Abstract
Immune reactions are stringently regulated and balanced by complex interactions of stimulating and suppressing mechanisms. Dysfunctions of this sophisticated immune regulatory network can lead to a variety of diseases such as autoimmunity, allergy, cancer, and pregnancy disorders. The rediscovery of suppressor T cells a decade ago—now designated as T regulatory cells—set off a huge avalanche of research activities leading to a multitude of preclinical and clinical studies. Herein, we give a comprehensive review about this research on T regulatory cells and the relevance of this suppressive T cell population for the development of innovative immune therapeutic strategies.
| Abbreviations | ||
| IL | = | interleukin |
| APC | = | antigen presenting cells |
| TGF | = | transforming growth factor |
| cAMP | = | cyclic AMP |
| MHC | = | major histocompatibility complex |
| GITR | = | glucocorticoid‐induced TNF receptor |
| CTLA | = | cytotoxic T lymphocyte antigen |
| TLR | = | toll‐like receptor |
| CCR5 | = | CC chemokine receptor 5 |
| IFN | = | interferon |
| NK | = | natural killer cells |
| TNF | = | tumor necrosis factor |
Introduction
History
The revelation of the thymus function and of thymus‐derived lymphocytes (T cells) during the 60s and 70s of the last century clearly indicated the vital importance of T cells for the regulation of an immune response Citation1,2. Besides the immune stimulating function of T cells it was quite obvious that a certain fraction of this population has suppressive properties and therefore was referred to as suppressor T cells Citation3,4. A multitude of studies convincingly demonstrated the crucial role of suppressor T cells for the maintenance of immunologic tolerance Citation5. Unfortunately, the characterization of I‐J determinants on suppressor T cells that subsequently could not be approved by genomic analyses discredited this field of immunology for a long time Citation6–9. Nevertheless, many studies using autoimmune and transplantation models consistently demonstrated that suppressor T cells exist although it was not possible to unequivocally characterize such cells. Finally, a ground‐breaking study by Sakaguchi and colleagues definitively identified suppressor T cells as CD4+CD25+ T cells that emerged from the thymus and could be isolated from the periphery of nonimmunized mice Citation10. In order to avoid the discredited expression suppressor T cells these cells were designated as T regulatory cells (Tregs) or more precisely as naturally occurring Tregs (nTregs). nTregs comprise 5%–10% of whole CD4+ T cells, and depletion of them before transfer of CD4+ T cells into T cell‐deficient mice resulted in a large panel of different autoimmune diseases Citation10. Thus, these findings indicate that a fraction of the positively selected peripheral CD4+ T cells is potentially autoreactive and requires being tolerized by nTregs. Initial analyses revealed that the reduction of interleukin (IL)‐2 expression in responder T cells is the main target of nTreg‐mediated suppression Citation11. Regarding their characterization, it should be noted that CD25 is not a unique membrane‐bound marker of nTregs but also expressed by activated conventional T cells. Thus, CD25 is not exclusively expressed by nTregs. Moreover, despite very intensive endeavors an exclusive surface marker for nTregs could not be identified yet, but the transcription repressor FoxP3 was found to be expressed selectively by murine nTregs Citation12,13.
In the beginning of 2000 it was shown that mutations in the gene encoding for FoxP3 were responsible not only for the phenotype seen in the so‐called scurfy mouse but also were the cause for IPEX (immune dysregulation, polyendocrinopathy, enteropathy, X‐linked syndrome) in humans Citation14–17. Recent studies clearly revealed that FoxP3 is not only a transcriptional repressor but also essential for the development and function of murine and human Tregs Citation12,Citation18,19. In contrast to mice, human conventional CD4+ and CD8+ T cells have been shown to upregulate FoxP3 mRNA and protein upon in vitro activation Citation20–24, suggesting a cell‐intrinsic role, e.g. in regulating cytokine production and proliferation in these cells rather than the conversion into adaptive regulatory T cells. Moreover, the existence of IPEX‐like individuals which lack mutations within the FoxP3 gene but are phenotypically not distinct from IPEX‐patients casts doubt on the role of FoxP3 in the development and function of human Tregs. On the other hand, the result that transduction with FoxP3 of human conventional CD4+ T cells leads to T cells with suppressive properties Citation25, resumes the discussion whether the mechanisms of nTreg development and function differ in humans.
The characterization of human Tregs as well as the description of various T cell populations with suppressive activities, provisionally termed induced Tregs (iTregs), in vitro and in vivo, eventually led to a multitude of studies, which resulted in a huge amount of data. This review is primarily focused on the function of nTregs/iTregs in several preclinical animal models and their potential exploitation for novel immune therapeutic strategies.
Treg subpopulations
It was clearly shown by thymectomy and by using T cell receptor transgenic mice that nTregs develop in the thymus to a discrete T cell population Citation10,Citation26. However, it is still a prominent question, at least with respect to humans, if and to what extent such cells with suppressive potencies can arise in the periphery Citation27. Indeed, there are several evidences for the peripheral induction of CD4+ T cells with suppressive properties (iTregs, ). However, in the human system, as CD25 and even FoxP3 are not reliable markers for human nTregs, it is a desirable approach to determine whether large numbers of iTregs are generated in humans. As it is therefore not possible to discriminate between nTregs and iTregs in the periphery of an individual, these populations are in the following referred to as Tregs.
Table I. Characteristics of naturally occurring (nTregs) and induced T regulatory cells (iTregs).
In vitro, conventional CD4+ T cells can be converted to so‐called Tr1 (T regulatory type 1) or Th3 (T helper type 3) cells by using different approaches. In general, all these approaches favor the activation of T cells under suboptimal or repressing conditions, e.g. repetitive stimulation with nonprofessional antigen presenting cells (APCs) or in the presence of immune‐suppressive drugs or cytokines like transforming growth factor (TGF)‐β or IL‐10. Tr1 cells were initially generated by chronic stimulation of conventional CD4+ T cells in the presence of IL‐10 Citation28. In addition, the in vitro treatment with a combination of vitamin D3 and dexamethasone resulted also in the generation of T cells with suppressive properties Citation29. Complementary to Tr1 cells, Th3 cells which produce preferentially TGF‐β have been first generated in vivo following oral tolerance induction Citation30. Besides these studies, it was recently shown that FoxP3+ iTregs develop from conventional CD4+ T cells after activation in the presence of TGF‐β and IL‐2 Citation31,32. Furthermore, there is growing evidence that cyclic AMP‐elevating agents, like prostaglandin E2 (PGE2) or the vasoactive intestinal peptide (VIP), not only induce the expression of FoxP3 in conventional T cells but also promote the generation of T cells with suppressive properties in vivo and in vitroCitation33–35. Finally, also dendritic cells (DC) can play a decisive role in the extrathymic induction of T cells with regulatory properties. The activation of conventional CD4+ T cells in vitro and in vivo with immature DC or with DC treated with immune‐suppressive cytokines (e.g. IL‐10, TGF‐β) results in the induction of anergic T cells with regulatory properties Citation36.
The number of iTreg family members is still growing and iTregs cannot be discriminated from nTregs by specific surface markers. However, some iTregs are distinct from nTregs as the suppressive potency of the former is generally based on immune‐suppressive cytokines (e.g. IL‐10 or TGF‐β) and not on cell contact‐dependent mechanisms as shown for nTregs. Furthermore, iTregs are heterogeneous in their ability to express CD25, glucocorticoid‐induced TNF receptor (GITR) and FoxP3. However, despite the heterogeneity of iTregs and their mechanism of action, the therapeutic potential of these peripherally generated iTregs is of great interest especially in the human system. As the isolation of large numbers of nTregs from the peripheral blood of individuals is challenging, the generation of great amounts of antigen‐specific iTregs ex vivo might represent a sanguine approach for the control of autoimmune responses.
