Abstract
Endothelium‐dependent relaxations are attributed to the release of various factors, such as nitric oxide, carbon monoxide, reactive oxygen species, adenosine, peptides and arachidonic acid metabolites derived from the cyclooxygenases, lipoxygenases, and cytochrome P450 monooxygenases pathways. The hyperpolarization of the smooth muscle cell can contribute to or be an integral part of the mechanisms underlying the relaxations elicited by virtually all these endothelial mediators. These endothelium‐derived factors can activate different families of K+ channels of the vascular smooth muscle. Other events associated with the hyperpolarization of both the endothelial and the vascular smooth muscle cells (endothelium‐derived hyperpolarizing factor (EDHF)‐mediated responses) contribute also to endothelium‐dependent relaxations. These responses involve an increase in the intracellular Ca2+ concentration of the endothelial cells followed by the opening of Ca2+‐activated K+ channels of small and intermediate conductance and the subsequent hyperpolarization of these cells. Then, the endothelium‐dependent hyperpolarization of the underlying smooth muscle cells can be evoked by direct electrical coupling through myoendothelial junctions and/or the accumulation of K+ ions in the intercellular space between the two cell types. These various mechanisms are not necessarily mutually exclusive and, depending on the vascular bed and the experimental conditions, can occur simultaneously or sequentially, or also may act synergistically.
| Abbreviations | ||
| 11,12,15‐THETA | = | 11,12,15‐trihydroxyeicosatrienoic acid |
| 12‐S‐HETE | = | 12‐(S)‐hydroxyeicosatetraenoic acid |
| 20‐HETE | = | 20‐hydroxyeicosatetraenoic acid |
| ANP | = | atrial natriuretic peptide |
| BKCa | = | large conductance calcium‐activated potassium channel |
| BNP | = | brain natriuretic peptide |
| CaV | = | voltage‐dependent calcium channel |
| CGRP | = | calcitonin gene‐related peptide |
| GIRK | = | G protein‐regulated inward‐rectifier potassium channel |
| CNP | = | C‐type natriuretic peptide |
| Cx | = | connexin |
| EDHF | = | endothelium‐derived hyperpolarizing factor |
| EET | = | epoxyeicosatrienoic acid |
| HO | = | heme‐oxygenase |
| IKCa | = | intermediate conductance calcium‐activated potassium channel |
| IP | = | receptor prostacyclin receptor |
| K2P | = | two‐pore domain potassium channel |
| KATP | = | ATP‐sensitive potassium channel |
| KCa | = | calcium‐activated potassium channel |
| KIR | = | inwardly rectifying potassium channel |
| KV | = | voltage‐dependent potassium channel |
| L‐NAME | = | L‐nitro‐arginine‐methyl‐ester |
| NPR‐C | = | natriuretic peptide receptor C subtype |
| PKA | = | protein kinase A |
| PKG | = | protein kinase G |
| SHR | = | spontaneously hypertensive rat |
| SKCa | = | small conductance calcium‐activated potassium channel |
| SERCA | = | sarcoplasmic reticulum Ca2+ ATPase |
| STOC | = | spontaneous transient outward current |
| TRPV4 | = | vanilloid transient receptor potential channel |
| VIP | = | vasoactive intestinal peptide |
Introduction
The endothelial cells, being at the interface of the blood and the vascular wall, are involved in many different physiological functions, including metabolism, angiogenesis, hemostasis, inflammation, synthesis and degradation of the extracellular matrix, as well as the regulation of vascular permeability and tone. The endothelium maintains the balance between inhibition and promotion of the proliferation and migration of smooth muscle cells, between prevention and stimulation of the adhesion and aggregation of the platelets, between thrombogenesis and fibrinolysis, as well as between vasodilatation and vasoconstriction. Upsetting this tightly regulated balance leads to endothelial dysfunction Citation1,2.
Endothelial cells control the tone of the underlying vascular smooth muscle cells by releasing various relaxing and contracting factors Citation3. Furchgott and Zawadzki Citation4 were the first to show that the presence of the endothelial lining is required to observe the relaxation of an isolated artery in response to acetylcholine. The term ‘endothelium‐dependent relaxations’ was then coined although other endothelium‐derived vasoactive substances had been identified previously (e.g. prostacyclin (PGI2), adenosine). Nevertheless, it was only after this seminal work that the essential role of the endothelial cells in controlling the tone of the underlying vascular smooth muscle was fully understood, and usually attributed to the production of nitric oxide (NO) by the endothelial NO synthase (NOS‐3) Citation1,Citation4. However, in numerous blood vessels from different species, including the human, endothelium‐dependent relaxations are still observed in the presence of inhibitors of cyclooxygenases and NO synthases. These relaxations are generally associated with the hyperpolarization of the vascular smooth muscle cells and were first attributed to a noncharacterized endothelial factor termed EDHF for endothelium‐derived hyperpolarizing factor (). The acronym ‘EDHF’ turned out to be confusing because it implies that a single diffusible substance, different from nitric oxide (NO) and PGI2, mediates this type of endothelium‐dependent relaxation. In fact, numerous endothelium‐derived factors, including NO and PGI2 themselves, but also reactive oxygen species, carbon monoxide, adenosine, peptides, and various metabolites of arachidonic acid can hyperpolarize the underlying smooth muscle. Additionally, another pathway, which does not involve the release of a factor but is associated with the hyperpolarization of both the endothelial and the vascular smooth muscle cells, contributes also to certain endothelium‐dependent relaxations Citation1,Citation5.
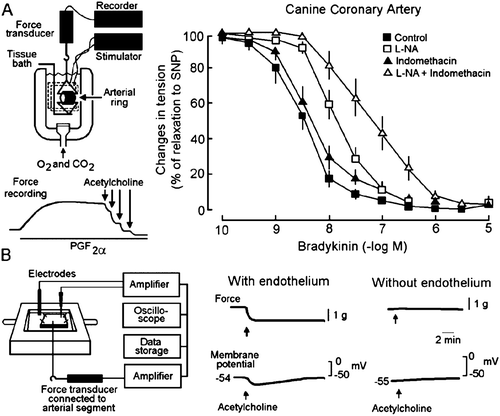
Figure 1. Endothelium‐dependent relaxation and endothelium‐dependent hyperpolarization. A: Left: The isometric tension developed by an arterial ring is studied in an organ chamber filled with thermostated and oxygenated physiological salt solution. The inset shows recording traces of an experiment reproducing the seminal observation first made by Furchgott and Zawadzki Citation4. An isolated ring of canine coronary artery with endothelium is contracted with prostaglandin F2α (PGF2α). The addition of acetylcholine produces a concentration‐dependent and endothelium‐dependent relaxation. Right: Bradykinin produces a concentration‐dependent and endothelium‐dependent relaxation in canine coronary arterial rings. The cyclooxygenase inhibitor, indomethacin, does not affect the relaxation to bradykinin while the NO synthase inhibitor L‐nitro‐arginine (L‐NA) produces a significant shift to the right of the concentration‐response curves, indicating a contribution of NO in the overall relaxation mechanism. The combination of the two inhibitors produces a further shift, indicating that prostacyclin contributes to the mechanism of bradykinin‐induced endothelium‐dependent relaxation when the NO synthase is inhibited. However, even in the presence of the two inhibitors, bradykinin still evokes the complete relaxation of the isolated arteries (modified from Citation210). SNP = sodium nitroprusside. B: Left: Changes in tension and in membrane potential are recorded in an isolated canine coronary artery with an intracellular microelectrode. Right: Acetylcholine produces a relaxation and a hyperpolarization of the vascular smooth muscle only when the endothelial cells are present (modified from Citation211).
The hyperpolarization of the smooth muscle cells is a powerful way to produce relaxation. It decreases not only Ca2+ influx by reducing the open probability of voltage‐dependent Ca2+ channel (CaV) but also the release of Ca2+ from intracellular stores by decreasing the turnover of intracellular phosphatidylinositol and the CaV‐dependent activation of the sarcoplasmic reticulum Citation1,Citation6,7.
Key messages
The hyperpolarization of vascular smooth muscle cells is a powerful way to produce a relaxation, and numerous endothelium‐derived factors hyperpolarize the underlying smooth muscle cells.
Another pathway, which does not involve the release of a factor (endothelium‐derived hyperpolarizing factor (EDHF)‐mediated response), is associated with the hyperpolarization of both the endothelial and the vascular smooth muscle cells and contributes also to endothelium‐dependent relaxations.
The gene deletion of each key molecular component involved in these EDHF‐mediated responses is associated with alterations in arterial blood pressure.
In humans, these responses occur in vivo and contribute to the regulation of basal blood flow and to the endothelium‐dependent relaxations in response to flow and certain neurohumoral mediators.
Arachidonic acid metabolites
Cyclooxygenases
PGI2 is the principal metabolite of arachidonic acid produced by cyclooxygenase in the endothelial cells Citation8. This prostaglandin, a preferential agonist of prostacyclin (IP) receptors, is a potent antithrombotic and antiplatelet agent as well as a vasodilator substance. Although several types of G proteins are likely to be coupled to the IP receptor, in vascular smooth muscle cells the Gs‐adenylyl‐cyclase‐cyclic adenosine monophosphate (cAMP)‐PKA pathway is a preponderant signaling system responsible for vascular relaxation Citation9. The relaxation to PGI2, or its synthetic analogues, is often associated with the concomitant hyperpolarization of the smooth muscle cells and can involve the opening of ATP‐sensitive (KATP) Citation10,11, voltage‐dependent (KV) Citation12, inwardly rectifying (KIR) Citation13, large conductance Ca2+‐activated K+ channels (BKCa) Citation14,15 and/or two‐pore domain (K2P) K+ channel family Citation16. The activation of K+ channels following IP receptor stimulation relies on cAMP‐dependent and ‐independent pathways. The latter pathway could be associated with the direct activation of the K+ channel by Gs protein Citation17.
Therefore, in numerous vascular beds, PGI2 can act as an endothelium‐derived hyperpolarizing substance Citation1,Citation11 (). However, inhibitors of cyclooxygenases abolish the basal and stimulated generation of PGI2, indicating that its release cannot account for the responses observed in the presence of the combination of inhibitors of NO synthase and cyclooxygenase.
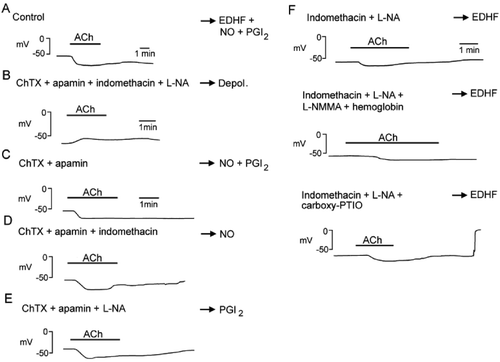
Figure 2. Endothelium‐dependent hyperpolarizations in the guinea‐pig carotid artery. A: Acetylcholine (ACh) induces an endothelium‐dependent hyperpolarization. B: In the presence of charybdotoxin (ChTX) plus apamin plus indomethacin plus L‐nitro‐arginine (L‐NA), acetylcholine no longer produces an endothelium‐dependent hyperpolarization but a small depolarization. Depol. = depolarization. C: In the presence of charybdotoxin (ChTX) plus apamin, acetylcholine produces an endothelium‐dependent hyperpolarization attributable to the release of NO and PGI2. PGI2 = prostacyclin. D: In the presence of charybdotoxin (ChTX) plus apamin plus indomethacin, acetylcholine produces an endothelium‐dependent hyperpolarization attributable to the release of NO. E: In the presence of charybdotoxin (ChTX) plus apamin plus L‐nitro‐arginine, acetylcholine produces an endothelium‐dependent hyperpolarization attributable to the release of PGI2. F: In the presence of indomethacin plus L‐nitro‐arginine (L‐NA), acetylcholine produces an EDHF‐mediated response. This response is not affected by the subsequent addition of a second NO synthase inhibitor, L‐monomethyl‐arginine (L‐NMMA) plus a NO scavenger, hemoglobin (middle trace), or another NO scavenger carboxy‐PTIO (lower trace), indicating that residual and stored NO are not involved.
Cytochrome P450
Epoxyeicosatrienoic acids (EETs), derived from cytochrome P450 2C or 2J epoxygenases, play an important role in endothelium‐dependent hyperpolarizations and relaxations in various blood vessels. In many arteries, inhibitors of cytochrome P450 monooxygenases or antisense oligonucleotides directed against cytochrome P450‐2C8‐9 inhibit endothelium‐dependent vasodilator responses that persist in the presence of inhibitors of NO synthases and cyclooxygenases Citation18,19. EETs can be involved in non‐NO‐, non‐PGI2‐mediated responses as a factor released by the endothelial cells or without being a diffusible factor per se, for instance by acting as essential endothelial intracellular second messengers Citation18. However, bradykinin, pulsatile stretch, and shear stress can release EETs, which diffuse toward and then hyperpolarize the vascular smooth muscle cells by activating BKCaCitation19–24. The mechanism by which EETs activate BKCa in vascular smooth muscle cells is not completely understood. They do not directly activate these channels as shown for other fatty acids Citation25. EETs may interact with an intracellular ‘receptor’ to activate adenosine diphosphate (ADP)‐ribosyltransferase. This enzyme catalyzes the ADP ribosylation of stimulatory G‐protein Gsα promoting the phosphorylation and the activation of the Slo1 α subunit of BKCaCitation26,27. EETs can also activate BKCa in much more indirect manners. Indeed, 11,12‐EET can increase the activity of heme‐oxygenase (HO) and the subsequent generation of carbon monoxide (CO) activates BKCaCitation28. Furthermore, EETs can promote Ca2+ entry in smooth muscle cells by activating cationic channels such as the vanilloid transient receptor potential channel (TRPV4). The Ca2+ influx through TRPV4 preferentially stimulates ryanodine receptors located on the sarcoplasmic reticulum increasing the frequency of unitary Ca2+ release events (Ca2+ sparks). The EETs‐induced Ca2+ sparks activate nearby sarcolemmal BKCa and increase the frequency of spontaneous transient outward currents (STOCs). This EETs‐dependent activation of the Ca2+‐signaling complex (TRPV4‐ryanodine receptors‐BKCa) hyperpolarizes and relaxes vascular smooth muscle cells Citation29.
