyocytes. Am J Physiol Lung Cell Mol Physiol 2002; 282: L1324–9
- Dougherty C. J., Kubasiak L. A., Frazier D. P., Li H., Xiong W. C., Bishopric N. H., et al. Mitochondrial signals initiate the activation of c‐Jun N‐terminal kinase (JNK) by hypoxia‐reoxygenation. FASEB J 2004; 18: 1060–70
- Lebuffe G., Schumacker P. T., Shao Z. H., Anderson T., Iwase H., Vanden Hoek T. L. ROS and NO trigger early preconditioning: relationship to mitochondrial KATP channel. Am J Physiol Heart Circ Physiol 2003; 284: H299–308
- Ning X. H., Chen S. H., Buroker N. E., Xu C. S., Li F. R., Li S. P., et al. Short‐cycle hypoxia in the intact heart: hypoxia‐inducible factor 1 signaling and the relationship to injury threshold. Am J Physiol Heart Circ Physiol 2007; 292: H333–41
- Hwang J. J., Hur K. C. Insulin cannot activate extracellular‐signal‐related kinase due to inability to generate reactive oxygen species in SK‐N‐BE(2) human neuroblastoma cells. Mol Cells 2005; 20: 280–7
- Schmelter M., Ateghang B., Helmig S., Wartenberg M., Sauer H. Embryonic stem cells utilize reactive oxygen species as transducers of mechanical strain‐induced cardiovascular differentiation. FASEB J 2006; 20: 1182–4
- Nelson K. K., Subbaram S., Connor K. M., Dasgupta J., Ha X. F., Meng T. C., et al. Redox‐dependent matrix metalloproteinase‐1 expression is regulated by JNK through Ets and AP‐1 promoter motifs. J Biol Chem 2006; 281: 14100–10
- Mehdi M. Z., Pandey N. R., Pandey S. K., Srivastava A. K. H2O2‐induced phosphorylation of ERK1/2 and PKB requires tyrosine kinase activity of insulin receptor and c‐Src. Antioxid Redox Signal 2005; 7: 1014–20
- Papaiahgari S., Zhang Q., Kleeberger S. R., Cho H. Y., Reddy S. P. Hyperoxia stimulates an Nrf2‐ARE transcriptional response via ROS‐EGFR‐PI3K‐Akt/ERK MAP kinase signaling in pulmonary epithelial cells. Antioxid Redox Signal 2006; 8: 43–52
- Gonzalez‐Santiago L., Suarez Y., Zarich N., Munoz‐Alonso M. J., Cuadrado A., Martinez T., et al. Aplidin induces JNK‐dependent apoptosis in human breast cancer cells via alteration of glutathione homeostasis, Rac1 GTPase activation, and MKP‐1 phosphatase downregulation. Cell Death Differ 2006; 13: 1968–81
- Jaubert A., Ichas F., Bresson‐Bepoldin L. Signaling Pathway Involved in the Pro‐Apoptotic Effect of Dopamine in the GH3 Pituitary Cell Line. Neuroendocrinology 2006; 83: 77–88
- Ito K., Hirao A., Arai F., Takubo K., Matsuoka S., Miyamoto K., et al. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat Med 2006; 12: 446–51
- Ford E., Hughes M. N., Wardman P. Kinetics of the reactions of nitrogen dioxide with glutathione, cysteine, and uric acid at physiological pH. Free Radical Biol Med 2002; 32: 1314–23
- Yaniv G., Shilkrut M., Larisch S., Binah O. Hydrogen peroxide predisposes neonatal rat ventricular myocytes to Fas‐mediated apoptosis. Biochem Biophys Res Commun 2005; 336: 740–6
- Balijepalli R. C., Foell J. D., Hall D. D., Hell J. W., Kamp T. J. Localization of cardiac L‐type Ca(2+) channels to a caveolar macromolecular signaling complex is required for beta(2)‐adrenergic regulation. Proc Natl Acad Sci U S A 2006; 103: 7500–05
- Nishi T., Shimizu N., Hiramoto M., Sato I., Yamaguchi Y., Hasegawa M., et al. Spatial redox regulation of a critical cysteine residue of NF‐kappa B in vivo. J Biol Chem 2002; 277: 44548–56
- Hainaut P., Mann K. Zinc binding and redox control of p53 structure and function. Antioxid Redox Signal 2001; 3: 611–23
- Liu H., Colavitti R., Rovira I. I., Finkel T. Redox‐dependent transcriptional regulation. Circ Res 2005; 97: 967–74