Suppressive mechanisms
Central to the understanding of nTregs is the molecular basis of their mechanism of action. Although great effort has been made to unravel these mechanisms, even the basics of this process are far from definitive Citation37. Surveying the hitherto performed in vitro and in vivo studies the picture gets even more disconcerting. In vitro it was clearly shown that nTregs exert their suppressive properties via a cell contact‐dependent mechanism which requires antigen‐specific T cell receptor stimulation but is independent from APCs Citation38. In contrast to the in vitro obtained results, the in vivo suppressive mechanism of nTregs remains controversial. In particular all hitherto performed in vitro studies clearly vote for the colocalization and stable T cell interactions between nTregs and effector T cells. However, recent studies using two‐photon laser‐scanning microscopy to directly visualize nTregs and effector T cells within the same lymph node suggested only short‐term interactions between nTregs and effector T cells Citation39. Moreover, Tang et al. showed that nTregs formed persistent cell contacts with dendritic cells (DC) preceding a shortened contact between the effector T cells and DC, assuming that a cell contact‐dependent mechanism of suppression by nTregs in vivo might preferentially target DC. Moreover, in vitro studies clearly failed to identify a soluble factor mediating the suppressive function of nTregs. However, in vivo there is growing evidence that TGF‐β and IL‐10 play an important role in the immune regulatory network orchestrated by nTregs. Thus, it is likely that in vivo nTregs provide the basis of peripheral tolerance by initially suppressing other T cells via cell contact‐dependent mechanisms, and simultaneously conveying suppressive properties to these T cells in a process termed infectious tolerance Citation40,41. The resulting iTregs were found to produce the suppressive cytokines TGF‐β and/or IL‐10. Hence, the secretion of these inhibitory cytokines by iTregs considerably expands the Treg‐prearranged suppressive network, thereby manifesting also far‐ranging peripheral tolerance. By virtue of all recent data, the molecular mechanisms underlying nTreg‐mediated suppression in vitro and in vivo require further investigation.
Key messages
The enhancement of Treg function is an innovative immunotherapeutic strategy for the treatment of autoimmune, allergic, and infectious diseases and for the prevention of transplant rejection and pregnancy disorders as well. Conversely, for treatment of cancer the transient abrogation of Treg function is a desirable approach.
Preclinical mouse models
Autoimmunity
Nearly 40 years ago neonatal thymectomy was found to lead to oophoritis suggesting that thymus‐derived cells can suppress autoimmunity Citation42. This study provided the first experimental evidence that thymocytes and their progeny are involved in the prevention of organ‐specific autoimmune diseases. Subsequently, the transfer of nTreg‐depleted CD4+ T cells in T cell‐deficient mice was found to result in the development of various autoimmune diseases, among them gastritis, oophoritis, and thyroiditis, indicating the crucial role of nTregs for the maintenance of peripheral tolerance Citation10. These findings led to a reevaluation of results from established autoimmune models and caused a huge wave of studies to clarify the role of nTregs in autoimmune diseases.
Inflammatory bowel disease
In a murine inflammatory bowel disease (IBD) model that corresponds to human diseases morbus Crohn and colitis ulcerosa it was shown that nTregs can prevent the induction of the disease Citation43. The analysis of the suppressive mechanism revealed that TGF‐β as well as IL‐10 were involved. However, the influence of IL‐10 was found to be rather complex depending on the IBD model used Citation44. IL‐10 could be produced by nTregs as well as by other hematopoietic cells among them Tr1 and Th2 cells. Very recently, it was shown that transferred nTregs can cure intestinal inflammation Citation45. The progeny of these nTregs were found to be selectively enriched in the colonic lamina propria and to express IL‐10 and FoxP3 indicating the important function of nTreg‐derived IL‐10 in vivo. TGF‐β also plays a crucial role but has not to be produced by nTregs themselves since TGF‐β‐deficient nTregs are fully functional after transfer in wild‐type mice Citation46. However, their suppressive capacity was inhibited when TGF‐β was neutralized indicating that in vivo this cytokine is provided by non‐Treg cells under the influence of nTregs Citation47. In addition, colitogenic T effector cells which cannot respond to TGF‐β cannot be controlled by nTregs, underscoring the importance of TGF‐β for Treg‐mediated suppression Citation48. Recently, it was published that CD4+FoxP3++ T cells which were generated in vitro in the presence of TGF‐β can prevent the induction of Th1‐mediated experimental colitis Citation49. In analogy to the preclinical studies, increased numbers of human Tregs could be identified in the lamina propria of patients suffering from morbus Crohn suggesting that in the human system Tregs can also play an important role for the regulation of IBD Citation50. Human Tregs from inflamed mucosa could be expanded in vitro indicating that such Tregs represent an attractive option for an adaptive immunotherapy of IBD Citation51.
Experimental autoimmune encephalomyelitis
Experimental autoimmune encephalomyelitis (EAE) is a model for multiple sclerosis and can be induced by immunizing mice with myelin oligodendrocyte glycoprotein (MOG), myelin basic protein (MPB), or proteolipid protein (PLP) leading to the development of encephalitogenic Th1 cells Citation52. Feeding MBP before induction of EAE could prevent outbreak of the disease via the induction of oral tolerance. Further analyses revealed that orally administered MBP induced the development of TGF‐β‐producing Th3 cells in mesenteric lymph nodes Citation30. Furthermore, depletion of CD25+ T cells could aggravate PLP‐induced EAE while the transfer of nTregs from naïve SJL (Swiss James Lambert) mice alleviated symptoms of the disease Citation53. Transferred Tregs from IL‐10‐deficient mice were ineffective suggesting that nTregs control EAE via an IL‐10‐dependent mechanism. These data imply that a complex interaction between nTregs, Tr1, and Th3 cells is required in combination with IL‐10‐ and TGF‐β‐dependent mechanisms to prevent autoimmune reactions directed to the central nervous system. Recently, it was found that administration of vasoactive intestinal peptide (VIP) to EAE mice results in the expansion of nTregs which inhibit encephalitogenic T cell activation. Interestingly, the suppressive mechanism of such nTregs was based partially on direct cell contact and on TGF‐β and IL‐10 as well Citation34. Hence, these data strongly suggest that EAE can be profoundly alleviated through preferential activation and recruitment of distinct Treg populations. Therefore, transient enhancement and expansion of T cells with suppressive potencies will certainly be a promising strategy for treatment of EAE.
Type 1 diabetes
An important animal model of human type 1 diabetes (T1D) is the nonobese diabetic mouse (NOD) which spontaneously develops autoimmunity that is caused by autoaggressive CD4+ and CD8+ T cells Citation54,55. These T effector cells ultimately destroy β cells of the pancreas. T1D could be induced by transfer of Th1 cells while Th2 cells were found to inhibit a diabetogenic response in NOD mice Citation56. Therefore, an initial strategy for treatment of T1D was the promotion of Th2 cells. Unfortunately, Th2 cells subsequently were also described to induce T1D indicating that other T cell populations have to be involved in the control of T1D Citation57. Meanwhile, it has been found that nTregs play a central role in NOD mice because it was shown that their decrease allows the development of T1D in such mice Citation58,59. Disruption of the CD28‐B7 interaction prevented the development of nTregs and resulted in exacerbated diabetes in NOD mice. Transfer of nTregs in CD28‐deficient NOD mice could prevent or at least delay diabetes. Thus, development of diabetes depends on the balance of diabetogenic T cells and nTregs and is regulated by costimulatory mechanisms. nTregs that had been antigen‐specifically expanded in vitro could very efficiently inhibit diabetes in an early phase of disease Citation60,61. Intranasal application of proinsulin in combination with intravenously administered anti‐CD3e F(ab')2 fragments induced iTregs and prevented autoimmune diabetes Citation62,63. Similar to other autoimmune models IL‐10 and TGF‐β are decisively involved in the suppressive mechanism of such iTregs. These data suggest a profound therapeutic potential for this combination of polyclonal T cell activation and antigen‐specific stimulation of iTregs and nTregs as well.