Therefore, in some vascular beds, especially but not exclusively in coronary arteries, and following appropriate stimulation, the endothelial cells can release EETs, which act as diffusible endothelium‐derived hyperpolarizing substances and contribute to relaxations resistant to inhibitors of NO synthases and cyclooxygenases. Additionally, cytochrome P450 metabolites play an important role in the regulation of kidney the circulation and contribute to the long‐term regulation of blood pressure and sodium homeostasis. EETs not only regulate regional blood flow, but possess anti‐inflammatory actions, improve insulin sensitivity and lipid metabolism that could protect the kidney vasculature from injury during renal and cardiovascular diseases and prevent/delay metabolic syndrome and atherogenesis Citation30,31.
However, the effects of EETs are specific for certain vascular beds and in many blood vessels EETs evoke either minor relaxations (or hyperpolarization) and do not contribute to endothelium‐dependent relaxations Citation1,Citation32,33.
Lipoxygenase
The lipoxygenase metabolites 12‐(S)‐hydroxyeicosatetraenoic acid (12‐S‐HETE) and 11,12,15‐trihydroxyeicosatrienoic acid (11,12,15‐THETA) can be released by the endothelial cells and evoke relaxation of the vascular smooth muscle by activating KCaCitation34,Citation36. However, the involvement of lipoxygenase derivatives in endothelium‐dependent relaxations appears restricted to a limited number of blood vessels Citation1,Citation37.
Endocannabinoids
In isolated blood vessels or in anaesthetized animals, endogeneous and exogeneous cannabinoids usually have vasodilator properties and are likely to play a role in the regulation of the cardiovascular system Citation38. However, the suggestion that anandamide could be an endothelium‐derived vasoactive factors released by neurohumoral mediators has not been substantiated Citation1,Citation39.
Gaseous mediators
Gaseous mediators are not involved like other neurohumoral mediators in classic ligand‐membrane‐bound receptor interactions. They readily cross lipid membranes and therefore diffuse homogeneously, in a nonpolarized manner from their production site. Nitric oxide can be considered as the first member of this new family of neurohumoral substances. Two other low‐molecular‐weight compounds can be included in this family: carbon monoxide and hydrogen sulfide Citation40.
Nitric oxide (NO)
NO is a potent vasodilator and a powerful inhibitor of platelet adhesion and aggregation. The relaxations elicited by NO are generally associated with the stimulation of the cytosolic soluble guanylyl cyclase and the subsequent activation of cyclic guanosine monophosphate (cGMP)‐dependent protein kinase (PKG). However, the mechanisms underlying the relaxations caused by NO also involve cyclic GMP‐independent mechanisms such as the stimulation of the uptake of cytosolic Ca2+ via the sarcoplasmic reticulum Ca2+ ATPase (SERCA). The combined decrease in the Ca2+ sensitivity of the contractile proteins and the lowering of intracellular free Ca2+ explain the powerful vasodilatory properties of NO Citation41,42. NO regulates the activity of various ion channels and, depending on the vascular beds, the hyperpolarizing effects of NO on vascular smooth muscle cells can substantially contribute to their relaxation. NO can activate BKCaCitation43,44 via a PKG‐dependent phosphorylation of the channel Citation45 or by a direct, cyclic‐GMP‐independent manner Citation46,47. NO can also activate KVCitation12,Citation48, KATPCitation49,50 and/or KIRCitation51. Beyond K+ conductances, NO also interacts with other ionic channels, such as chloride and cationic channels, and can therefore further influence the membrane potential of the smooth muscle cells Citation1.
In contrast to inhibitors of cyclooxygenase, inhibitors of NO synthase do not necessarily fully inhibit the production of NO. Thus, in their presence, residual NO can be produced by the endothelial cells and contributes to the relaxation and/or hyperpolarization of the underlying vascular smooth muscle Citation52,53. Furthermore, NO can also be stored and released independently of the activation of NO synthase. Two such stores have been described: the photosensitive store Citation54,55 and the adventitial dinitrosyl nonheme iron complexes and S‐nitrosated proteins Citation56,57.
Therefore, it is obvious that the role of NO as an endothelium‐derived hyperpolarizing substance has certainly been underestimated assuming that the presence of an inhibitor of NO synthases rules out its contribution. A non‐NO‐, non‐PGI2‐mediated response should be reported not only when a relaxation and/or hyperpolarization is recorded in the combined presence of inhibitors of cyclooxygenases and NO synthases, but when this response is still observed with the additional presence of a NO scavenger Citation58 ().
Carbon monoxide (CO)
The predominant biological source of CO is from the heme degradation by heme‐oxygenase (HO), which generates biliverdin, free ferrous iron, and carbon monoxide. Three isoforms of HO have been described HO‐1 (inducible isoform), HO‐2 (constitutive isoform), and HO‐3, nearly devoid of catalytic activity Citation59. Both HO‐1 and HO‐2 are expressed in vascular smooth muscle and endothelial cells Citation40,Citation60. In many physiopathological situations the HO‐CO pathway compensates for the decrease NO bioavailability Citation59.
CO is a potent vasodilator in most, but not all, vascular beds. The mechanisms of CO‐induced vasodilatation involve the stimulation of soluble guanylate cyclase, the inhibition of cytochrome P450, and/or the activation of K+ channels Citation54,Citation59. CO and CO‐donors activate smooth muscle BKCa, KATPCitation61, and/or KV Citation62. CO induces the cGMP‐dependent activation of BKCa but also directly increases the open probability of this potassium channel Citation1,Citation59. CO augments the apparent Ca2+ sensitivity of the α subunit of BKCa, enhancing the probability of eliciting STOCs in response to Ca2+ sparks Citation63. Additionally, CO, like NO, can also indirectly activate BKCa by inhibiting the cytochrome P450‐dependent synthesis of 20‐HETE, the endogenous vasoconstrictor and tonic inhibitor of BKCaCitation64. However, CO is also a tonic inhibitor of NOS by binding to its prosthetic heme and can contribute to endothelial dysfunction Citation65.
An endothelial production of CO, contributing to endothelium‐dependent relaxations in response to neurohumoral substances, has been demonstrated only in a limited number of arteries Citation1,Citation66,67.
Hydrogen sulfide (H2S)
Two main enzymes are responsible for the production of hydrogen sulfide (H2S), cystathionine β‐synthase and cystathionine γ‐lyase and both use L‐cysteine as substrate. The physiological cardiovascular effects of H2S are linked to the latter enzyme, which in the vascular wall is expressed mostly in smooth muscle but is virtually absent in endothelial cells Citation40. Therefore, H2S does not qualify as an endothelium‐derived mediator.
Reactive oxygen species
Hydrogen peroxide (H2O2)
H2O2, depending on the blood vessel, the presence of the endothelium, the experimental conditions, or the concentrations studied, possesses dilator or constrictor properties. The endothelial production of hydrogen peroxide (H2O2) can be involved in endothelium‐dependent relaxations in response to agonists and flow Citation68,69 or compensate for the decreased production of NO Citation70. In some arteries, endothelium‐derived H2O2 evokes the hyperpolarization and relaxation of smooth muscle cells Citation71–73, and in vivo this reactive oxygen species could play an important role in coronary autoregulation Citation74 and a cardioprotective role during ischemia‐reperfusion injury Citation75.
The endothelium‐independent relaxations of arterial myocytes to H2O2 involve multiple pathways, direct effects such as the activation of guanylyl cyclase and/or K+ channels, and indirect effects via the metabolism of arachidonic acid either through cyclooxygenases or lipoxygenases Citation76. H2O2 produces the hyperpolarization of the vascular smooth muscle cells by activating BKCaCitation77,78, KATPCitation79, KVCitation80, and/or KIR channels Citation81. The activation of smooth muscle KV by H2O2 involves redox‐sensitive elements, thiol groups, on the channel itself or on associated regulatory proteins Citation80. The activation of BKCa by H2O2 is evoked by a direct action on the channel as well as by a soluble guanylyl cyclase‐dependent effect Citation77. However, H2O2 can also be a potent inhibitor of BKCa by altering two distinct cysteines situated on its α subunit Citation82.
Taken in conjunction, the available evidence suggests that H2O2 has multiple effects on the vascular cells. Nevertheless, as this reactive oxygen species is produced by the endothelial cells and in some arteries evokes hyperpolarization and relaxation of smooth muscle cells, H2O2 can be an endothelium‐derived hyperpolarizing mediator Citation73. However, H2O2 does not always hyperpolarize vascular smooth muscle cells Citation83, and its relaxing effect is not necessarily associated with hyperpolarization of the vascular smooth muscle cells or/and the opening of K+ channels Citation84. Furthermore, in many arteries EDHF‐mediated responses cannot be attributed to the generation of H2O2Citation1,Citation76.
Other reactive oxygen species
Hydroxyl radicals are endothelium‐derived relaxing factors in mouse cerebral arterioles Citation85. In vascular smooth muscle cells, superoxide anions may activate KCa in a cGMP‐independent manner Citation79, whereas peroxynitrite can in some arteries activate KATP and/or KCa, possibly by a cGMP‐dependent mechanism Citation86.
Vasoactive peptides
Endothelial cells can theoretically synthesize numerous vasoactive peptides. Among them C‐type natriuretic peptide (CNP), vasoactive intestinal peptide (VIP), calcitonin gene‐related peptide (CGRP), and adrenomedullin cause relaxation of vascular smooth muscle cells.
CNP
The natriuretic peptide family includes four distinct peptides, atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), CNP and urodilatin which are involved in the control of body fluid homeostasis and blood pressure. In contrast to ANP and BNP that are synthesized predominantly in the heart, CNP is produced in the central nervous system but has also been detected in peripheral endothelial cells and to a lesser extent in vascular smooth muscle cells Citation87,88. CNP evokes relaxations and hyperpolarizations of arterial and venous smooth muscle cells Citation89,90. This natriuretic peptide induces the accumulation of cGMP and opens BKCaCitation91–93.
Additionally, in the rat mesenteric artery, endothelium‐derived CNP could activate natriuretic peptide receptor (NPR)‐C subtype present in smooth muscle. Hyperpolarization of the smooth muscle cell would be evoked via the cyclic‐GMP‐independent activation of a G protein‐regulated inward‐rectifier K+ channel (GIRK) Citation94,95. This latter hypothesis, based on pharmacological evidence, suggesting that CNP is an endothelium‐derived hyperpolarizing substance, should be examined with caution and awaits confirmation with experiments associating electrophysiological and molecular biology techniques Citation53,Citation96,97. In the isolated guinea‐pig carotid artery, endothelium‐dependent hyperpolarizations do not rely on the release of CNP Citation98.
Other peptides
Adrenomedullin, CGRP, VIP and pituitary adenylyl cyclase‐activating polypeptide are extremely potent vasodilator peptides, and the mechanism underlying the relaxation often involves the hyperpolarization of the vascular smooth muscle cells. However, there is very little evidence so far that these peptides can be released acutely by endothelial cells. Since the three latter are well established neurotransmitters, they could be considered as ‘nerve‐derived hyperpolarizing factors’ Citation1.
Adenosine
Adenosine, an endogenous vasodilator released from contracting skeletal muscle and myocardium, contributes to exercise hyperemia and is an important regulator of coronary blood flow especially during ischemia Citation99,100. Endothelial cells can also release adenine nucleotides and adenosine, but their contribution appears modest when compared to that of cardiac or skeletal muscle cells. Endothelial cells, in response to various stimuli including hypoxia and increases in shear stress, release mainly adenosine triphosphate (ATP), but this nucleotide is rapidly transformed into adenosine by ectonucleotidases. However, in most blood vessels there is little evidence that adenosine plays a major role as an endothelium‐derived relaxing or hyperpolarizing factor Citation1.
Endothelium‐dependent hyperpolarizations (EDHF‐mediated responses)
EDHF‐mediated responses are endothelium‐dependent relaxations resistant to inhibitors of NO synthases and cyclooxygenases, which do not involve one of the mediators mentioned above (residual or stored NO, EETs, CNP, H2O2, and so on). These relaxations are produced by agonists that stimulate G protein‐coupled receptors, by substances that increase the endothelial intracellular Ca2+ concentration and by physical stimuli such as shear stress Citation1,Citation58.
Involvement of potassium channels
EDHF‐mediated responses are insensitive to specific blockers of KATP and BKCa. In most blood vessels, they involve the activation of small conductance KCa (SKCa), blocked by apamin, scyllatoxin, or UCL 1684, and/or intermediate conductance KCa (IKCa), blocked by charybdotoxin or TRAM‐34 Citation33,Citation50,Citation101,102 (). In freshly isolated vascular smooth muscle cells from various species, IKCa and SKCa are not or very poorly expressed whereas functional BKCa are constitutively present Citation103,104. In contrast, both SKCa (and especially the SK3 α subunit) and IKCa (IK1) are expressed in freshly isolated endothelial cells from various arteries. These channels are activated by substances that elicit EDHF‐mediated responses, and their activation produces the hyperpolarization of the endothelial cells Citation105–109. Thus, the two K+ channels involved in EDHF‐mediated responses, SKCa and IKCa, are located on the plasma membrane of the endothelial cells.