- Tell G., Pines A., Paron I., D'Elia A., Bisca A., Kelley M. R., et al. Redox effector factor‐1 regulates the activity of thyroid transcription factor 1 by controlling the redox state of the N transcriptional activation domain. J Biol Chem 2002; 277: 14564–74
- Malik G., Gorbounov N., Das S., Gurusamy N., Otani H., Maulik N., et al. Ischemic preconditioning triggers nuclear translocation of thioredoxin and its interaction with Ref‐1 potentiating a survival signal through the PI‐3‐kinase‐Akt pathway. Antioxid Redox Signal 2006; 8: 2101–9
- Hsieh M. M., Hegde V., Kelley M. R., Deutsch W. A. Activation of APE/Ref‐1 redox activity is mediated by reactive oxygen species and PKC phosphorylation. Nucleic Acids Res 2001; 29: 3116–22
- Ziel K. A., Grishko V., Campbell C. C., Breit J. F., Wilson G. L., Gillespie M. N. Oxidants in signal transduction: impact on DNA integrity and gene expression. FASEB J 2005; 19: 387–94
- Wang N., Stemerman M. B. Ref‐1 and transcriptional control of endothelial apoptosis. Circ Res 2001; 88: 1223–5
- Hall J. L., Wang X., Adamson Van, Zhao Y., Gibbons G. H. Overexpression of Ref‐1 inhibits hypoxia and tumor necrosis factor‐induced endothelial cell apoptosis through nuclear factor‐kappab‐independent and ‐dependent pathways. Circ Res 2001; 88: 1247–53
- Stempien‐Otero A., Karsan A., Cornejo C. J., Xiang H., Eunson T., Morrison R. S., et al. Mechanisms of hypoxia‐induced endothelial cell death: role of p53 in apoptosis. J Biol Chem 1999; 274: 8039–45
- Piret J. P., Mottet D., Raes M., Michiels C. Is HIF‐1alpha a pro‐ or an anti‐apoptotic protein?. Biochem Pharmacol 2002; 64: 889–92
- Lee M. J., Kim J. Y., Suk K., Park J. H. Identification of the hypoxia‐inducible factor 1 alpha‐responsive HGTD‐P gene as a mediator in the mitochondrial apoptotic pathway. Mol Cell Biol 2004; 24: 3918–27
- Hess J., Angel P., Schorpp‐Kistner M. AP‐1 subunits: quarrel and harmony among siblings. J Cell Sci 2004; 17: 5965–73
- Graham B., Gibson S. B. The two faces of NFkappaB in cell survival responses. Cell Cycle 2005; 4: 1342–5
- Lavin M. F., Gueven N. The complexity of p53 stabilization and activation. Cell Death Differ 2006; 13: 941–50
- Ordway J. M., Eberhart D., Curran T. Cysteine 64 of Ref‐1 is not essential for redox regulation of AP‐1 DNA binding. Mol Cell Biol 2003; 23: 4257–66
- Iwasaki K., Mackenzie E. L., Hailemariam K., Sakamoto K., Tsuji Y. Hemin‐mediated regulation of an antioxidant‐responsive element of the human ferritin H gene and role of Ref‐1 during erythroid differentiation of K562 cells. Mol Cell Biol 2006; 26: 2845–56
- Levonen A. L., Landar A., Ramachandran A., Ceaser E. K., Dickinson D. A., Zanoni G., et al. Cellular mechanisms of redox cell signalling: role of cysteine modification in controlling antioxidant defences in response to electrophilic lipid oxidation products. Biochem J 2004; 378: 373–82
- He X., Chen M. G., Lin G. X., Ma Q. Arsenic induces NAD(P)H:quinone oxidoreductase I by disrupting the NRF2/KEAP1/CUL3 complex and recruiting NRF2/MAF to are enhancer. J Biol Chem 2006; 281: 23620–31
- Eggler A. L., Liu G., Pezzuto J. M., van Breemen R. B., Mesecar A. D. Modifying specific cysteines of the electrophile‐sensing human Keap1 protein is insufficient to disrupt binding to the Nrf2 domain Neh2. Proc Natl Acad Sci U S A 2005; 102: 10070–5
- Leonard M. O., Kieran N. E., Howell K., Burne M. J., Varadarajan R., Dhakshinamoorthy S., et al. Reoxygenation‐specific activation of the antioxidant transcription factor Nrf2 mediates cytoprotective gene expression in ischemia‐reperfusion injury. FASEB J 2006; 20: 2624–6