Transplantation/pregnancy
Transplantation
About half a century ago G. Snell discovered the major histocompatibility complex (MHC) and elucidated its importance for tissue transplantation originally using a tumor transplant model Citation64,65. Simultaneously, P. Medawar and colleagues—inspired by R. Owen's work on vascular anastomoses between bovine twins—demonstrated that skin graft tolerance can be induced by neonatal transfer of spleen cells prepared from the strain of the graft Citation66,67. This finding indicated that transplantation tolerance can be actively acquired. Subsequently, treatment with an antilymphocyte serum enables the survival of skin heterografts implying the involvement of T cells in transplantation tolerance Citation68. The description of suppressor T cells at that time also suggested a crucial role of such T cells for acceptance of transplants via manipulation of the host immune system Citation4,Citation69. Alternatively, immunosuppressive agents like the calcineurin inhibitor cyclosporine A (CSA) were found to significantly improve the early survival of transplants. The analysis of mechanism of action revealed that treatment with CSA induced suppressive CD4+ T cells Citation70. In analogy, the treatment of adult mice with a combination of nonlytic anti‐CD4 and anti‐CD8 antibodies led also to tolerant T cells which, in addition, conveyed their suppressive properties to naïve T cells. Hence, these findings which were paraphrased by the expressions bystander suppression, linked suppression and infectious tolerance explained the observation that no further immune suppressive treatment was required for permanent transplantation tolerance Citation71,72. Subsequently, these data were doubtlessly corroborated in different models of transplantation tolerance indicating a crucial role for nTregs as well as iTregs Citation73–77. In a study using skin allotransplants nTregs and most possibly iTregs could also be identified in the transplant Citation78. After retransplantation of the skin graft to a second host these Tregs prevented rejection antigen‐specifically without hindrance of rejection of third party skin. These results demonstrate that nTregs and/or iTregs are directly active in a populated graft far beyond the lymphoid tissue. For therapeutic applications it is important that large numbers of murine nTregs can be obtained by activation and expansion in vitro. Such expanded nTregs were found to significantly inhibit a disease in a murine graft versus host disease (GVHD) model Citation79. Very recently human Treg cell lines could be generated from cord blood cells which strongly inhibited mixed lymphocyte reactions in vitro underscoring the therapeutic potency of human Tregs for the induction of transplantation tolerance Citation80.
Pregnancy
The maternal immune system has to accept a semiallogeneic fetus during pregnancy and tolerate the paternal alloantigens. Hence, pregnancy represents a state of immunological tolerance Citation81,82. Abortion‐prone mice were found to harbor significantly higher numbers of Th1 cells and simultaneously lower frequencies of nTregs as compared to females with normal pregnancy Citation83. In addition, abortion could be inhibited by transfer of nTregs from normal pregnant females, indicating the essential role of nTregs for the maintenance of transplantation tolerance during pregnancy. In addition, the number of human Tregs was elevated during early pregnancy in decidua and circulation, peaked in the second trimester, and declined postpartumCitation84–87. Thus, these data suggest that human Tregs in analogy to murine nTregs can prevent pregnancy disorders via suppression of antipaternal immune responses.
Cancer
Besides their beneficial role in protecting the host from autoimmune responses, nTregs may unfortunately suppress adequate antitumor immune responses. Hence, in murine tumor models, transient depletion of nTregs by anti‐CD25 monoclonal antibodies (mAb) in vivo can elicit an antitumor response through tumorantigen‐specific cytotoxic T lymphocytes (CTL) and tumorantigen‐nonspecific CD4−CD8− cytotoxic cells with natural killer (NK)‐like activity Citation88,89. Administration of the immunomodulator cytotoxic T lymphocyte antigen (CTLA)‐4‐Ig which acts as a competitive inhibitor of CD28‐B7 costimulation can profoundly enhance this antitumor response. These findings emphasize the hypothesis that likewise as self‐antigens, tumor cells are protected by nTregs. Moreover, tumors seem to be able to actively exploit the suppressive capacity of nTregs to escape an effective antitumor immune response. In this context, transfer of nonfractionated draining lymph node cells from tumor‐bearing mice 9 days post tumor inoculation resulted in a robust antitumor immune response against established tumors. In contrast, the same transfer on day 12 post tumor inoculation seldom prevented lethal tumor progression. As the lymph node cells from the tumor‐draining lymph node 12 days post tumor inoculation showed a four‐fold increase in cell numbers, this experiment suggested that preferentially T cells with regulatory properties were induced or that the number of Tregs was strongly increased at the tumor site Citation90.
Furthermore, tumor‐derived IL‐10/TGF‐β as well as tumor‐induced endogenous IL‐10/TGF‐β locally favor the conversion of conventional CD4+ T cells to a T cell with regulatory properties analogous to iTregs Citation91,92. In addition, solid tumors may produce chemokines (CCL22) that preferentially attract nTregs, which constitutively express several chemokine receptors Citation93. Both mechanisms—induction of iTregs and/or attraction of nTregs—will prevent efficient antitumor responses. Thus, transient depletion of nTregs and iTregs is a promising therapeutic approach to enable a curative and long‐lasting antitumor immune response. However, abrogation of tumor‐mediated suppression by depletion of Tregs is a potentially risky strategy since only little is known about the repopulation of nTregs and iTregs upon depletion. Moreover, as also tumor‐specific T effector cells may express CD25, the use of anti‐CD25 mAbs requires sophisticated kinetics and dosage studies during the course of an antitumor immune response. Recently, Treg‐affecting antitumor therapeutic strategy was ameliorated by application of nondepleting anti‐GITR mAb Citation94. Injection of agonistic anti‐GITR mAb into tumor‐bearing mice led to a strong antitumor response that was further improved by the simultaneous application of nondepleting anti‐CTLA‐4 mAb. Although the authors suggested that administration of anti‐GITR mAb primarily could abrogate the suppressive potency of nTregs, it is more likely that stimulation via GITR leads to a strong coactivation of antitumor‐specific T effector cells, which thereby escaped nTreg‐mediated suppression. However, this concept is not easily transferable into the human system, since the function of human GITR is not well characterized, and cross‐linking of GITR poses the risk of a polyclonal T effector cell stimulation potentially causing fatal autoimmunity.
Besides the application of anti‐CD25 mAb, administration of cyclophosphamide was assumed to primarily reduce the number of nTregs Citation95,96. Alternatively to direct manipulation of nTregs and T effector cell activity, immunologic suppression can be broken by conditioning of mature DC with specific toll‐like receptor (TLR)‐ligands. Several approaches clearly demonstrate that such a treatment, based on the usage of in vitro conditioned DC or through direct application of TLR ligands in vivo (that is to say reanimating an approach that was elaborated and applied by Busch, Fehleisen, and Coley a century ago Citation97–100) can neutralize or overcome the suppressive effects of nTregs leading to strong cytolytic activities and antitumor responses Citation101–103. Using a murine model of established tumor tolerance, it was found that a persistent TLR signal is needed in combination with a DC‐based vaccine for an effective antitumor response Citation104. As outlined below in the ‘Clinical translation’ section, Tregs were identified in many patients suffering from different tumors suggesting that the transient impairment of immune suppression by Tregs should profoundly improve specific tumor therapies.