In anesthetized rats, the acute administration of charybdotoxin plus apamin does not affect basal arterial blood pressure but attenuates the decrease in blood pressure produced by acetylcholine or ghrelin Citation110,111. Similarly, in the mesenteric and hindlimb vascular beds, the combination of the two toxins does not affect the basal conductance but inhibits the vasodilatation evoked by acetylcholine or bradykinin, indicating that EDHF‐mediated responses play a physiological role in the regulation of vascular resistance Citation112.
Beyond endothelial hyperpolarization
In the vascular wall, gap junctions link smooth muscle with other smooth muscle cells, endothelial with other endothelial cells, and in many blood vessels smooth muscle with endothelial cells. The number of myoendothelial gap junctions increases with a reduction in the size of the artery Citation113, a phenomenon that parallels the contribution of the EDHF‐mediated responses to endothelium‐dependent relaxations Citation114,115. Furthermore, the presence of these heterocellular gap junctions is associated with EDHF‐mediated responses Citation116,117. Endothelium and smooth muscle cells can communicate via these myoendothelial gap junctions physically, as Ca2+ can diffuse from one cell type to another Citation118,119, and electrically, since depolarization and hyperpolarization are conducted bidirectionally from one cell type to the other Citation120–126. The connexins (Cx) 37, 40, and 43 are the predominant isoforms of gap junction proteins expressed in the vascular wall. Depending on the blood vessel, they can be involved in myoendothelial gap junction communication Citation127–130. Blockers of gap junctions abolish or partially inhibit EDHF‐like responses in many arteries in vitroCitation127,Citation131–136 and in vivoCitation137. Furthermore, in the rat mesenteric artery, antibodies directed against Cx40, when loaded selectively in the endothelial cells block EDHF‐mediated responses Citation138. In this preparation, SKCa colocalize with Cx37, Cx40, and Cx43 and are adjacent to endothelial cell gap junctions, whereas IKCa colocalize with Cx37 and Cx40 in myoendothelial gap junctions Citation139. Therefore, the transmission of endothelial hyperpolarization to the underlying smooth muscle via myoendothelial gap junctions is likely to play a predominant role in EDHF‐mediated responses in many blood vessels. Additionally, the endothelial cell lining is a privileged low‐resistance pathway for conducted vasodilatation/hyperpolarization along the length of the vessel Citation124,125.
An additional mechanism to achieve hyperpolarization and relaxation of the underlying vascular smooth muscle cells is also directly linked to the hyperpolarization of the endothelial cells. Indeed, the activation of endothelial IKCa and SKCa causes an efflux of K+ ions from the intracellular compartment toward the extracellular space. K+ ions accumulate in the intercellular space between endothelial and smooth muscle cells. A moderate increase in the extracellular K+ concentration (1 to 15 mM) can provoke the relaxation of vascular smooth muscle cells Citation140 by activating KIRCitation141 and the Na+/K+ pump Citation142. Indeed, a unique feature of KIR channels is the action of extracellular potassium on its gating. A moderate increase in potassium concentration, in the range of 1 to 20 mM, enhances potassium efflux through KIR at physiologically relevant potentials Citation143. The activation of KIR and the Na+/K+ pump overcomes the small depolarizing effects linked to the increase in potassium ions per se and the net resultant is hyperpolarization and thus relaxation of the smooth muscle cells. The KIR channel can be regarded as a metabolic sensor producing vasodilatation and increases in blood flow when potassium accumulates in the circulation during neuronal activity or exercise for instance. Therefore, accumulation of K+ ions in the intercellular space may contribute to the mechanism of EDHF‐mediated responses. This hypothesis was demonstrated successfully in the hepatic and mesenteric arteries of the rat Citation144. Edwards et al. provided evidence that the targets of apamin and charybdotoxin are located on the endothelial cells, that potassium ions accumulate in the intercellular space between endothelial and smooth muscle cells, and that the potassium efflux associated with the activation of endothelial IKCa and SKCa channels produced hyperpolarization by activating both KIR and the Na+/K+ pump on the smooth muscle cells. A role for the accumulation of K+ ions in the intercellular space was then confirmed in many different arteries Citation145–152. Nevertheless, this hypothesis has been highly controversial, possibly because the experimental conditions, and especially the presence or not of contractile agents, can markedly influence the contribution of K+ ions in the overall mechanisms underlying endothelial‐dependent hyperpolarizations. Contractile agonists stimulate receptors on vascular smooth muscle cells, increase intracellular calcium concentration, and depolarize the cells. The rise in intracellular calcium activates BKCa while the depolarization activates KV as braking mechanisms. Potassium effluxes through these two channels, and K+ ions accumulate in the intercellular space. This ‘potassium cloud’ activates KIR and Na+/K+‐ATPase, preventing the effects of any subsequent rise of potassium during endothelium‐dependent hyperpolarization Citation153.
The KIR channel, most likely involved in K+ ion‐induced hyperpolarization in rat arteries, is composed of the Kir2.1 α subunits Citation154–156. However, in some blood vessels, K+ does not evoke or inconsistently produces relaxations and hyperpolarizations Citation126,Citation134,135,Citation157,158. Therefore, in these blood vessels the contribution of K+ ions in EDHF‐mediated responses must be, if anything, minimal.
The involvements of gap junctions and K+ ions are not necessarily mutually exclusive. The relative proportion of each mechanism almost certainly depends on numerous parameters including the state of activation of the vascular smooth muscle, the density of myoendothelial gap junctions, and the level of the expression of the appropriate isoforms of Na+/K+‐ATPase and/or KIR on the vascular smooth muscle Citation1,Citation58 ().
Figure 3. EDHF‐mediated responses. The activation of endothelial receptors (R) and the shear stress exerted by the flowing blood increase endothelial [Ca2+]i through calcium entry (TRP = transient receptor potential channel) and calcium release from internal stores (SR = sarcoplasmic reticulum). This activates endothelial KCa channels and produces endothelial hyperpolarization which is conducted through myoendothelial gap junctions to the underlying vascular smooth muscle. Cyclic‐AMP enhances the electrotonic conduction via gap junctions. Additionally, accumulation of potassium ions in the intercellular space can hyperpolarize the smooth muscle cells by activating KIR and Na+/K+‐ATPase. The presence of a ‘potassium cloud’ can markedly affect the contribution of K+ ions to EDHF‐mediated responses. Contractile agonists that stimulate their receptor on vascular smooth muscle cells increase intracellular calcium (Ca2+) concentration and depolarize the cells. The rise in intracellular calcium activates BKCa while the depolarization activates KV as a braking mechanism. Potassium effluxes through these two channels and accumulates in the intercellular space (potassium cloud). Potassium ions activate KIR and Na+/K+‐ATPase, preventing the effects of any subsequent rise of potassium during endothelium‐dependent hyperpolarization. The relative proportion of each mechanism depends on numerous parameters, including the state of activation of the vascular smooth muscle, the density of myoendothelial gap junctions, and the level of the expression of the appropriate isoforms of Na+/K+‐ATPase and/or KIR. BK = bradykinin; SP = substance P; SLIGRL‐NH2 = peptide agonist of proteinase‐activated recepor‐2; P3 = inositol trisphosphate; TRPV4 = transient receptor potential channel vanilloid 4; AC = adenylyl cyclase; αGA = glycyrrhetinic acid derivatives; CX = connexin; 4‐AP = 4‐aminopyridine; IbTX = iberiotoxin; ChTX = charybdotoxin; SK3 = small conductance calcium‐activated potassium channel formed by SK3 α subunits; IK1 = intermediate conductance calcium‐activated potassium channel formed by IK1 α subunits; Kir2.1 = Inward rectifying potassium channel constituted of Kir2.1 α subunits; KV = voltage‐gated potassium channels; BKCa = large conductance calcium‐activated potassium channels (modified from Citation58); ACh = acetylcholine.
![Figure 3. EDHF‐mediated responses. The activation of endothelial receptors (R) and the shear stress exerted by the flowing blood increase endothelial [Ca2+]i through calcium entry (TRP = transient receptor potential channel) and calcium release from internal stores (SR = sarcoplasmic reticulum). This activates endothelial KCa channels and produces endothelial hyperpolarization which is conducted through myoendothelial gap junctions to the underlying vascular smooth muscle. Cyclic‐AMP enhances the electrotonic conduction via gap junctions. Additionally, accumulation of potassium ions in the intercellular space can hyperpolarize the smooth muscle cells by activating KIR and Na+/K+‐ATPase. The presence of a ‘potassium cloud’ can markedly affect the contribution of K+ ions to EDHF‐mediated responses. Contractile agonists that stimulate their receptor on vascular smooth muscle cells increase intracellular calcium (Ca2+) concentration and depolarize the cells. The rise in intracellular calcium activates BKCa while the depolarization activates KV as a braking mechanism. Potassium effluxes through these two channels and accumulates in the intercellular space (potassium cloud). Potassium ions activate KIR and Na+/K+‐ATPase, preventing the effects of any subsequent rise of potassium during endothelium‐dependent hyperpolarization. The relative proportion of each mechanism depends on numerous parameters, including the state of activation of the vascular smooth muscle, the density of myoendothelial gap junctions, and the level of the expression of the appropriate isoforms of Na+/K+‐ATPase and/or KIR. BK = bradykinin; SP = substance P; SLIGRL‐NH2 = peptide agonist of proteinase‐activated recepor‐2; P3 = inositol trisphosphate; TRPV4 = transient receptor potential channel vanilloid 4; AC = adenylyl cyclase; αGA = glycyrrhetinic acid derivatives; CX = connexin; 4‐AP = 4‐aminopyridine; IbTX = iberiotoxin; ChTX = charybdotoxin; SK3 = small conductance calcium‐activated potassium channel formed by SK3 α subunits; IK1 = intermediate conductance calcium‐activated potassium channel formed by IK1 α subunits; Kir2.1 = Inward rectifying potassium channel constituted of Kir2.1 α subunits; KV = voltage‐gated potassium channels; BKCa = large conductance calcium‐activated potassium channels (modified from Citation58); ACh = acetylcholine.](/cms/asset/167fbc72-80ef-4740-8777-af2a5e8d2113/iann_a_248977_f0003_b.gif)
Genetically modified animals
In NOS‐3 knockout mice, EDHF‐mediated responses play a compensatory role for the absence of endothelial NO Citation159 (), and this adaptation to NO synthase deletion is gender‐specific Citation160. Similarly, in resistance arteries from double‐knockout mice for NOS‐3 and cyclooxygenase‐1 (COX‐1), endothelium‐dependent relaxations are preserved by an EDHF‐mediated mechanism, while in arteries from the males the endothelium‐dependent relaxations are impaired severely. In those female mice, the double deletion of NOS‐3 and COX‐1 does not affect mean arterial blood pressure, while the corresponding males are hypertensive Citation161.
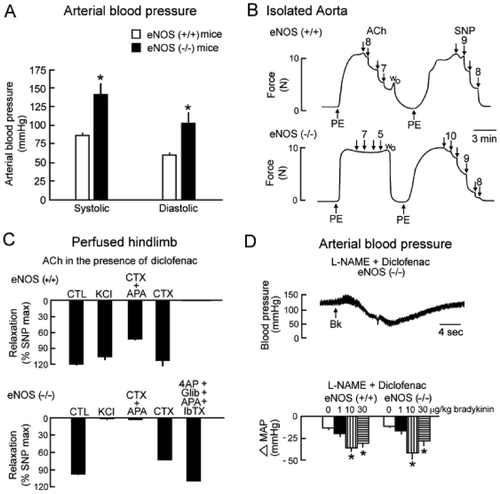
Figure 4. EDHF‐mediated responses in two strains of mice, wild‐type (C57BL/6), and NOS‐3 knockout mice (eNOS(−/−)). A: NOS‐3 knockout mice are hypertensive. B: Acetylcholine (ACh) produces an endothelium‐dependent relaxation in the isolated aortic rings of wild‐type mice (eNOS(+/+)) but no longer in that of NOS‐3 knockout mice (eNOS(−/−)). However, the NO donor and endothelium‐independent vasodilator sodium nitroprusside (SNP) produces a relaxation in the aorta from both strains. These results confirm the deletion of the NOS‐3 gene and demonstrate that, in a large blood vessel such as the aorta, NO is the preponderant endothelium‐derived relaxing factor released by acetylcholine. C: Acetylcholine induces a vasodilatation in the hindlimb of both wild‐type and NOS‐3 knockout mice. In the wild‐type mice, this vasodilatation is unaffected by a depolarizing solution (KCl) and partially inhibited by the combination of charybdotoxin (CTX) and apamin (apa) but not by charybdotoxin alone, indicating that an EDHF‐mediated response may contribute to the mechanism of vasodilatation but is not the preponderant mechanism. In the NOS‐3 knockout mice, the acetylcholine‐induced vasodilatation is preserved and is blocked by KCl or the combination of charybdotoxin plus apamin, partially inhibited by charybdotoxin alone and unaffected by the combination of iberiotoxin (IbTX), apamin, glibenclamide (Glib) and 4‐aminopyridine (4AP). These results indicate that acetylcholine induces an endothelium‐dependent vasodilatation showing the hallmarks of EDHF‐mediated responses. The EDHF‐mediated response compensates the disruption of the NOS‐3 gene. D: Original recording of arterial blood pressure in an anaesthetized NOS‐3 knockout mouse treated with an inhibitor of cyclooxygenase, diclofenac, and an inhibitor of NO synthase, L‐arginine‐methyl‐ester (L‐NAME). The intravenous administration of the endothelium‐dependent vasodilator bradykinin induces a transient decrease in arterial blood pressure. The summary bar graphs show the concentration‐dependent changes in mean arterial pressure (MAP) evoked by administration of bradykinin in wild‐type and eNOS(−/−) mice under control conditions (top graphs) or after treatment with inhibitors of cyclooxygenases and NO synthases (lower graphs). The amplitude of the hypotensive response induced by bradykinin is similar in NOS‐3 knockout and wild‐type animals. In both strains, the effect of bradykinin is unaffected by the presence of inhibitors of cyclooxygenases and NO synthases. These data strongly suggest that EDHF‐mediated responses play an essential role in the hypotensive effect of bradykinin (modified from Citation159). CTL = control.