Infection
Although Tregs seem to have far‐reaching beneficial effects on our health by influencing autoimmunity, transplantation, and even allergy, these cells always act as suppressors of immune responses, leading not only to salutary but also to detrimental effects. For example, inhibition of antimicrobial T effector cells by nTregs may lead on the one hand to severe and chronic infections, but on the other hand nTregs can prevent collateral damage of host tissue caused by vigorous antimicrobial immune responses Citation105,106. A very well investigated indicative of this ambivalent role of nTregs is their involvement in gastrointestinal homeostasis Citation107. During gastrointestinal inflammation commensal gut bacteria can enhance the pathology of such infections. However, in different mouse models of colitis nTregs play a crucial role in preventing excessive immune reactions Citation18,Citation108. In addition, in an infectious mouse model using Leishmania major it was found that these parasites can be totally eradicated in the absence of nTregs Citation109. However, these mice did not develop a protective memory response. Obviously, in this infectious model long‐lasting immunity of the host is based on a compromise that depends on a persisting low‐level parasitic infection, which is enabled by a limited nTreg‐mediated suppression. Moreover, nTregs, migrating in response to CCR5 ligands, rapidly accumulate within chronic dermal sites of Leishmania major infection Citation110. Apparently, nTregs favor low‐level parasite persistence by a partial suppression of T effector cells. Thus, in the course of an antimicrobial immune response, nTregs play a very delicate and sophisticated role. During the initial phase, nTregs should ideally not inhibit an antimicrobial immune response in order to immediately allow for a full‐blown response. Recent data strongly suggest that this is accomplished by two mechanisms, induced by the invading microbes themselves. DC especially conditioned by microbial TLR‐ligands penetrate the intrinsic suppressive shield of nTregs by strongly costimulating the proliferation of T effector cells and nTregs as well Citation111–113. Moreover, secretion of growth factors (e.g. IL‐2) by T effector cells potentiates the suppressive capacity of nTregs and their survival/proliferation Citation114. Simultaneously, TLR2 ligands directly induce the proliferation of nTregs thereby transiently concealing their suppressive potency Citation115. In the late phase of the response—after clearance of the microbial stimulators—DC remain in a nonactivated stage and the expanded nTregs display again their suppressive properties for effector T cells and DC as well. In addition, according to results from in vitro studies, demonstrating that preactivated nTregs have a much higher suppressive capacity than freshly isolated nTregs, expanded nTregs in the late phase have even stronger suppressive properties as compared to nTregs during initiation of the microbial infection Citation38,Citation116. Very recently, it was suggested that nTregs can not only suppress but actively kill their target cells Citation117. Thus, the successful combat of a microbial infection is a result of a dynamic regulation and modulation of Tregs, T effector cells and DC under the influence of microbial components, in particular TLR ligands.
Collectively, all preclinical models presented herein convincingly indicate that nTregs as well as iTregs are very promising targets for innovative therapeutic strategies to improve treatment of autoimmune and infectious diseases, cancer, transplant rejection, and pregnancy disorders. summarizes these preclinical data and illustrates future prospects for clinical applications. However, translation to the human system—as discussed below—certainly bears high risks and therefore requires great additional efforts to further characterize the distinct Treg populations, clarify their development and interrelationship, and to study their relations to other hematopoietic cells.
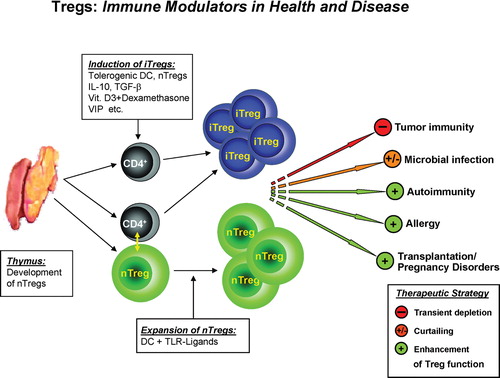
Figure 1. Tregs—immune modulators in health and disease. Naturally occurring CD4+CD25+ Tregs (nTregs) develop directly from CD4+ T cell precursors during positive selection in the thymus. Induced Tregs (iTregs) develop from naïve conventional CD4+ T cells either as a result of cell contact‐dependent interaction with nTregs or under the influence of suppressive agents like IL‐10, TGF‐β, dexamethasone, vitamin D3, vasoactive intestinal peptide, tolerogenic DC, and potentially other inhibitory mechanisms. Modulation of Treg (nTregs/iTregs) function is a promising intervention strategy to either improve responses in tumor and microbial diseases or suppress those unwanted in autoimmunity, allergy, transplantation, and pregnancy disorders. The transient depletion of Tregs as well as their manipulation, especially via toll‐like receptor (TLR) ligands, allow a transient reduction of Treg activity and enforce antitumor responses and immunity against viral infections. On the other hand their selective activation could diminish chronic pathological immune and autoimmune responses.
Clinical translation
As mentioned above, Tregs contribute to many immune‐mediated diseases either by inhibiting effective immune responses (e.g. cancer) or by failing to control unwanted immune responses in case of autoimmunity, allergy, certain infections, or transplantation. Therefore, manipulation of Treg activity becomes an attractive approach for immune intervention strategies.
Several evidences from cancer patients suggest that increased Treg activity is associated with an ineffective antitumor immune response. Enhanced numbers of Tregs have been observed in patients with lung, breast, pancreatic, and skin cancer either in peripheral blood or in the tumor itself Citation118–121. These Tregs inhibit proliferation and interferon (IFN)‐γ production of CD4+ and CD8+ effector cells and block cell‐mediated cytotoxicity of NK cells. Furthermore, the tumor tissue itself can favor the induction of Tregs by the production of suppressive cytokines such as IL‐10 and TGF‐β that convert immunostimulatory DC into tolerogenic APC. These DC further enhance the immunosuppressive state in cancer patients Citation122–124. The greatest barrier for Treg‐specific immunointervention of cancer is the lack of specific markers for targeting of human Tregs and to distinguish Tregs from activated effector T cells. Depletion of CD25+ Tregs by anti‐CD25 mAb, for example, includes the risk to deplete also tumor‐specific CD25+ T effector cells. Furthermore, IL‐10‐producing iTregs induced during tumor development by mechanisms of infectious tolerance or by tolerogenic DC induced by the tumor microenvironment might not be affected by this strategy. Therefore, anticancer therapies should target both nTreg and iTreg populations. An attractive alternative to direct manipulation of Treg activity in cancer patients comes from experiments showing that strong inflammatory conditions can break immunological tolerance. It has been demonstrated that conditioning of mature DC with specific TLR ligands can neutralize or overcome Treg‐mediated immunosuppression leading to strong antitumor responses Citation36,Citation103,Citation113,Citation125. Thus, for novel anticancer immunotherapeutic strategies, transient depletion or functional inactivation of Tregs in combination with an effective, antigen‐specific immune stimulation by conditioned tumorantigen‐presenting DC is a promising therapeutic objective.
Several studies have investigated whether human autoimmune diseases are associated with defective function of Tregs. In patients with multiple sclerosis, type I diabetes, psoriasis, and rheumatoid arthritis, isolated CD4+CD25+ T cells showed a reduced suppressive activity in vitro suggesting a defect in Treg function rather than reduced ratios of Tregs that contributes to the disease in these patients Citation126–130. However, these results might also reflect the inability to distinguish between activated CD4+CD25+ T effector cells and CD25+ Tregs in the periphery and/or increased migration of functional active Tregs to the inflamed tissues. Additionally, increasing evidence exists that autoimmune diseases can develop as a consequence of an enhanced resistance of autoaggressive T cells to regulation. For example, a recent study in patients with rheumatoid arthritis showed that isolated Tregs from these patients displayed an anergic phenotype and suppressed proliferation of T effector cells in vitro. In contrast, they were unable to inhibit cytokine release of activated T cells and production of proinflammatory cytokines by monocytes in a TNF‐α‐dependent manner Citation130. Furthermore, Tregs derived from patients were unable to convey a suppressive phenotype to CD4+ T effector cells. Interestingly, in patients that received an anti‐TNF‐α therapy, these defects appeared to be restored, suggesting that anti‐inflammatory therapies can also restore the functional activities of Tregs. Additionally, as the ratios of Tregs are unchanged or increased at the side of autoimmune lesions, therapeutic intervention strategies on the basis of Treg manipulation should be concentrated to restoration of Treg activity rather than simply increasing Treg numbers. Anti‐inflammatory drugs that enhance Treg activity and might also restore the sensibility of T effector cells to regulation have been documented. Well established examples are glucocorticoids, which are able to induce anti‐inflammatory cytokines such as IL‐10 and to modify human Treg activity Citation131,132. Furthermore, immunosuppressive drugs can induce tolerogenic DC that are able to convey nonregulatory CD4+ T helper cells into IL‐10‐producing iTregs. Although most effects of the drugs are nonspecific, anti‐inflammatory treatment might improve the efficacy and functional activity of Tregs as shown for anti‐TNF‐α therapy in patients with rheumatoid arthritis. More recent reports showed the potential of another nonspecific therapy of autoimmunity using non‐Fc‐binding anti‐CD3 antibodies in patients with new‐onset type I diabetes Citation133,134. Patients treated with anti‐CD3 mAb early after disease onset maintained an enhanced β cell function and required lower doses of insulin compared to the control group for up to 18 months. Although the mechanisms of action are not fully known, anti‐CD3‐treated patients showed a decrease of autoaggressive T cells together with a TGF‐β‐depending increase of Tregs Citation135. The oldest example of an efficient immunotherapy is the antigen‐specific desensitization of allergic patients. Injection of increasing concentrations of specific allergen in selected patient groups is able to induce antigen‐specific immunological tolerance. Two recent studies Citation136,137 provide evidence that this tolerance is associated with the induction allergen‐specific IL‐10‐producing iTregs.
Modulation of imbalanced immunity in immune‐mediated pathologies by directed manipulation of Treg activity has the potential to work therapeutically. However, many questions remain, and strategies that specifically and efficiently target Tregs in vivo must be defined or optimized.
Acknowledgements
The authors are grateful to Drs Karen Lingnau and Matthias Klein for critical reading of this manuscript and helpful discussions. This work was supported by the Deutsche Forschungsgemeinschaft grant SFB548‐A6 (to T. Bopp, E. Schmitt) and grant SFB548‐A8 (to H. Jonuleit).
Notes
1. It is difficult, especially in the human system, to decide whether ex vivo isolated Tregs belong to the nTreg or iTreg pool. Therefore, in case of doubt, we used the general word Tregs instead of iTregs or nTregs.
References
- Miller J. F. Immunological function of the thymus. Lancet 1961; 2: 748–9
- Miller J. F., Mitchell G. F. Cell to cell interaction in the immune response. I. Hemolysin‐forming cells in neonatally thymectomized mice reconstituted with thymus or thoracic duct lymphocytes. J Exp Med 1968; 128: 801–20
- Gershon R. K., Cohen P., Hencin R., Liebhaber S. A. Suppressor T cells. J Immunol 1972; 108: 586–90
- Gershon R. K., Kondo K. Cell interactions in the induction of tolerance: the role of thymic lymphocytes. Immunology 1970; 18: 723–37
- Gershon R. K. A disquisition on suppressor T cells. Transplant Rev 1975; 26: 170–85
- Okumura K., Takemori T., Tokuhisa T., Tada T. Specific enrichment of the suppressor T cell bearing I‐J determinants: parallel functional and serological characterizations. J Exp Med 1977; 146: 1234–45
- Tada T., Taniguchi M., David C. S. Properties of the antigen‐specific suppressive T‐cell factor in the regulation of antibody response of the mouse. IV. Special subregion assignment of the gene(s) that codes for the suppressive T‐cell factor in the H‐2 histocompatibility complex. J Exp Med 1976; 144: 713–25
- Kronenberg M., Steinmetz M., Kobori J., Kraig E., Kapp J. A., Pierce C. W., et al. RNA transcripts for I‐J polypeptides are apparently not encoded between the I‐A and I‐E subregions of the murine major histocompatibility complex. Proc Natl Acad Sci U S A 1983; 80: 5704–8
- Steinmetz M., Minard K., Horvath S., McNicholas J., Srelinger J., Wake C., et al. A molecular map of the immune response region from the major histocompatibility complex of the mouse. Nature 1982; 300: 35–42
- Sakaguchi S., Sakaguchi N., Asano M., Itoh M., Toda M. Immunologic self‐tolerance maintained by activated T cells expressing IL‐2 receptor alpha‐chains (CD25). Breakdown of a single mechanism of self‐tolerance causes various autoimmune diseases. J Immunol 1995; 155: 1151–64
- Thornton A. M., Shevach E. M. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med 1998; 188: 287–96
- Khattri R., Cox T., Yasayko S. A., Ramsdell F. An essential role for Scurfin in CD4+CD25+ T regulatory cells. Nat Immunol 2003; 4: 337–42
- Schubert L. A., Jeffery E., Zhang Y., Ramsdell F., Ziegler S. F. Scurfin (FOXP3) acts as a repressor of transcription and regulates T cell activation. J Biol Chem 2001; 276: 37672–9
- Chatila T. A., Blaeser F., Ho N., Lederman H. M., Voulgaropoulos C., Helms C., et al. JM2, encoding a fork head‐related protein, is mutated in X‐linked autoimmunity‐allergic disregulation syndrome. J Clin Invest 2000; 106: R75–81
- Brunkow M. E., Jeffery E. W., Hjerrild K. A., Paeper B., Clark L. B., Yasayko S. A., et al. Disruption of a new forkhead/winged‐helix protein, scurfin, results in the fatal lymphoproliferative disorder of the scurfy mouse. Nat Genet 2001; 27: 68–73
- Wildin R. S., Ramsdell F., Peake J., Faravelli F., Casanova J. L., Buist N., et al. X‐linked neonatal diabetes mellitus, enteropathy and endocrinopathy syndrome is the human equivalent of mouse scurfy. Nat Genet 2001; 27: 18–20
- Bennett C. L., Christie J., Ramsdell F., Brunkow M. E., Ferguson P. J., Whitesell L., et al. The immune dysregulation, polyendocrinopathy, enteropathy, X‐linked syndrome (IPEX) is caused by mutations of FOXP3. Nat Genet 2001; 27: 20–1
- Hori S., Nomura T., Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science 2003; 299: 1057–61
- Fontenot J. D., Gavin M. A., Rudensky A. Y. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol 2003; 4: 330–6
- Walker M. R., Kasprowicz D. J., Gersuk V. H., Benard A., Van Landeghen M., Buckner J. H., et al. Induction of FoxP3 and acquisition of T regulatory activity by stimulated human CD4+. J Clin Invest 2003; 112: 1437–43
- Morgan M. E., van Bilsen J. H., Bakker A. M., Heemskerk B., Schilham M. W., Hartgers F. C., et al. Expression of FOXP3 mRNA is not confined to CD4+CD25+ T regulatory cells in humans. Hum Immunol 2005; 66: 13–20
- Allan S. E., Crome S. Q., Crellin N. K., Passerini L., Steiner T. S., Bacchetta R., et al. Activation‐induced FOXP3 in human T effector cells does not suppress proliferation or cytokine production. Int Immunol 2007; 19: 345–54
- Wang J., Ioan‐Facsinay A., van der Voort E. I., Huizinga T. W., Toes R. E. Transient expression of FOXP3 in human activated nonregulatory CD4+ T cells. Eur J Immunol 2007; 37: 129–38
- Roncador G., Brown P. J., Maestre L., Hue S., Martinez‐Torrecuadrada J. L., Ling K. L., et al. Analysis of FOXP3 protein expression in human CD4+CD25+ regulatory T cells at the single‐cell level. Eur J Immunol 2005; 35: 1681–91
- Yagi H., Nomura T., Nakamura K., Yamazaki S., Kitawaki T., Hori S., et al. Crucial role of FOXP3 in the development and function of human CD25+CD4+ regulatory T cells. Int Immunol 2004; 16: 1643–56
- Jordan M. S., Boesteanu A., Reed A. J., Petrone A. L., Holenbeck A. E., Lerman M. A., et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self‐peptide. Nat Immunol 2001; 2: 301–6
- Chatenoud L., Bach J. F. Adaptive human regulatory T cells: myth or reality?. J Clin Invest 2006; 116: 2325–7
- Roncarolo M. G., Bacchetta R., Bordignon C., Narula S., Levings M. K. Type 1 T regulatory cells. Immunol Rev 2001; 182: 68–79
- Adorini L., Giarratana N., Penna G. Pharmacological induction of tolerogenic dendritic cells and regulatory T cells. Semin Immunol 2004; 16: 127–34
- Chen Y., Kuchroo V. K., Inobe J., Hafler D. A., Weiner H. L. Regulatory T cell clones induced by oral tolerance: suppression of autoimmune encephalomyelitis. Science 1994; 265: 1237–40
- Chen W., Jin W., Hardegen N., Lei K. J., Li L., Marinos N., et al. Conversion of peripheral CD4+CD25‐ naive T cells to CD4+CD25+ regulatory T cells by TGF‐beta induction of transcription factor Foxp3. J Exp Med 2003; 198: 1875–86