Transgenic mice harboring genetically targeted alleles for the SK3 channel, in which SK3 gene expression can be experimentally controlled by dietary doxycycline, have been engineered Citation162. In those transgenic mice, the suppression of SK3 expression in the endothelial cells depolarizes both the endothelial and the vascular smooth muscle cells, reduces the diameter of resistance vessels in situ, and increases arterial blood pressure Citation108. Disruption of the IK1 gene reduces the hyperpolarization of endothelial and smooth muscle cells in response to acetylcholine. The associated vasodilatation in the carotid artery and in resistance vessels is also diminished because of a substantial reduction in EDHF‐mediated responses. Moreover, the IK1 deletion also led to a significant increase in arterial blood pressure and to mild left ventricular hypertrophy Citation109. These results confirm that in mice the endothelial SKCa and IKCa are fundamental determinants of endothelial hyperpolarization and EDHF signaling, and indicate that they actively control vascular tone and the overall regulation of the circulation. In mice that lack Kir2.1, but not in those deleted for Kir2.2 genes, KIR currents are absent, and stimulation with moderate increase in K+ concentration does not produce relaxation, indicating that Kir2.1 gene expression is required for KIR currents and K+‐induced dilatation. Kir2.1(−/−) animals die 8–12 hours after birth preventing the acquisition of more information on cardiovascular parameters such as arterial blood pressure Citation154.
Connexins 37 and 40 are the predominant gap junction proteins in the endothelial cells of the mouse Citation163. Connexin 40 proteins are involved in endothelial homocellular gap junctions and also in heterocellular gap junctions linking endothelial cells not only to smooth muscle cells but also to renin‐producing juxtaglomerular cells. The presence of the latter gap junction communication is required in order to maintain the calcium‐dependent inhibitor effects of angiotensin II and that of intrarenal pressure on renin secretion and synthesis, suggesting that the endothelium is strongly involved in the regulation of the renin system. Mice deficient for connexin 40 are hypertensive. However, the control of renin production only partially explained the hypertension in connexin 40 knockout mice Citation164. The arterioles of these animals also exhibit a reduced spread of dilatation in response to endothelium‐dependent vasodilators and irregular arteriolar vasomotion Citation165–168. Mice subjected to specific deletion of endothelial connexin 43 do not present major alterations in arterial blood pressure Citation169,170, possibly because this connexin is not the major one expressed in murine endothelial cells.
These results show that deletion of each key molecular component of EDHF‐mediated responses is associated with hemodynamic alterations suggesting that this endothelial pathway contributes to the overall regulation of arterial blood pressure.
Endothelium‐dependent responses resistant to inhibitors of NO‐synthases and cyclooxygenases in human blood vessels
Isolated blood vessels
In isolated human vascular preparations, endothelium‐dependent responses resistant to inhibitors of NO synthases and cyclooxygenases are observed in coronary (), internal mammary, radial, renal, and cerebral arteries, as well as in resistance arteries from various vascular beds and in forearm veins Citation1. As in animal arteries, the contribution of EDHF‐mediated responses appears significantly more important in smaller than in large human arteries Citation171,172.
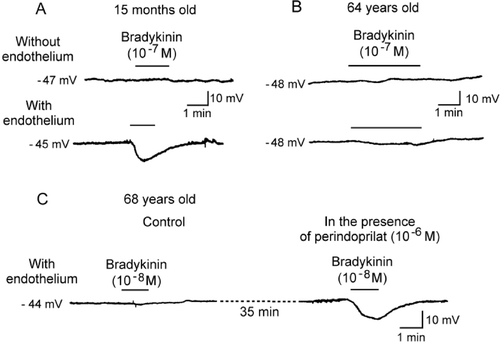
Figure 5. Endothelium‐dependent hyperpolarizations in isolated human coronary arteries (in the presence of inhibitors of cyclooxygenases and NO synthases). A: In coronary arteries taken from a young patient (15 months old, congenital heart disease), bradykinin produces an endothelium‐dependent hyperpolarization. B: In coronary arteries taken from an older patient (64 years old, ischemic heart disease) the endothelium‐dependent hyperpolarization to bradykinin is reduced markedly when compared to that observed in the juvenile patient. C: In coronary arteries taken from a patient (68 years old, ischemic cardiomyopathy) the endothelium‐dependent hyperpolarization to bradykinin is virtually absent. The presence of the angiotensin‐converting enzyme inhibitor, perindoprilat, restores the endothelium‐dependent hyperpolarization (modified from Citation1).
These hyperpolarizations or/and relaxations resistant to inhibitors of NO synthases and cyclooxygenases can be attributed to the activation of K+ channels as they are blocked by elevated K+ concentrations. These responses are independent of KATP activation and in general are minimally or not at all affected by charybdotoxin, iberiotoxin, or apamin given alone but are blocked either by nonspecific K+ channel inhibitors or by the combination of charybdotoxin plus apamin. The various hypotheses formulated for the identification of these responses are as follows: 1) EETs derived from cytochrome P450 monooxygenase in coronary, internal mammary, and subcutaneous arteries Citation173–176; 2) H2O2 in coronary and mesenteric arteries Citation68,Citation72; and 3) EDHF‐mediated responses with a preferential involvement of Cx43‐dependent myoendothelial gap junction communication in subcutaneous arteries Citation126,Citation128,Citation177 and a preferential involvement of K+ ions in interlobar renal and thyroid arteries Citation152,Citation178. CNP also relaxes and hyperpolarizes human subcutaneous resistance arteries by a cyclic GMP‐dependent activation of BKCaCitation179, but its contribution to endothelium‐dependent responses has not yet been demonstrated. To the best of our knowledge, there is no conclusive evidence for a major role of lipoxygenase in the endothelium‐dependent relaxations/hyperpolarizations of human blood vessels. Contractile lipoxygenase derivatives, generated in response to various agonists, such as angiotensin II, arachidonic acid, acetylcholine, or bradykinin, can play a role in many blood vessels, but they do not originate from the endothelial cells Citation180.
Thus, the in vitro data available demonstrate that endothelium‐dependent responses resistant to inhibitors of NO‐synthases and cyclooxygenases are observed in isolated human blood vessels and that the characteristics of these responses are similar to those observed in most animal studies ().
Table I. Nonexhaustive summary of potential mechanisms underlying endothelium‐dependent hyperpolarizations/relaxations resistant to the combined presence of inhibitors of NO‐synthases and cyclooxygenases.
Intact organism
An EDHF‐mediated mechanism is often suggested to explain vasodilatations resistant to inhibitors of NO synthase. However, many human studies do not involve the administration of an inhibitor of cyclooxygenases. Furthermore, complete blockade of NO synthase is difficult to obtain (or to demonstrate), and/or a nonendothelial effect of the vasodilators such as a direct effect on the smooth muscle cells or an inhibitory effect on the sympathetic system cannot be excluded easily. In the human, apamin plus charybdotoxin cannot be administered for obvious ethical reasons. However, studies especially designed to study EDHF‐mediated responses have shown convincingly that in the forearm vasculature, basal forearm blood flow, flow‐induced vasodilatation, as well as the increases in blood flow in response to bradykinin, substance P, and to a lesser extent acetylcholine involve a non‐PGI2‐, non‐NO‐mediated mechanism that depends on activation of K+ channels Citation181–188.
The mechanism underlying the non‐NO‐, non‐PGI2‐mediated responses is still controversial. The endothelial production of a cytochrome P450 metabolite has been suggested to play a role in the regulation of the basal diameter and flow‐induced vasodilatation of the radial artery Citation187,188 as well as in exercise‐induced increases in skeletal muscle blood flow Citation189. Similarly, in hypertensive patients, a sulfaphenazole‐sensitive component contributes to the increase in forearm blood flow evoked by the administration of bradykinin and to a lesser extent that of acetylcholine Citation190. By contrast, other studies have shown that in the forearm of healthy volunteers and patients with various pathologies, the cytochrome P450 inhibitor sulfaphenazole does not influence basal tone or does not reduce bradykinin‐induced vasodilatations when given alone or in the presence of inhibitors of cyclooxygenase and NO synthase Citation191–193. Furthermore, in patients with coronary artery disease, inhibition of cytochrome P450 2C9 paradoxically improves the endothelium‐dependent, nitric oxide‐mediated vasodilatation Citation193, most likely because cytochrome P450 2C9 generates superoxide anions which reduce the bioavailability of NO Citation194. The discrepancies between these various studies can possibly be explained by two different phenomena. Residual NO is an important issue in human studies performed in vivo where typically only relatively low doses of one of the weakest inhibitors of NO synthase (L‐NMMA) can be administered. Furthermore, the lack of selectivity of the available inhibitors of cytochrome P450 makes it difficult to determine whether or not the release of EETs underlies non‐NO‐, non‐PGI2‐mediated responses. For instance, inhibitors of cytochrome P450 of the imidazole family block various populations of K+ channels and especially IKCa, the K+ channel involved in EDHF‐mediated responses Citation195.
Therefore, in humans, responses resistant to the inhibition of NO synthases and cyclooxygenases can occur in vivo, and they contribute to the regulation of basal blood flow as well as to the endothelium‐dependent dilatations in response to flow and certain neurohumoral mediators.
Endothelium‐dependent responses resistant to inhibitors of NO synthases and cyclooxygenases: endothelial dysfunction and therapeutic interventions
Endothelial dysfunction, observed in various cardiovascular diseases, is associated with a decrease of NO synthesis and/or a loss of its biological activities. However, alteration of the EDHF pathway can also contribute to these endothelial dysfunctions or conversely can compensate the loss of NO bioavailability Citation1,2, Citation196.
The alteration of EDHF‐mediated responses has been reported with aging and various pathological conditions (hypertension, atherosclerosis, hypercholesterolemia, heart failure, ischemia‐reperfusion, angioplasty, eclampsia, diabetes, sepsis) Citation1,Citation196. However, again, depending on the model or the vascular bed studied, marked differences can be observed. Hypertension per se does not produce a consistent depression of the EDHF‐mediated responses. For instance, in the mesenteric artery of the spontaneously hypertensive rat (SHR), the impairment of the endothelium‐dependent relaxations is attributed to a marked attenuation of the EDHF component and a concomitant production of cyclooxygenase‐derived contractile prostanoids (endothelium‐derived contractile factors, EDCF) with no or little alteration in the production of NO, whereas, in the model of L‐arginine‐methyl‐ester (L‐NAME)‐induced hypertension and in the same vascular bed, an increase in EDHF‐mediated responses may compensate the inhibition of NO production Citation1. In human with essential hypertension, the mechanism underlying the endothelial dysfunction is also linked to oxidative stress and the activation of cyclooxygenase, which reduces availability of NO. In the forearm vascular bed, the presence of an EDHF‐like pathway, possibly a cytochrome P450‐dependent mechanism, compensates the decreased NO bioavailability in order to sustain endothelium‐dependent vasodilatation Citation190,Citation197. This compensatory pathway can be depressed by additional aggravating factors such as hyperhomocysteinemia Citation197. In myometrial arterioles from preeclamptic mothers, the upregulation of the EDHF‐mediated responses observed in normal pregnancy does not occur Citation198. The question remains whether the impairment of EDHF‐mediated responses contributes to the genesis of the syndrome or is a consequence of the hypertensive process.
Hypercholesterolemia is generally associated with a preserved or an enhanced contribution of EDHF‐mediated responses that compensate for the decrease in NO‐mediated relaxation Citation199. The resistance of endothelium‐dependent hyperpolarizations to hypercholesterolemia has been demonstrated in arteries from rabbit Citation200, SHR Citation201, apolipoprotein E‐deficient (ApoE‐/‐) mice Citation202–204 and dyslipidemic transgenic mice expressing human apolipoprotein B (hApoB+/+) Citation205. However, in isolated gastroepiploic arteries from atherosclerotic patients, endothelium‐dependent hyperpolarizations are inhibited Citation172. The prolonged duration of hypercholesterolemia and the severity of the atherosclerotic process in the human may contribute to the degree of dysfunction of the EDHF pathway Citation1.
Conversely, EDHF‐mediated responses are depressed in various models of type I and type II diabetes, with the exception of some murine models Citation196. In patients with type I diabetes under good glycemic control and without albuminuria, endothelial function appears normal, and both the NO‐ and the EDHF‐mediated responses are preserved. However, in patients with microalbuminuria, impairment of the endothelium‐dependent vasodilatation is observed Citation206. In these patients and in patients with type II diabetes, whether or not the various components of the endothelium‐dependent vasodilatation are differentially affected by the disease is not yet known Citation1,Citation206.
No drug is available which has been designed to target EDHF‐mediated responses. Nevertheless, therapeutic interventions, with beneficial effects on the cardiovascular system such as angiotensin‐converting enzyme inhibitors, antagonists of angiotensin receptors, and phosphodiesterase‐3 inhibitors Citation1,Citation207 can restore these responses, suggesting that the improvement of the EDHF pathway contributes to the observed beneficial effect. Similarly, various so‐called nonpharmacological therapeutic strategies, including exercise and supplementation with estrogens, omega‐3 polyunsaturated fatty acids, polyphenol derivatives, potassium and/or calcium, help to reverse endothelial dysfunction including blunted EDHF‐mediated responses. Whether or not the improvement of these EDHF‐mediated responses contributes to the beneficial effects of these dietary maneuvers can be suspected but is far from being demonstrated Citation1, Citation198.
Conclusions and perspectives
Endothelial cells control the tone of the underlying vascular smooth muscle by releasing numerous vasoactive substances, including NO, reactive oxygen species, potassium ions, and metabolites of arachidonic acid (e.g. prostacyclin, EETs, lipoxygenase derivatives). Furthermore, the endothelial monolayer behaves as a conductive tissue propagating an electrical signal along the axis of the blood vessel by means of homocellular gap junctions and throughout the vascular wall itself by means of myoendothelial gap junctions. Endothelium‐dependent relaxations, independent of the production of NO and prostacyclin, probably play an important role in cardiovascular physiology in numerous animal species and in the human. They can act as a backup system when NO is inhibited or reduced, but this is not necessarily the case.
However, it is often difficult to reach a conclusion as to the true importance of endothelium‐dependent hyperpolarizations because of the use of unspecific pharmacological tools and the lack of electrophysiological measurements. The mechanism underlying endothelium‐responses must be carefully dissected in order to be properly identified. The synthesis of more selective compounds such as nonpeptidic inhibitors and activators of SKCa and IKCaCitation208,209, may in the future allow the selective blockade/activation of EDHF‐mediated responses and hence the proper determination of their physiological role in the human circulation.
The improvement or restoration of EDHF responses has not been, yet, the direct purpose of any pharmaceutical effort. Activating or increasing the expression of endothelial TRP, IKCa and/or SKCa channels, and smooth muscle KIR and/or specific isoform(s) of Na+/K+‐ATPase, as well as facilitating myoendothelial communication (rotigaptide‐like drugs, phosphodiesterase‐3 inhibitors) and increasing the expression of appropriate connexins (Cx40, Cx43, Cx37) represent new potential targets.
References
- Félétou M., Vanhoutte P. M. EDHF: the complete story. Taylor & Francis, CRC press, Boca Raton, Fl 2005
- Félétou M., Vanhoutte P. M. Endothelial dysfunction: a multifaceted disorder (The Wiggers Award Lecture). Am J Physiol Heart Circ Physiol 2006; 291: H985–1002
- Furchgott R. F., Vanhoutte P. M. Endothelium‐derived relaxing and contracting factors. FASEB J 1989; 3: 2007–18
- Furchgott R. F., Zawadzki J. V. The obligatory role of the endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 1980; 288: 373–6
- Busse R., Edwards G., Félétou M., Fleming I., Vanhoutte P. M., Weston A. H. Endothelium‐dependent hyperpolarization, bringing the concepts together. Trends Pharmacol Sci 2002; 23: 374–80
- Itoh T., Seki N., Suzuki S., Ito S., Kajikuri J., Kuriyama H. Membrane hyperpolarisation inhibits agonist‐induced synthesis of inositol 1,4,5‐trisphosphate in rabbit mesenteric artery. J Physiol 1992; 451: 307–28
- del Valle‐Rodriguez A., Lopez‐Barneo J., Urena J. Ca2+ channel‐sarcoplasmic reticulum coupling: a mechanism of arterial myocyte contraction without Ca2+ influx. EMBO J 2003; 22: 4337–45
- Moncada S., Vane J. R. Pharmacology and endogenous roles of prostaglandin endoperoxides, thromboxane A2 and prostacyclin. Pharmacol Rev 1979; 30: 293–331
- Smyth E. M., FitzGerald G. A. Human prostacyclin receptor. Vitam Horm 2002; 65: 149–65
- Corriu C., Félétou M., Edwards G., Weston A. H., Vanhoutte P. M. Differential effects of Prostacyclin and Iloprost in the isolated carotid artery of the guinea‐pig. Eur J Pharmacol 2001; 426: 89–94
- Parkington H. C., Coleman H. A., Tare M. Prostacyclin and endothelium‐dependent hyperpolarizations. Pharmacol Res 2004; 49: 509–14
- Li P. L., Zou A. P., Campbell W. B. Regulation of potassium channels in coronary arterial smooth muscle by endothelium‐derived vasodilators. Hypertension 1997; 29: 262–7
- Orie N. N., Fry C. H., Clapp L. H. Evidence that inward rectifier K+ channels mediate relaxation by the PGI2 receptor agonist cicaprost via a cyclic AMP‐independent mechanism. Cardiovasc Res 2006; 69: 107–15
- Schubert R., Serebryakov N. V., Engel H., Hopp H. H. Iloprost activates KCa channels of vascular smooth muscle cells: role of cyclic‐AMP‐dependent proteine kinase. Am J Physiol 1996; 271: C1203–11
- Clapp L. H., Turcato S., Hall S., Baloch M. Evidence that Ca2+‐activated K+ channels play a major role in mediating the vascular effects of iloprost and cicaprost. Eur J Pharmacol 1998; 356: 215–24
- Olschewski A., Li Y., Tang B., Hanze J., Eul B., Bohle R. M., et al. Impact of TASK‐1 in human pulmonary artery smooth muscle cells. Circ Res 2006; 98: 1072–80
- Yamaki F., Kaga M., Horinouchi T., Tanaka H., Koike K., Shigenobu K., et al. MaxiK channel‐mediated relaxation of guinea‐pig aorta following stimulation of IP receptor with beraprost via cyclic AMP‐dependent and ‐independent mechanisms. Naunyn Schmiedebergs Arch Pharmacol 2001; 364: 538–50
- Fleming I. Cytochrome P450 epoxygenases as EDHF synthase(s). Pharmacol Res 2004; 49: 525–33
- Fisslthaler B., Popp R., Kiss L., Potente M., Harder D. R., Fleming I., et al. Cytochrome P4540 2C is an EDHF synthase in coronary arteries. Nature 1999; 401: 493–7
- Popp R., Bauersachs J., Sauer E., Hecker M., Fleming I., Busse R. A transferable, β‐naphtoflavone‐inducible, hyperpolarizing factor is synthesized by native and cultured porcine coronary endothelial cells. J Physiol 1996; 497: 699–709
- Campbell W. B., Gebremedhin D., Pratt P. F., Harder D. R. Identification of epoxyeicosatrienoic acids as endothelium‐derived hyperpolarizing factor. Circ Res 1996; 78: 415–23
- Gauthier K. M., Edwards E. M., Falck J. R., Reddy D. S., Campbell W. B. 14,15‐Epoxyeicosatrienoic acid represents a transferable endothelium‐dependent relaxing factor in bovine coronary arteries. Hypertension 2005; 45: 666–71
- Huang A., Sun D., Jacobson A., Carroll M. A., Kalck J. R., Kaley G. Epoxyeicosatrienoic acids are released to mediate shear stress‐dependent hyperpolarization of arteriolar smooth muscle. Circ Res 2005; 96: 376–83
- Weston A. H., Félétou M., Vanhoutte P. M., Falck J. R., Campbell W. B., Edwards G. Endothelium‐dependent hyperpolarizations induced by bradykinin in the vasculature; clarification of the role of epoxyeicosatrienoic acids. Br J Pharmacol 2005; 145: 775–84
- Ordway R. W., Walsh J. V., Singer J. J. Arachidonic acid and other fatty acids directly activates potassium channels in vascular smooth muscle cells. Science 1989; 244: 1176–9
- Li P. L., Campbell W. B. Epoxyeicosatrienoic acids activate K+ channels in coronary smooth muscle through a guanine nucleotide binding protein. Circ Res 1997; 80: 877–84
- Li P. L., Chen C. L., Bortell R., Campbell W. B. Epoxyeicosatrienoic acid stimulates endogenous mono‐ADP‐ribosylation in bovine coronary artery smooth muscle. Circ Res 1999; 85: 349–56
- Sacerdoti D., Bolognesi M., Di Pascoli M., Gatta A., McGiff J. C., Schwartzman M. L., et al. Rat mesenteric arterial dilator response to 11,12‐epoxyeicosatrienoic acid is mediated by activating heme oxygenase. Am J Physiol Heart Circ Physiol 2006; 291: H1999–2002
- Earley S., Heppner T. J., Nelson M. T., Brayden J. E. TRPV4 forms a novel Ca2+‐signaling complex with ryanodine receptors and BKCa channels. Circ Res 2005; 97: 1270–9
- Imig J. D. Epoxide hydrolase and epoxygenase metabolites as therapeutic targets for renal diseases. Am J Physiol Renal Physiol 2005; 289: F496–503
- Larsen B. T., Campbell W. B., Gutterman D. D. Beyond vasodilatation: non‐vasomotor roles of epoxyeicosatrienoic acids in the cardiovascular system. Trends Pharmacol Sci 2007; 28: 32–8
- Corriu C., Félétou M., Canet E., Vanhoutte P. M. Inhibitors of the cytochrome P450‐monooxygenase and endothelium‐dependent hyperpolarisations in the guinea‐pig isolated carotid artery. Br J Pharmacol 1996; 117: 607–10
- Chataigneau T., Félétou M., Duhault J., Vanhoutte P. M. Epoxyeicosatrienoic acids, potassium channel blockers and endothelium‐dependent hyperpolarisation in the guinea‐pig carotid artery. Br J Pharmacol 1998; 123: 574–80
- Zink M. H., Oltman C. L., Lu T., Katakam P. V., Kaduce T. L., Lee H., et al. 12‐lipoxygenase in porcine coronary circulation: implications for coronary vasoregulation. Am J Physiol 2001; 280: H693–704
- Pfister S. L., Spitzbarth N., Nithipatikom K., Edgemont W., Falk J. R., Campbell W. B. Identification of 11,14,15‐ and 11,12,15‐trihydroxyeicosatrienoic acids as endothelium‐derived relaxing factors of rabbit aorta. J Biol Chem 1998; 273: 30879–87
- Tang X., Holmes B. B., Nithipatikom K., Hillard C. J., Kuhn H., Campbell W. B. Reticulocyte 15‐lipoxygenase‐I is important in acetylcholine‐induced endothelium‐dependent vasorelaxation in rabbit aorta. Arterioscler Thromb Vasc Biol 2006; 26: 78–84
- Quignard J. F., Chataigneau T., Corriu C., Edwards G., Weston A. H., Félétou M., et al. Endothelium‐dependent hyperpolarization and lipoxygenase derived metabolites of arachidonic acid in the carotid artery of the guinea‐pig. J Cardiovasc Pharmacol 2002; 40: 467–77
- Randall M. D., Kendall D. A., O'Sullivan S. The complexities of the cardiovascular actions of cannbinoids. Br J Pharmacol 2004; 142: 20–6
- Chataigneau T., Félétou M., Thollon C., Villeneuve N., Vilaine J. P., Duhault J., et al. Cannabinoid CB1 receptor and endothelium‐dependent hyperpolarisation in guinea‐pig carotid, rat mesenteric and porcine coronary arteries. Br J Pharmacol 1998; 123: 968–74
- Wang R. Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter?. FASEB J 2002; 16: 1792–8
- Carvajal J. A., Germain A. M., Huidobro‐Toro J. P., Weiner C. P. Molecular mechanism of cGMP‐mediated smooth muscle relaxation. J Cell Physiol 2000; 184: 409–20
- Cohen R. A., Adachi T. Nitric‐oxide‐induced vasodilatation: regulation by physiologic s‐glutathiolation and pathologic oxidation of the sarcoplasmic endoplasmic reticulum calcium ATPase. Trends Cardiovasc Med 2006; 16: 109–14
- Robertson B. E., Schubert R., Hescheler J., Nelson M. T. Cyclic‐GMP‐dependent protein kinase activates Ca‐activated K channels in cerebral artery smooth muscle cells. Am J Physiol 1993; 65: C299–303
- Quignard J. F., Félétou M., Corriu C., Chataigneau T., Edwards G., Weston A. H., et al. 3‐Morpholinosydnonimine (SIN‐1) and K+ Channels in Smooth Muscle Cells of the Rabbit and Guinea‐Pig Carotid Arteries. Eur J Pharmacol 2000; 399: 9–16
- Archer S. L., Huang J. M. C., Hampl V., Nelson D. P., Shultz P. J., Weir E. K. Nitric oxide and cyclic‐GMP cause vasorelaxation by activation of a charybdotoxin‐sensitive K channel by cyclic‐GMP‐dependent protein kinase. Proc Natl Acad Sci USA 1994; 91: 7583–7