- Fantini M. C., Becker C., Monteleone G., Pallone F., Galle P. R., Neurath M. F. Cutting edge: TGF‐beta induces a regulatory phenotype in CD4+CD25‐ T cells through Foxp3 induction and down‐regulation of Smad7. J Immunol 2004; 172: 5149–53
- Delgado M., Chorny A., Gonzalez‐Rey E., Ganea D. Vasoactive intestinal peptide generates CD4+CD25+ regulatory T cells in vivo. J Leukoc Biol 2005; 78: 1327–38
- Fernandez‐Martin A., Gonzalez‐Rey E., Chorny A., Ganea D., Delgado M. Vasoactive intestinal peptide induces regulatory T cells during experimental autoimmune encephalomyelitis. Eur J Immunol 2006; 36: 318–26
- Baratelli F., Lin Y., Zhu L., Yang S. C., Heuze‐Vourc'h N., Zeng G., et al. Prostaglandin E2 induces FOXP3 gene expression and T regulatory cell function in human CD4+ T cells. J Immunol 2005; 175: 1483–90
- Jonuleit H., Schmitt E., Steinbrink K., Enk A. H. Dendritic cells as a tool to induce anergic and regulatory T cells. Trends Immunol 2001; 22: 394–400
- Shevach E. M. CD4+ CD25+ suppressor T cells: more questions than answers. Nat Rev Immunol 2002; 2: 389–400
- Thornton A. M., Shevach E. M. Suppressor effector function of CD4+CD25+ immunoregulatory T cells is antigen nonspecific. J Immunol 2000; 164: 183–90
- Tang Q., Adams J. Y., Tooley A. J., Bi M., Fife B. T., Serra P., et al. Visualizing regulatory T cell control of autoimmune responses in nonobese diabetic mice. Nat Immunol 2006; 7: 83–92
- Jonuleit H., Schmitt E., Kakirman H., Stassen M., Knop J., Enk A. H. Infectious tolerance: human CD25(+) regulatory T cells convey suppressor activity to conventional CD4(+) T helper cells. J Exp Med 2002; 196: 255–60
- Stassen M., Fondel S., Bopp T., Richter C., Muller C., Kubach J., et al. Human CD25+ regulatory T cells: two subsets defined by the integrins alpha 4 beta 7 or alpha 4 beta 1 confer distinct suppressive properties upon CD4+ T helper cells. Eur J Immunol 2004; 34: 1303–11
- Nishizuka Y., Sakakura T. Thymus and reproduction: sex‐linked dysgenesia of the gonad after neonatal thymectomy in mice. Science 1969; 166: 753–5
- Powrie F. Immune regulation in the intestine: a balancing act between effector and regulatory T cell responses. Ann N Y Acad Sci 2004; 1029: 132–41
- Annacker O., Asseman C., Read S., Powrie F. Interleukin‐10 in the regulation of T cell‐induced colitis. J Autoimmun 2003; 20: 277–9
- Uhlig H. H., Coombes J., Mottet C., Izcue A., Thompson C., Fanger A., et al. Characterization of Foxp3+CD4+CD25+ and IL‐10‐Secreting CD4+CD25+ T Cells during Cure of Colitis. J Immunol 2006; 177: 5852–60
- Kullberg M. C., Hay V., Cheever A. W., Mamura M., Sher A., Letterio J. J., et al. TGF‐beta1 production by CD4+ CD25+ regulatory T cells is not essential for suppression of intestinal inflammation. Eur J Immunol 2005; 35: 2886–95
- Maloy K. J., Salaun L., Cahill R., Dougan G., Saunders N. J., Powrie F. CD4+CD25+ T(R) cells suppress innate immune pathology through cytokine‐dependent mechanisms. J Exp Med 2003; 197: 111–9
- Fahlen L., Read S., Gorelik L., Hurst S. D., Coffman R. L., Flavell R. A., et al. T cells that cannot respond to TGF‐beta escape control by CD4(+)CD25(+) regulatory T cells. J Exp Med 2005; 201: 737–46
- Fantini M. C., Becker C., Tubbe I., Nikolaev A., Lehr H. A., Galle P., et al. Transforming growth factor beta induced FoxP3+ regulatory T cells suppress Th1 mediated experimental colitis. Gut 2006; 55: 671–80
- Makita S., Kanai T., Oshima S., Uraushihara K., Totsuka T., Sawada T., et al. CD4+CD25bright T cells in human intestinal lamina propria as regulatory cells. J Immunol 2004; 173: 3119–30
- Kelsen J., Agnholt J., Hoffmann H. J., Romer J. L., Hvas C. L., Dahlerup J. F. FoxP3(+)CD4(+)CD25(+) T cells with regulatory properties can be cultured from colonic mucosa of patients with Crohn's disease. Clin Exp Immunol 2005; 141: 549–57
- Zamvil S. S., Steinman L. The T lymphocyte in experimental allergic encephalomyelitis. Annu Rev Immunol 1990; 8: 579–621
- Zhang X., Koldzic D. N., Izikson L., Reddy J., Nazareno R. F., Sakaguchi S., et al. IL‐10 is involved in the suppression of experimental autoimmune encephalomyelitis by CD25+CD4+ regulatory T cells. Int Immunol 2004; 16: 249–56
- Tisch R., McDevitt H. Insulin‐dependent diabetes mellitus. Cell. 1996; 85: 291–7
- Delovitch T. L., Singh B. The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity 1997; 7: 727–38
- King C., Sarvetnick N. Organ‐specific autoimmunity. Curr Opin Immunol 1997; 9: 863–71
- Poulin M., Haskins K. Induction of diabetes in nonobese diabetic mice by Th2 T cell clones from a TCR transgenic mouse. J Immunol 2000; 164: 3072–8
- Salomon B., Lenschow D. J., Rhee L., Ashourian N., Singh B., Sharpe A., et al. B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity 2000; 12: 431–40
- Salomon B., Rhee L., Bour‐Jordan H., Hsin H., Montag A., Soliven B., et al. Development of spontaneous autoimmune peripheral polyneuropathy in B7‐2‐deficient NOD mice. J Exp Med 2001; 194: 677–84
- Masteller E. L., Warner M. R., Tang Q., Tarbell K. V., McDevitt H., Bluestone J. A. Expansion of functional endogenous antigen‐specific CD4+CD25+ regulatory T cells from nonobese diabetic mice. J Immunol 2005; 175: 3053–9
- Tarbell K. V., Yamazaki S., Olson K., Toy P., Steinman R. M. CD25+ CD4+ T cells, expanded with dendritic cells presenting a single autoantigenic peptide, suppress autoimmune diabetes. J Exp Med 2004; 199: 1467–77
- Every A. L., Kramer D. R., Mannering S. I., Lew A. M., Harrison L. C. Intranasal vaccination with proinsulin DNA induces regulatory CD4+ T cells that prevent experimental autoimmune diabetes. J Immunol 2006; 176: 4608–15
- Bresson D., Togher L., Rodrigo E., Chen Y., Bluestone J. A., Herold K. C., et al. Anti‐CD3 and nasal proinsulin combination therapy enhances remission from recent‐onset autoimmune diabetes by inducing Tregs. J Clin Invest 2006; 116: 1371–81
- Snell G. D., Higgins G. F. Alleles at the histocompatibility‐2 locus in the mouse as determined by tumor transplantation. Genetics 1951; 36: 306–10
- Snell G. D. The genetics of transplantation. J Natl Cancer Inst. 1953; 14: 691–700
- Billingham R. E., Brent L., Medawar P. B. Actively acquired tolerance of foreign cells. Nature 1953; 172: 603–6
- Owen R. D. Immunogenetic consequences of vascular anastomoses between bovine twins. Science 1945; 102: 400–01
- Lance E. M., Medawar P. B. Survival of skin heterografts under treatment with antilymphocytic serum. Lancet 1968; 1: 1174–6
- Gershon R. K., Kondo K. Infectious immunological tolerance. Immunology 1971; 21: 903–14
- Hall B. M., Jelbart M. E., Gurley K. E., Dorsch S. E. Specific unresponsiveness in rats with prolonged cardiac allograft survival after treatment with cyclosporine. Mediation of specific suppression by T helper/inducer cells. J Exp Med 1985; 162: 1683–94
- Qin S., Cobbold S. P., Pope H., Elliott J., Kioussis D., Davies J., et al. “Infectious” transplantation tolerance. Science 1993; 259: 974–7
- Waldmann H., Cobbold S. Regulating the immune response to transplants. A role for CD4+ regulatory cells?. Immunity 2001; 14: 399–406
- Yin D., Fathman C. G. CD4‐positive suppressor cells block allotransplant rejection. J Immunol 1995; 154: 6339–45
- Davies J. D., Martin G., Phillips J., Marshall S. E., Cobbold S. P., Waldmann H. T cell regulation in adult transplantation tolerance. J Immunol 1996; 157: 529–33
- Honey K., Cobbold S. P., Waldmann H. CD40 ligand blockade induces CD4+ T cell tolerance and linked suppression. J Immunol 1999; 163: 4805–10
- Arima T., Lehmann M., Flye M. W. Induction of donor specific transplantation tolerance to cardiac allografts following treatment with nondepleting (RIB 5/2) or depleting (OX‐38) anti‐CD4 mAb plus intrathymic or intravenous donor alloantigen. Transplantation 1997; 63: 284–92
- Zhai Y., Shen X. D., Lehmann M., Busuttil R., Volk H. D., Kupiec‐Weglinski J. W. T cell subsets and in vitro immune regulation in “infectious” transplantation tolerance. J Immunol 2001; 167: 4814–20
- Graca L., Cobbold S. P., Waldmann H. Identification of regulatory T cells in tolerated allografts. J Exp Med 2002; 195: 1641–6
- Taylor P. A., Lees C. J., Blazar B. R. The infusion of ex vivo activated and expanded CD4(+)CD25(+) immune regulatory cells inhibits graft‐versus‐host disease lethality. Blood 2002; 99: 3493–9
- Godfrey W. R., Spoden D. J., Ge Y. G., Baker S. R., Liu B., Levine B. L., et al. Cord blood CD4(+)CD25(+)‐derived T regulatory cell lines express FoxP3 protein and manifest potent suppressor function. Blood 2005; 105: 750–8
- Tafuri A., Alferink J., Moller P., Hammerling G. J., Arnold B. T cell awareness of paternal alloantigens during pregnancy. Science 1995; 270: 630–3
- Woodruff M. F. Transplantation immunity and the immunological problem of pregnancy. Proc R Soc Lond B Biol Sci 1958; 148: 68–75
- Zenclussen A. C., Gerlof K., Zenclussen M. L., Sollwedel A., Bertoja A. Z., Ritter T., et al. Abnormal T‐cell reactivity against paternal antigens in spontaneous abortion: adoptive transfer of pregnancy‐induced CD4+CD25+ T regulatory cells prevents fetal rejection in a murine abortion model. Am J Pathol 2005; 166: 811–22
- Saito S., Sasaki Y., Sakai M. CD4(+)CD25high regulatory T cells in human pregnancy. J Reprod Immunol 2005; 65: 111–20
- Sasaki Y., Sakai M., Miyazaki S., Higuma S., Shiozaki A., Saito S. Decidual and peripheral blood CD4+CD25+ regulatory T cells in early pregnancy subjects and spontaneous abortion cases. Mol Hum Reprod 2004; 10: 347–53
- Heikkinen J., Mottonen M., Alanen A., Lassila O. Phenotypic characterization of regulatory T cells in the human decidua. Clin Exp Immunol 2004; 136: 373–8
- Somerset D. A., Zheng Y., Kilby M. D., Sansom D. M., Drayson M. T. Normal human pregnancy is associated with an elevation in the immune suppressive CD25+ CD4+ regulatory T‐cell subset. Immunology 2004; 112: 38–43
- Onizuka S., Tawara I., Shimizu J., Sakaguchi S., Fujita T., Nakayama E. Tumor rejection by in vivo administration of anti‐CD25 (interleukin‐2 receptor alpha) monoclonal antibody. Cancer Res 1999; 59: 3128–33
- Sutmuller R. P., van Duivenvoorde L. M., van Elsas A., Schumacher T. N., Wildenberg M. E., Allison J. P., et al. Synergism of cytotoxic T lymphocyte‐associated antigen 4 blockade and depletion of CD25(+) regulatory T cells in antitumor therapy reveals alternative pathways for suppression of autoreactive cytotoxic T lymphocyte responses. J Exp Med 2001; 194: 823–32
- Peng L., Kjaergaard J., Plautz G. E., Awad M., Drazba J. A., Shu S., et al. Tumor‐induced L‐selectinhigh suppressor T cells mediate potent effector T cell blockade and cause failure of otherwise curative adoptive immunotherapy. J Immunol 2002; 169: 4811–21
- Gerlini G., Tun‐Kyi A., Dudli C., Burg G., Pimpinelli N., Nestle F. O. Metastatic melanoma secreted IL‐10 down‐regulates CD1 molecules on dendritic cells in metastatic tumor lesions. Am J Pathol 2004; 165: 1853–63
- Van Belle P., Rodeck U., Nuamah I., Halpern A. C., Elder D. E. Melanoma‐associated expression of transforming growth factor‐beta isoforms. Am J Pathol 1996; 148: 1887–94
- Curiel T. J., Coukos G., Zou L., Alvarez X., Cheng P., Mottram P., et al. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 2004; 10: 942–9
- Ko K., Yamazaki S., Nakamura K., Nishioka T., Hirota K., Yamaguchi T., et al. Treatment of advanced tumors with agonistic anti‐GITR mAb and its effects on tumor‐infiltrating Foxp3+CD25+CD4+ regulatory T cells. J Exp Med 2005; 202: 885–91
- Ghiringhelli F., Larmonier N., Schmitt E., Parcellier A., Cathelin D., Garrido C., et al. CD4+CD25+ regulatory T cells suppress tumor immunity but are sensitive to cyclophosphamide which allows immunotherapy of established tumors to be curative. Eur J Immunol 2004; 34: 336–44
- Ercolini A. M., Ladle B. H., Manning E. A., Pfannenstiel L. W., Armstrong T. D., Machiels J. P., et al. Recruitment of latent pools of high‐avidity CD8(+) T cells to the antitumor immune response. J Exp Med 2005; 201: 1591–602
- Fehleisen F. Über die Züchtung der Erysipelkokken auf künstlichem Nährboden und die Übertragbarkeit auf den Menschen. Dtsch Med Wochenschau 1882; 8: 553
- Hobohm U. Fever and cancer in perspective. Cancer Immunol Immunother 2001; 50: 391–6
- Busch W. Aus der Sitzung der medizinischen Sektion vom 13 November 1867. Berl Klin Wochenschr 1868; 5: 137
- Coley W. B. A preliminary note on the treatment of inoperable sarcoma by the toxic product of erysipelas. Postgraduate 1893; 8: 278
- Goldszmid R. S., Idoyaga J., Bravo A. I., Steinman R., Mordoh J., Wainstok R. Dendritic cells charged with apoptotic tumor cells induce long‐lived protective CD4+ and CD8+ T cell immunity against B16 melanoma. J Immunol 2003; 171: 5940–7
- Rechtsteiner G., Warger T., Osterloh P., Schild H., Radsak M. P. Cutting edge: priming of CTL by transcutaneous peptide immunization with imiquimod. J Immunol 2005; 174: 2476–80
- Warger T., Osterloh P., Rechtsteiner G., Fassbender M., Heib V., Schmid B., et al. Synergistic activation of dendritic cells by combined Toll‐like receptor ligation induces superior CTL responses in vivo. Blood 2006; 108: 544–50