- Bolotina V. M., Najibi S., Palacino J. J., Pagano P. J., Cohen R. A. Nitric oxide directly activates calcium‐dependent potassium channels in vascular smooth muscle cells. Nature 1994; 368: 850–3
- Mistry D. K., Garland C. J. Nitric oxide (NO)‐induced activation of large conductance Ca2+‐dependent K+ channels (BKCa) in smooth muscle cells isolated from the rat mesenteric artery. Br J Pharmacol 1998; 124: 1131–40
- Yuan X. J., Tod M. L., Rubin L. J., Blaustein M. P. NO hyperpolarizes pulmonary artery smooth muscle cells and decreases the intracellular Ca2+ concentration by activating voltage‐gated K+ channels. Proc Natl Acad Sci USA 1996; 93: 10489–94
- Parkington H. C., Tare M., Tonta M. A., Coleman H. A. Stretch revealed three components in the hyperpolarisation of guinea‐pig coronary artery in response to acetylcholine. J Physiol 1993; 465: 459–76
- Corriu C., Félétou M., Canet E., Vanhoutte P. M. Endothelium‐derived factors and hyperpolarisations of the isolated carotid artery of the guinea‐pig. Br J Pharmacol 1996; 119: 959–64
- Schubert R., Krien U., Wulfsen I., Schiemann D., Lehmann G., Ulfig N., et al. Nitric oxide donor sodium nitroprusside dilates rat small arteries by activation of inward rectifier potassium channels. Hypertension 2004; 43: 891–6
- Cohen R. A., Plane F., Najibi S., Huk I., Malinski T., Garland C. J. Nitric oxide is the mediator of both endothelium‐dependent relaxation and hyperpolarisation of the rabbit carotid artery. Proc Natl Acad Sci USA 1997; 94: 4193–8
- Ge Z. D., Zhang X. H., Fung P. C., He G. W. Endothelium‐dependent hyperpolarization and relaxation: resistance to N(G)‐nitro‐L‐arginine and indomethacin in coronary circulation. Cardiovasc Res 2000; 46: 547–56
- Furchgott R. F., Jothianandan D. Endothelium‐dependent and ‐independent vasodilatation involves cyclic GMP: relaxation induced by nitric oxide, carbon monoxide and light. Blood Vessels 1991; 28: 52–61
- Chauhan S., Rahman A., Nilsson H., Clapp L., MacAllister R., Ahluwalia A. NO contributes to EDHF‐like responses in rat small arteries: role of NO stores. Cardiovasc Res 2003; 57: 207–16
- Muller B., Kleschyov A. L., Alencar J. L., Vanin A., Stoclet J. C. Nitric oxide transport and storage in the cardiovascular system. Ann N Y Acad Sci 2002; 962: 131–9
- Batenburg W. W., De Vries R., Saxena P. R., Danser A. H. L‐S nitrosothiols: endothelium‐derived hyperpolarizing factors in porcine coronary arteries. J Hypertens 2004; 22: 1927–36
- Félétou M., Vanhoutte P. M. EDHF: where are we now?. Arterioscler Thromb Vasc Biol 2006; 26: 1215–25
- Wu L., Wang R. Carbon monoxide: endogenous production, physiological functions, and pharmacological applications. Pharmacol Rev 2005; 57: 585–630
- Durante W. Carbon monoxide and bile pigments: surprising mediators of vascular function. Vasc Med 2002; 7: 195–202
- Foresti R., Hammad J., Clark J. E., Johnson T. R., Mann B. E., Friebe A., et al. Vasoactive properties of CORM‐3, a novel water‐soluble carbon monoxide‐releasing molecule. Br J Pharmacol 2004; 142: 453–60
- Barbe C., Dubuis E., Rochetaing A., Kreher P., Bonnet P., Vandier C. A 4‐AP‐sensitive current is enhanced by chronic carbon monoxide exposure in coronary artery myocytes. Am J Physiol Heart Circ Physiol 2002; 282: H2031–8
- Jaggar J. H., Leffler C. W., Cheranov S. Y., Tcheranova D. E. S., Cheng X. Carbon monoxide dilates cerebral arterioles by enhancing the coupling of Ca2+ sparks to Ca2+‐activated K+ channels. Circ Res 2002; 91: 610–7
- Roman R. J. P450 Metabolites of arachidonic acid in the control of cardiovascular function. Physiol Rev 2002; 82: 131–85
- Durante W., Johnson F. K., Johnson R. A. Role of carbon monoxide in cardiovascular function. J Cell Mol Med 2006; 10: 672–86
- Zakhary R., Gaine S. P., Dinerman J. L., Ruat M., Flavahan N. A., Snyder S. H. Heme oxygenase 2: endothelial and neuronal localization and role in endothelium‐dependent relaxation. Proc Natl Acad Sci U S A 1996; 93: 795–8
- Baragatti B., Brizzi F., Barogi S., Laubach V. E., Sodini D., Shesely E. G., et al. Interactions between NO, CO and an endothelium‐derived hyperpolarizing factor (EDHF) in maintaining patency of the ductus arteriosus in the mouse. Br J Pharmacol 2007; 151: 54–62
- Miura H., Bosnjak J. J., Ning G., Saito T., Miura M., Gutterman D. D. Role for hydrogen peroxide in flow‐induced dilation of human coronary arterioles. Circ Res 2003; 92: e31–40
- Hatoum O. A., Binion D. G., Miura H., Telford G., Otterson M. F., Gutterman D. D. Role of hydrogen peroxide in ACh‐induced dilation of human submucosal intestinal microvessels. Am J Physiol Heart Circ Physiol 2005; 288: H48–54
- Cosentino F., Katusic Z. S. Tetrahydrobiopterin and dysfunction of endothelial nitric oxide synthase in coronary arteries. Circulation 1995; 91: 139–44
- Matoba Y., Shimokawa H., Nakashima M., Hirakawa Y., Mukai Y., Hirano K., et al. Hydrogen peroxide is an endothelium‐derived hyperpolarizing factor in mice. J Clin Invest 2000; 106: 1521–30
- Matoba Y., Shimokawa H., Kubota H., Morikawa K., Fujiki T., Kunihiro I., et al. Hydrogen peroxide is an endothelium‐derived hyperpolarizing factor in human mesenteric artery. Biochem Biophys Res Commun 2002; 290: 909–13
- Shimokawa H., Matoba T. Hydrogen peroxide as an endothelium‐derived hyperpolarizing factor. Pharmacol Res 2004; 49: 543–9
- Yada T., Shimokawa H., Hiramatsu O., Kajita T., Shigeto F., Goto M., et al. Hydrogen peroxide, an endogenous endothelium‐derived hyperpolarizing factor, plays an important role in coronary autoregulation in vivo. Circulation 2003; 107: 1040–5
- Yada T., Shimokawa H., Hiramatsu O., Haruna Y., Morita Y., Kashihara N., et al. Cardioprotective role of endogenous hydrogen peroxide during ischemia‐reperfusion injury in canine coronary microcirculation in vivo. Am J Physiol Heart Circ Physiol 2006; 291: H1138–46
- Ellis A., Triggle C. R. Endothelium‐derived reactive oxygen species: their relationship to endothelium‐dependent hyperpolarization and vascular tone. Can J Physiol Pharmacol 2003; 81: 1013–28
- Hayabuchi Y., Nakaya Y., Matsukoa S., Kuroda Y. Hydrogen peroxide‐induced vascular relaxation in porcine coronary arteries is mediated by Ca2+‐activated K+ channels. Heart Vessels 1998; 13: 9–17
- Thengchaisri N., Kuo L. Hydrogen peroxide induces endothelium‐dependent and ‐independent coronary arteriolar dilation: role of cyclooxygenase and potassium channels. Am J Physiol 2003; 285: H2255–63
- Wei E. P., Kontos H. A., Beckman J. S. Mechanisms of cerebral vasodilation by superoxide, hydrogen peroxide, and peroxynitrite. Am J Physiol 1996; 271: H1262–6
- Rogers P. A., Dick G. M., Knudson J. D., Focardi M., Bratz I. N., Swafford A. N., Jr., et al. H2O2‐induced redox‐sensitive coronary vasodilation is mediated by 4‐aminopyridine‐sensitive K+ channels. Am J Physiol Heart Circ Physiol 2006; 291: H2473–82
- Iida Y., Katusic Z. S. Mechanisms of cerebral arterial relaxations to hydrogen peroxide. Stroke 2000; 31: 2224–30
- Tang X. D., Garcia M. L., Heinemann S. H., Hoshi T. Reactive oxygen species impair Slo1 BK channel function by altering cysteine‐mediated calcium sensing. Nat Struct Mol Biol 2004; 11: 171–8
- Gluais P., Edwards G., Weston A. H., Vanhoutte P. M., Félétou M. Hydrogen peroxide and the endothelium‐dependent hyperpolarization of the guinea‐pig isolated carotid artery. Eur J Pharmacol 2005; 513: 219–24
- Chaytor A. T., Edwards D. H., Bakker L. M., Griffith T. M. Distinct hyperpolarizing and relaxant roles for gap junctions and endothelium‐derived H2O2 in NO‐independent relaxations of rabbit arteries. Proc Natl Acad Sci U S A 2003; 100: 15212–7
- Rosenblum W. I. Hydroxyl radical mediates the endothelium‐dependent relaxation produced by bradykinin in mouse cerebral arterioles. Circ Res 1987; 61: 601–3
- Ohashi M., Faraci F., Heistad D. Peroxynitrite hyperpolarizes smooth muscle and relaxes internal carotid artery in rabbit via ATP‐sensitive K+ channels. Am J Physiol Heart Circ Physiol 2005; 289: H2244–50
- Koller K. J., Goeddel D. V. Molecular biology of the natriuretic peptides and their recptors. Circulation 1992; 86: 1081–8
- Kelsall C. J., Chester A. H., Sarathchandra P., Singer D. R. Expression and localization of C‐type natriuretic peptide in human vascular smooth muscle cells. Vascul Pharmacol 2006; 45: 368–73
- Stingo A. J., Clavell A. L., Heublein D. M., Wei C. M., Pittlekow M. R., Burnett J. C., Jr. Presence of C‐type natriuretic peptide in cultured human endothelial cells and plasma. Am J Physiol 1992; 263: H1318–21
- Honing M. L., Smits P., Morrison P. J., Burnett J. C., Jr., Rabelink T. J. C‐type natriuretic peptide‐induced vasodilation is dependent on hyperpolarization in human forearm resistance vessels. Hypertension 2001; 37: 1179–83
- Wei C. M., Hu S., Miller V. M., Burnett J. C., Jr. Vascular actions of C‐type natriuretic peptide in isolated porcine coronary arteries and coronary vascular smooth muscle cells. Biochem Biophys Res Comm 1994; 205: 765–71
- Banks M., Wei C. M., Kim C. H., Burnett J. C., Jr., Miller V. M. Mechanism of relaxations to C‐type natriuretic peptide in veins. Am J Physiol 1996; 271: H1907–11
- Suga S., Itoh H., Komatsu Y., Ogawa Y., Hama N., Yoshimasa T., et al. Cytokine‐induced C‐type natriuretic peptide (CNP) secretion from vascular endothelial cells—evidence for CNP as a novel autocrine/paracrine regulator from endothelial cells. Endocrinology 1993; 133: 3038–41
- Chauhan S. D., Nilsson H., Ahluwalia A., Hobbs A. J. Release of C‐type natriuretic peptide accounts for the biological activity of endothelium‐derived hyperpolarizing factor. Proc Natl Acad Sci U S A 2003; 100: 1426–31
- Villar I. C., Panayiotou C. M., Sheraz A., Madhani M., Scotland R. S., Nobles M., et al. Definitive role for natriuretic peptide receptor‐C in mediating the vasorelaxant activity of C‐type natriuretic peptide and endothelium‐derived hyperpolarising factor. Cardiovasc Res 2007; 74: 515–25
- Ahluwalia A., Hobbs A. J. Endothelium‐derived C‐type natriuretic peptide: more than just a hyperpolarizing factor. Trends Pharmacol Sci 2005; 26: 162–7
- Sandow S. L., Tare M. C‐type natriuretic peptide: a new endothelium‐derived hyperpolarizing factor?. Trends Pharmacol Sci 2007; 28: 61–7
- Leuranguer V., Verbeuren T., Félétou M. C‐type natriuretic peptide (CNP) is not an EDHF in the guinea‐pig carotid artery. Fundam Clin Pharmacol 2007; 21: S1
- Shepherd J. T., Vanhoutte P. M. The Human Cardiovascular System. Facts and Concepts. Raven Press, New York 1979
- Berne R. M. The role of adenosine in the regulation of coronary blood flow. Circ Res 1980; 47: 807–13
- Garland C. J., Plane F. Relative importance of endothelium‐derived hyperpolarizing factor for the relaxation of vascular smooth muscle in different arterial beds. Endothelium‐Derived Hyperpolarizing Factor, P. M Vanhoutte. Harwood Academic Publishers, Amsterdam 1996; 173–9
- Gluais P., Edwards G., Weston A. H., Falck J. R., Vanhoutte P. M., Félétou M. SKCa and IKCa in the endothelium‐dependent hyperpolarization of the guinea‐pig isolated carotid artery. Br J Pharmacol 2005; 144: 477–85
- Neylon C. B., Lang R. J., Fu Y., Bobik A., Reinhart P. H. Molecular cloning and characterization of the intermediate‐conductance Ca(2+)‐activated K(+) channel in vascular smooth muscle: relationship between K(Ca) channel diversity and smooth muscle function. Circ Res 1999; 85: 33–43
- Quignard J. F., Félétou M., Edwards G., Duhault J., Weston A. H., Vanhoutte P. M. Role of endothelial cells hyperpolarization in EDHF‐mediated responses in the guinea‐pig carotid artery. Br J Pharmacol 2000; 129: 1103–12