- Yang Y., Huang C. T., Huang X., Pardoll D. M. Persistent Toll‐like receptor signals are required for reversal of regulatory T cell‐mediated CD8 tolerance. Nat Immunol 2004; 5: 508–15
- Rouse B. T., Suvas S. Regulatory cells and infectious agents: detentes cordiale and contraire. J Immunol 2004; 173: 2211–5
- Suvas S., Azkur A. K., Kim B. S., Kumaraguru U., Rouse B. T. CD4+CD25+ regulatory T cells control the severity of viral immunoinflammatory lesions. J Immunol 2004; 172: 4123–32
- Powrie F., Read S., Mottet C., Uhlig H., Maloy K. Control of immune pathology by regulatory T cells. Novartis Found Symp 2003; 252: 92–8
- Mottet C., Uhlig H. H., Powrie F. Cutting edge: cure of colitis by CD4+CD25+ regulatory T cells. J Immunol 2003; 170: 3939–43
- Belkaid Y., Piccirillo C. A., Mendez S., Shevach E. M., Sacks D. L. CD4+CD25+ regulatory T cells control Leishmania major persistence and immunity. Nature 2002; 420: 502–7
- Yurchenko E., Tritt M., Hay V., Shevach E. M., Belkaid Y., Piccirillo C. A. CCR5‐dependent homing of naturally occurring CD4+ regulatory T cells to sites of Leishmania major infection favors pathogen persistence. J Exp Med 2006; 203: 2451–60
- Iwasaki A., Medzhitov R. Toll‐like receptor control of the adaptive immune responses. Nat Immunol 2004; 5: 987–95
- Pasare C., Medzhitov R. Toll pathway‐dependent blockade of CD4+CD25+ T cell‐mediated suppression by dendritic cells. Science 2003; 299: 1033–6
- Pasare C., Medzhitov R. Toll‐dependent control mechanisms of CD4 T cell activation. Immunity 2004; 21: 733–41
- Thornton A. M., Donovan E. E., Piccirillo C. A., Shevach E. M. Cutting edge: IL‐2 is critically required for the in vitro activation of CD4+CD25+ T cell suppressor function. J Immunol 2004; 172: 6519–23
- Sutmuller R. P., den Brok M. H., Kramer M., Bennink E. J., Toonen L. W., Kullberg B. J., et al. Toll‐like receptor 2 controls expansion and function of regulatory T cells. J Clin Invest 2006; 116: 485–94
- Stassen M., Jonuleit H., Muller C., Klein M., Richter C., Bopp T., et al. Differential regulatory capacity of CD25+ T regulatory cells and preactivated CD25+ T regulatory cells on development, functional activation, and proliferation of Th2 cells. J Immunol 2004; 267–74, 173
- Zhao D. M., Thornton A. M., Dipaolo R. J., Shevach E. M. Activated CD4+CD25+ T cells selectively kill B lymphocytes. Blood 2006; 107: 3925–32
- Woo E. Y., Chu C. S., Goletz T. J., Schlienger K., Yeh H., Coukos G., et al. Regulatory CD4(+)CD25(+) T cells in tumors from patients with early‐stage non‐small cell lung cancer and late‐stage ovarian cancer. Cancer Res 2001; 61: 4766–72
- Wolf A. M., Wolf D., Steurer M., Gastl G., Gunsilius E., Grubeck‐Loebenstein B. Increase of regulatory T cells in the peripheral blood of cancer patients. Clin Cancer Res 2003; 9: 606–12
- Antony P. A., Piccirillo C. A., Akpinarli A., Finkelstein S. E., Speiss P. J., Surman D. R., et al. CD8+ T cell immunity against a tumor/self‐antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol 2005; 174: 2591–601
- Gray C. P., Arosio P., Hersey P. Association of increased levels of heavy‐chain ferritin with increased CD4+ CD25+ regulatory T‐cell levels in patients with melanoma. Clin Cancer Res 2003; 9: 2551–9
- Enk A. H., Jonuleit H., Saloga J., Knop J. Dendritic cells as mediators of tumor‐induced tolerance in metastatic melanoma. Int J Cancer 1997; 73: 309–16
- Rosenberg S. A. Shedding light on immunotherapy for cancer. N Engl J Med 2004; 350: 1461–3
- O'Neill D. W., Adams S., Bhardwaj N. Manipulating dendritic cell biology for the active immunotherapy of cancer. Blood 2004; 104: 2235–46
- Pasare C., Medzhitov R. Toll‐like receptors: balancing host resistance with immune tolerance. Curr Opin Immunol 2003; 15: 677–82
- Viglietta V., Baecher‐Allan C., Weiner H. L., Hafler D. A. Loss of functional suppression by CD4+CD25+ regulatory T cells in patients with multiple sclerosis. J Exp Med 2004; 199: 971–9
- Cao D., Malmstrom V., Baecher‐Allan C., Hafler D., Klareskog L., Trollmo C. Isolation and functional characterization of regulatory CD25brightCD4+ T cells from the target organ of patients with rheumatoid arthritis. Eur J Immunol 2003; 33: 215–23
- Lindley S., Dayan C. M., Bishop A., Roep B. O., Peakman M., Tree T. I. Defective suppressor function in CD4(+)CD25(+) T‐cells from patients with type 1 diabetes. Diabetes 2005; 54: 92–9
- Sugiyama H., Gyulai R., Toichi E., Garaczi E., Shimada S., Stevens S. R., et al. Dysfunctional blood and target tissue CD4+CD25high regulatory T cells in psoriasis: mechanism underlying unrestrained pathogenic effector T cell proliferation. J Immunol 2005; 174: 164–73
- Ehrenstein M. R., Evans J. G., Singh A., Moore S., Warnes G., Isenberg D. A., et al. Compromised function of regulatory T cells in rheumatoid arthritis and reversal by anti‐TNFalpha therapy. J Exp Med 2004; 200: 277–85
- Xystrakis E., Kusumakar S., Boswell S., Peek E., Urry Z., Richards D. F., et al. Reversing the defective induction of IL‐10‐secreting regulatory T cells in glucocorticoid‐resistant asthma patients. J Clin Invest 2006; 116: 146–55
- Karagiannidis C., Akdis M., Holopainen P., Woolley N. J., Hense G., Ruckert B., et al. Glucocorticoids upregulate FOXP3 expression and regulatory T cells in asthma. J Allergy Clin Immunol 2004; 114: 1425–33
- Keymeulen B., Vandemeulebroucke E., Ziegler A. G., Mathieu C., Kaufman L., Hale G., et al. Insulin needs after CD3‐antibody therapy in new‐onset type 1 diabetes. N Engl J Med 2005; 352: 2598–608
- Herold K. C., Gitelman S. E., Masharani U., Hagopian W., Bisikirska B., Donaldson D., et al. A single course of anti‐CD3 monoclonal antibody hOKT3gamma1(Ala‐Ala) results in improvement in C‐peptide responses and clinical parameters for at least 2 years after onset of type 1 diabetes. Diabetes 2005; 54: 1763–9
- Belghith M., Bluestone J. A., Barriot S., Megret J., Bach J. F., Chatenoud L. TGF‐beta‐dependent mechanisms mediate restoration of self‐tolerance induced by antibodies to CD3 in overt autoimmune diabetes. Nat Med 2003; 9: 1202–8
- Akdis M., Verhagen J., Taylor A., Karamloo F., Karagiannidis C., Crameri R., et al. Immune responses in healthy and allergic individuals are characterized by a fine balance between allergen‐specific T regulatory 1 and T helper 2 cells. J Exp Med 2004; 199: 1567–75
- Bellinghausen I., Knop J., Saloga J. The role of interleukin 10 in the regulation of allergic immune responses. Int Arch Allergy Immunol 2001; 126: 97–101