- Köhler R., Degenhardt C., Kühn M., Runkel N., Paul M., Hoyer J. Expression and function of endothelial Ca2+‐activated K+ channels in human mesenteric artery—A single cell reverse transcriptase‐polymerase chain reaction and electrophysiological study in situ. Circ Res 2000; 87: 496–503
- Burnham M. P., Bychkov R., Félétou M., Richards G. R., Vanhoutte P. M., Weston A. H., et al. Characterization of an apamin‐sensitive small conductance Ca2+‐activated K+ channel in porcine coronary artery endothelium: relevance to EDHF. Br J Pharmacol 2002; 135: 1133–43
- Bychkov R., Burnham M. P., Richards G. R., Edwards G., Weston A. H., Félétou M., et al. Characterization of a Charybdotoxin‐Sensitive Intermediate Conductance Ca2+‐Activated K+ Channel in Porcine Coronary Endothelium: Relevance to EDHF. Br J Pharmacol 2002; 138: 1346–54
- Taylor M. S., Bonev A. D., Gross T. P., Eckman D. M., Brayden J. E., Bond C. T., et al. Altered expression of small‐conductance Ca2+‐activated K+ (SK3) channels modulate arterial tone and blood pressure. Circ Res 2003; 93: 124–31
- Si H., Heyken W. T., Wolfle S. E., Tysiac M., Schubert R., Grgic I., et al. Impaired Endothelium‐Derived Hyperpolarizing Factor‐Mediated Dilations and Increased Blood Pressure in Mice Deficient of the Intermediate‐Conductance Ca2+‐Activated K+ Channel. Circ Res 2006; 99: 537–44
- Shinde U. A., Desai K. M., Yu C., Gopalakrishnan V. Nitric oxide synthase inhibition exaggerates the hypotensive response to ghrelin: role of calcium‐activated potassium channels. J Hypertens 2005; 23: 779–84
- Desai K. M., Gopalakrishnan V., Hiebert L. M., McNeill J. R., Wilson T. W. EDHF‐mediated rapid restoration of hypotensive response to acetylcholine after chronic, but not acute, nitric oxide synthase inhibition in rats. Eur J Pharmacol 2006; 546: 120–6
- Parkington H. C., Chow J. A., Evans R. G., Coleman H. A., Tare M. Role for endothelium‐derived hyperpolarizing factor in vascular tone in rat mesenteric and hindlimb circulations in vivo. J Physiol 2002; 542: 929–37
- Sandow S. L., Hill C. E. Incidence of myo‐endothelial gap junctions in the proximal and distal mesenteric arteries of the rat is suggestive of a role in endothelium‐derived hyperpolarizing factor‐mediated responses. Circ Res 2000; 86: 341–6
- Hwa J. J., Ghibaudi L., Williams P., Chaterjee M. Comparison of acetylcholine‐dependent relaxation in large and small arteries of rat mesenteric vascular bed. Am J Physiol 1994; 266: H952–8
- Shimokawa H., Yasutake H., Fujii K., Owada M. K., Nakaike R., Fukumoto Y., et al. The importance of the hyperpolarizing mechanism increases as the vessel size decrease in endothelium‐dependent relaxations in rat mesenteric circulation. J Cardiovasc Pharmacol 1996; 28: 703–11
- Sandow S. L., Tare M., Coleman H. A., Hill C. E., Parkington H. C. Involvement of myoendothelial gap junctions in the action of endothelium‐derived hyperpolarizing factor. Circ Res 2002; 90: 1108–13
- Dora K. A., Sandow S., Gallagher N. T., Takano H., Rummery N. M., Hill C. E., et al. Myoendothelial gap junctions may provide the pathway for EDHF in mouse mesenteric artery. J Vasc Res 2003; 40: 480–90
- Dora K. A., Doyle M. P., Duling B. R. Elevation of intracellular calcium in smooth muscle causes endothelial cell generation of NO in arterioles. Proc Natl Acad Sci U S A 1997; 94: 6529–34
- Yashiro Y., Duling B. R. Integrated Ca(2+) signaling between smooth muscle and endothelium of resistance vessels. Circ Res 2000; 87: 1048–54
- Beny J. L., Pacicca C. Bidirectional electrical communication between smooth muscle and endothelial cells in the pig coronary artery. Am J Physiol 1994; 266: H1465–72
- Marchenko S. M., Sage S. O. Smooth muscle cells affect endothelial membrane potential in rat aorta. Am J Physiol 1994; 267: H804–11
- Yamamoto Y., Fukuta H., Nakahira Y., Suzuki H. Blockade by 18β‐glycyrrhetinic acid of intercellular electrical coupling in guinea‐pig arterioles. J Physiol 1998; 511: 501–18
- Yamamoto Y., Imaeda K., Suzuki H. Endothelium‐dependent hyperpolarization and intercellular electrical coupling in guinea‐pig mesenteric arterioles. J Physiol 1999; 514: 505–13
- Emerson G. G., Segal S. S. Electrical coupling between endothelial cells and smooth muscle cells in hamster feed arteries: role in vasomotor control. Circ Res 2000; 87: 474–9
- Emerson G. G., Segal S. S. Endothelial cell pathway for conduction of hyperpolarization and vasodilation along hamster feed artery. Circ Res 2000; 86: 94–100
- Coleman H. A., Tare M., Parkington H. C. EDHF is not K+ but may be due to spread of current from the endothelium in guinea‐pig arterioles. Am J Physiol 2001; 280: H2478–83
- Griffith T. M. Endothelium‐dependent smooth muscle hyperpolarization: do gap junctions provide a unifying hypothesis?. Br J Pharmacol 2004; 141: 881–903
- Lang N. N., Luksha L., Newby D., Kublickiene K. Connexin 43 Mediates Endothelium‐Derived Hyperpolarising Factor Induced Vasodilatation in Subcutaneous Resistance Arteries from Healthy Pregnant Women. Am J Physiol Heart Circ Physiol 2006; 292: H1026–32
- Haddock R. E., Grayson T. H., Brackenbury T. D., Meaney K. R., Neylon C. B., Sandow S. L., et al. Endothelial coordination of cerebral vasomotion via myoendothelial gap junctions containing connexins 37 and 40. Am J Physiol Heart Circ Physiol 2006; 291: H2047–56
- Matchkov V. V., Rahman A., Bakker L. M., Griffith T. M., Nilsson H., Aalkjaer C. Analysis of effects of connexin‐mimetic peptides in rat mesenteric small arteries. Am J Physiol Heart Circ Physiol 2006; 291: H357–67
- Chaytor A. Y., Evans W. H., Griffith T. M. Central role of heterocellular gap junction communication in endothelium‐dependent relaxations of rabbit arteries. J Physiol 1998; 508: 561–73
- Chaytor A. Y., Martin P. E., Edwards D. H., Griffith T. M. Gap junctional communication underpins EDHF‐type relaxations evoked by ACh in the rat hepatic artery. Am J Physiol 2001; 280: H2441–50
- Dora K. A., Martin P. E., Chaytor A. T., Evans W. H., Garland C. J., Griffith T. M. Role of heterocellular Gap junctional communication in endothelium‐dependent smooth muscle hyperpolarization: inhibition by a connexin‐mimetic peptide. Biochem Biophys Res Commun 1999; 254: 27–31
- Edwards G., Félétou M., Gardener M. J., Thollon C., Vanhoutte P. M., Weston A. H. Role of gap junctions in the responses to EDHF in rat and guinea‐pig small arteries. Br J Pharmacol 1999; 128: 1788–94
- Edwards G., Thollon C., Gardener M. J., Félétou M., Vilaine J. P., Vanhoutte P. M., et al. Role of gap junctions and EETs in endothelium‐dependent hyperpolarization of porcine coronary artery. Br J Pharmacol 2000; 129: 1145–62
- Berman R. S., Martin P. E. M., Evans W. H., Griffith T. M. Relative contributions of NO and gap junctional communication to endothelium‐dependent relaxations of rabbit resistance arteries vary with vessel size. Microvasc Res 2002; 63: 115–28
- De Vriese A. S., Van de Voorde J., Lameire N. H. Effects of connexin‐mimetic peptides on nitric oxide synthase‐ and cyclooxygenase‐independent renal vasodilatation. Kidney Int 2002; 61: 177–85
- Mather S., Dora K. A., Sandow S. L., Winter P., Garland C. J. Rapid endothelial cell‐selective loading of connexin 40 antibody blocks endothelium‐derived hyperpolarizing factor dilation in rat small mesenteric arteries. Circ Res 2005; 97: 399–407
- Sandow S. L., Neylon C. B., Chen M. X., Garland C. J. Spatial separation of endothelial small‐ and intermediate‐conductance calcium‐activated potassium channels (K(Ca)) and connexins: possible relationship to vasodilator function?. J Anat 2006; 209: 689–98
- Hendrickx H., Casteels R. Electrogenic sodium pump in arterial smooth muscle cells. Pflugers Arch 1974; 346: 299–306
- Nelson M. T., Quayle J. M. Physiological roles and properties of potassium channels in arterial smooth muscle. Am J Physiol 1995; 268: C799–822
- Prior H. M., Webster N., Quinn K., Beech D. J., Yates M. S. K(+)‐induced dilation of a small renal artery: no role for inward rectifier K+ channels. Cardiovasc Res 1998; 37: 780–90
- Knot H. J., Zimmermann P. A., Nelson M. T. Extracellular K(+)‐induced hyperpolarizations and dilatations of rat coronary and cerebral arteries involve inward rectifier K(+) channels. J Physiol 1996; 492: 419–30
- Edwards G., Dora K. A., Gardener M. J., Garland C. J., Weston A. H. K+ is an endothelium‐derived hyperpolarizing factor in rat arteries. Nature 1998; 396: 269–72
- Edwards G., Gardener M. J., Félétou M., Brady G., Vanhoutte P. M., Weston A. H. Further investigation of endothelium‐derived hyperpolarizing factor (EDHF) in rat hepatic artery: studies using 1‐EBIO and ouabain. Br J Pharmacol 1999; 128: 1064–70
- Dora K. A., Garland C. J. Properties of smooth muscle hyperpolarization and relaxation to K+ in the rat isolated mesenteric artery. Am J Physiol 2001; 280: H2424–9
- McGuire J. J., Hollenberg M. D., Andrade‐Gordon P., Triggle C. R. Multiple mechanisms of vascular smooth muscle relaxation by the activation of proteinase‐activated receptor 2 in mouse mesenteric arterioles. Br J Pharmacol 2002; 135: 155–69
- Beny J. L., Schaad O. An evaluation of potassium ions as endothelium‐derived hyperpolarizing factor in porcine coronary arteries. Br J Pharmacol 2000; 131: 965–73
- Nelli S., Wilson W. S., Laidlaw H., Llano A., Middleton S., Price A. G., et al. Evalauation of potassium ions as the endothelium‐derived hyperpolarizing factor (EDHF) in the bovine coronary artery. Br J Pharmacol 2003; 139: 982–8
- Randriamboavonjy V., Busse R., Fleming I. 20‐HETE‐induced contraction of small coronary arteries depends on the activation of Rho‐kinase. Hypertension 2003; 41: 801–6
- Büssemaker E., Wallner C., Fisslthaler B., Fleming I. The Na+K+‐ATPase is a target for an EDHF displaying characteristics similar to potassium ions in the porcine renal interlobar arteries. Br J Pharmacol 2002; 137: 647–54
- Büssemaker E., Popp R., Binder J., Busse R., Fleming I. Characterization of the endothelium‐derived hyperpolarizing factor (EDHF) response in the human interlobar artery. Kidney Int 2003; 63: 1749–55
- Richards G. R., Weston A. H., Burnham M. P., Félétou M., Vanhoutte P. M., Edwards G. Suppression of K(+)‐induced hyperpolarization by phenylephrine in rat mesenteric artery: relevance to studies of endothelium‐derived hyperpolarizing factor. Br J Pharmacol 2001; 134: 1–5
- Bradley K. K., Jaggar J. H., Bonev A. D., Heppner T. J., Flynn E. R., Nelson M. T., et al. Kir2.1 encodes the inward rectifier potassium channel in rat arterial smooth muscle cells. J Physiol 1999; 515: 639–51
- Zaritsky J. J., Eckman D. M., Wellman G. C., Nelson M. T., Schwarz T. L. Targeted disruption of Kir2.1 and Kir2.2 genes reveal the essential role of the inwardly rectifying K+ current in K+‐mediated vasodilation. Circ Res 2000; 87: 160–6
- Edwards G., Richards G. R., Gardener M. J., Félétou M., Vanhoutte P. M., Weston A. H. Role of the inward‐rectifier K+ channel and Na+/K+‐ATPase in the hyperpolarization to K+ in rat mesenteric arteries. EDHF 2002, P. M Vanhoutte. Taylor & Francis, London 2003; 309–17
- Quignard J. F., Félétou M., Duhault J., Vanhoutte P. M. Potassium ions as endothelium‐derived hyperpolarizing factors in the isolated carotid artery of the guinea‐pig. Br J Pharmacol 1999; 127: 27–34
- Dong H., Jiang Y., Cole W. C., Triggle C. R. Comparison of the pharmacological properties of EDHF‐mediated vasorelaxation in the guinea‐pig cerebral and mesenteric resistance vessels. Br J Pharmacol 2000; 130: 1983–91
- Brandes R. P., Schmitz‐Winnenthal F. H., Félétou M., Gödecke A., Huang P. L., Vanhoutte P. M., et al. An endothelium‐derived hyperpolarizing factor distinct from NO and prostacyclin is a major endothelium‐dependent vasodilator in resistance vessels of wild type and endothelial NO synthase knock‐out mice. Proc Natl Acad Sci U S A 2000; 97: 9747–52
- Wu Y., Huang A., Sun D., Falck J. R., Koller A., Kaley G. Gender‐specific compensation for the lack of NO in the mediation of flow‐induced arteriolar dilation. Am J Physiol Heart Circ Physiol 2001; 280: H2456–61
- Scotland R. S., Madhani M., Chauhan S., Moncada S., Andresen J., Nilsson H., et al. Investigation of vascular responses in endothelial nitric oxide synthase/cyclooxygenase‐1 double knock‐out mice. Key role for endothelium‐derived hyperpolarizing factor in the regulation of blood pressure in vivo. Circulation 2005; 111: 796–803
- Bond C. T., Sprengel R., Bissonnette J. M., Kaufmann W. A., Pribnow D., Neelands T., et al. Respiratory and parturition affected by conditional overexpression of the Ca2+‐activated K+ channel subunit, SK3. Science 2000; 289: 1942–6
- Simon A. M., McWhorter A. R. Decreased intercellular dye‐transfer and downregulation of non‐ablated connexins in aortic endothelium deficient in connexin37 or connexin40. J Cell Sci 2003; 116: 2223–36
- Wagner C., de Wit C., Kurtz L., Grunberger C., Kurtz A., Schweda F. Connexin40 is essential for the pressure control of renin synthesis and secretion. Circ Res 2007; 100: 556–63
- Simon A. M., McWhorter A. R. Vascular abnormalities in mice lacking the endothelial gap junction protein connexin37 and connexin40. Dev Biol 2002; 251: 206–20
- De Wit C., Roos F., Bolz S. S., Kirchhoff S., Kruge O., Willecke K., et al. Impaired conduction of vasodilatation along arterioles in connexin 40‐deficient mice. Circ Res 2000; 86: 649–55
- De Wit C., Roos F., Bolz S. S., Pohl U. Lack of vascular connexin 40 is associated with hypertension and irregular arteriolar vasomotion. Physiol Genomics 2003; 13: 169–77
- Figueroa X. F., Paul D. L., Simon A. M., Goodenough D. A., Day K. H., Damon D. N., et al. Central role of connexin 40 in the propagation of electrically activated vasodilation in mouse cremasteric arterioles in vivo. Circ Res 2003; 92: 793–800
- Theis M., de Wit C., Shlaeger T. M., Eckardt D., Kruger O., Doring B., et al. Endothelium‐specific replacement of the connexin 43 coding region by a lacZ reporter gene. Genesis 2001; 29: 1–13
- Liao Y., Day K. H., Damon D. N., Duling B. R. Endothelial cell‐specific knockout of connexin 43 causes hypotension and bradycardia in mice. Proc Natl Acad Sci U S A 2001; 98: 9989–94
- Woolfson R. G., Poston L. Effect of NG‐monomethyl‐L‐arginine on endothelium‐dependent relaxation of human subcutaneous resistance arteries. Clin Sci London 1990; 79: 273–8
- Urakami‐Harasawa L., Shimokawa H., Nakashima M., Egashira K., Takeshita A. Importance of endothelium‐derived hyperpolarizing factor in human arteries. J Clin Invest 1997; 100: 2793–9
- Miura H., Gutterman D. D. Human coronary arteriolar dilation to arachidonic acid depends on cytochrome P450 monooxygenase and Ca2+‐activated K+ channels. Circ Res 1998; 83: 501–7
- Miura H., Wachtel R. E., Liu Y., Loberiza F. R., Jr., Saito T., Miura M., et al. Flow‐induced dilation of human coronary arterioles: important role of Ca(2+)‐activated K(+) channels. Circulation 2001; 103: 1992–8
- Coats P., Johnston F., MacDonald J., McMurray J. J., Hillier C. Endothelium‐derived hyperpolarizing factor: identification and mechanisms of action in human subcutaneous resistance arteries. Circulation 2001; 103: 1702–8
- Archer S. L., Gragasin F. S., Wu X., Wang S., McMurthry S., Kim D. H., et al. Endothelium‐derived hyperpolarizing factor in human internal mammary artery is 11,12‐epoxieicosatrienoic acid and causes relaxation by activating BK(Ca) channels. Circulation 2003; 107: 769–76
- Luksha L., Nisell H., Kublickiene K. The mechanism of EDHF‐mediated responses in subcutaneous small arteries of healthy pregnant women. Am J Physiol 2004; 286: R1102–9
- Torondel B., Vila J. M., Segarra G., Lluch P., Medina P., Martinez‐Leon J., et al. Endothelium‐dependent responses in human isolated thyroid arteries from donors. J Endocrinol 2004; 181: 379–84
- Garcha R. S., Hughes A. D. CNP, but not ANP or BNP, relax human isolated subcutaneous resistance arteries by an action involving cyclic GMP and BKCa channels. J Renin Angiotensin Aldosterone Syst 2006; 7: 87–91
- Stanke‐Labesque F., Devillier P., Bedouch P., Cracowski J. L., Chavanon O., Bessard G. Angiotensin II‐induced contractions in human internal mammary artery: effects of cyclooxygenase and lipoxygenase inhibition. Cardiovasc Res 2000; 47: 376–83
- Tagawa H., Shimokawa H., Tagawa T., Kuroiwa‐Matsumoto M., Hirooka Y., Takeshita A. Short‐term estrogen augments both nitric oxide‐mediated and non‐nitric oxide‐mediated endothelium‐dependent vasodilation in postmenopausal women. J Cardiovasc Pharmacol 1997; 30: 481–8
- Honing M. L., Smits P., Morrison P. J., Rabelink T. J. Bradykinin‐induced vasodilatation of human forearm resistance vessels is primarily mediated by endothelium‐dependent hyperpolarization. Hypertension 2000; 35: 1314–8
- Katz S. D., Krum H. Acetylcholine‐mediated vasodilatation in the forearm circulation of patients with heart failure: indirect evidence for the role of endothelium‐derived hyperpolarizing factor. Am J Physiol 2001; 87: 1089–92
- Halcox J. P., Narayanan S., Cramer‐Joyce L., Mincemoyer R., Quyyumi A. A. Characterization of endothelium‐derived hyperpolarizing factor in the human forearm microcirculation. Am J Physiol 2001; 280: H2470–7
- Inokuchi K., Hirooka Y., Shimokawa H., Sakai K., Kishi T., Ito K., et al. Role of endothelium‐derived hyperpolarizing factor in human forearm circulation. Hypertension 2003; 42: 919–24
- Bellien J., Joannides R., Iacob M., Arnaud P., Thuillez C. Calcium‐activated potassium channels and NO regulate human peripheral conduit artery mechanics. Hypertension 2005; 46: 210–6
- Bellien J., Joannides R., Iacob M., Arnaud P., Thuillez C. Evidence for a basal release of a cytochrome‐related endothelium‐derived hyperpolarizing factor in the radial artery in humans. Am J Physiol Heart Circ Physiol 2006; 290: H1347–52
- Bellien J., Iacob M., Gutierrez L., Isabelle M., Lahary A., Thuillez C., et al. Crucial role of NO and endothelium‐derived hyperpolarizing factor in human sustained conduit artery flow‐mediated dilatation. Hypertension 2006; 48: 1088–94
- Hillig T., Krustrup P., Fleming I., Osada T., Saltin B., Hellsten Y. Cytochrome P450 2C9 plays an important role in the regulation of exercise‐induced skeletal muscle blood flow and oxygen uptake in humans. J Physiol 2003; 546: 307–14
- Taddei S., Versari D., Cipriano A., Ghiadoni L., Galetta F., Franzoni F., et al. Identification of a cytochrome P450 2C9‐derived endothelium‐derived hyperpolarizing factor in essential hypertensive patients. J Am Coll Cardiol 2006; 48: 508–15
- Passauer J., Bussemaker E., Lassig G., Pistrosch F., Fauler J., Gross P., et al. Baseline blood flow and bradykin‐induced vasodilator responses in the human forearm are insensitive to the cytochrome P450 2C9 (CYP2C9) inhibitor sulphaphenazole. Clin Sci 2003; 105: 513–8
- Passauer J., Pistrosch F., Lassig G., Herbrig K., Bussemaker E., Gross P., et al. Nitric oxide‐ and EDHF‐mediated arteriolar tone in uremia is unaffected by selective inhibition of vascular cytochrome P450 2C9. Kidney Int 2005; 67: 1907–12
- Fichtlscherer S., Dimmeler S., Breuer S., Busse R., Zeiher A. M., Fleming I. Inhibition of cytochrome P450 2C9 improves endothelium‐dependent, nitric oxide‐mediated vasodilatation in patients with coronary diseases. Circulation 2004; 109: 178–83
- Fleming I., Michaelis U. R., Bredenkotter D., Fisslthaler B., Dehghani F., Brandes R. P., et al. Endothelium‐derived hyperpolarizing factor synthase (cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circ Res 2001; 88: 44–51
- Alvarez J., Montero M., Garcia‐Sancho J. High affinity inhibition of Ca2+‐dependent K+ channels by cytochrome P‐450 inhibitors. J Biol Chem 1992; 267: 11789–93
- Félétou M., Vanhoutte P. M. EDHF: new therapeutic targets?. Pharmacol Res 2004; 49: 565–80
- Taddei S., Virdis A., Ghiadoni L., Sudano I., Salvetti A. Endothelial dysfunction in hypertension. J Cardiovasc Pharmacol 2001; 38: S11–4
- Kenny L. C., Baker P. N., Kendall D. A., Randall M. D., Dunn W. R. Different mechanisms of endothelial‐dependent vasodilator responses in human myometrial small arteries in normal pregnancy and pre‐eclampsia. Clin Sci 2002; 103: 67–73
- Selemidis S., Cocks T. M. Endothelium‐dependent hyperpolarization as a remote anti‐atherogenic mechanism. Trends Pharmacol Sci 2002; 23: 213–20
- Brandes R. P., Behra A., Lebherz C., Boger R. H., Bode‐Boger S. M., Phivthong‐Ngam L., et al. N(G)‐nitro‐L‐arginine‐ and indomethacin‐resistant endothelium‐dependent relaxation in the rabbit renal artery: effect of hypercholestorelemia. Atherosclerosis 1997; 135: 49–55
- Kagota S., Tamashiro A., Yamaguchi Y., Nakamura K., Kunimoto M. Excessive salt or cholesterol intake alters the balance among endothelium‐derived factors released from renal arteries in spontaneously hypertensive rats. J Cardiovasc Pharmacol 1999; 34: 533–9
- Morikawa K., Matoba T., Kubota H., Hatanaka M., Fujiki T., Takahashi S., et al. Influence of diabetes mellitus, hypercholesterolemia, and their combination on EDHF‐mediated responses in mice. J Cardiovasc Pharmacol 2005; 45: 485–90
- Wolfle S. E., de Wit C. Intact endothelium‐dependent dilation and conducted responses in resistance vessels of hypercholesterolemic mice in vivo. J Vasc Res 2005; 42: 475–82
- Ding H., Hashem M., Wiehler W. B., Lau W., Martin J., Reid J., et al. Endothelial dysfunction in the streptozotocin‐induced diabetic apoE‐deficient mouse. Br J Pharmacol 2005; 146: 1110–8
- Krummen S., Falck J. R., Thorin E. Two distinct pathways account for EDHF‐dependent dilatation in the gracilis artery of dyslipidaemic hApoB+/+ mice. Br J Pharmacol 2005; 145: 264–70
- De Vriese A. S., Verbeuren T. J., Van de Voorde J., Lameire N. H., Vanhoutte P. M. Endothelial dysfunction in diabetes. Br J Pharmacol 2000; 130: 963–74
- Matsumoto T., Kobayashi T., Wakabayashi K., Kamata K. Cilostazol improves endothelium‐derived hyperpolarizing factor‐type relaxation in mesenteric arteries from diabetic rats. Am J Physiol Heart Circ Physiol 2005; 289: H1933–40
- Wulff H., Miller M. J., Haensel W., Grissner S., Cahalan M. D., Chandy K. G. Design of potent and selective inhibitor of the intermediate‐conductance Ca2+‐activated K+ channel, IKCa1: a potential immunosuppressant. Proc Natl Acad Sci U S A 2000; 97: 8151–6
- Liegeois J. F., Mercier F., Graulich A., Graulich‐Lorge F., Scuvee‐Moreau J., Setin V. Modulation of small conductance calcium‐activated potassium (SK) channels: a new challenge in medicinal chemistry. Curr Med Chem 2003; 10: 625–47
- Corriu C., Félétou M., Puybasset L., Bea M. L., Berdeaux A., Vanhoutte P. M. Endothelium‐dependent hyperpolarization in isolated arteries taken from animals treated with NO‐synthase inhibitors. J Cardiovasc Pharmacol 1998; 32: 944–50
- Félétou M., Vanhoutte P. M. Endothelium‐dependent hyperpolarisation of canine coronary smooth muscle. Br J Pharmacol 1988; 93: 515–24
- Coleman H. A., Tare M., Parkington H. C. K+ currents underlying the action of endothelium‐derived hyperpolarizing factor in guinea‐pig, rat and human blood vessels. J Physiol 2001; 531: 359–73
- Liu C., Ngai C. Y., Huang Y., Ko W. H., Wu M., He G. W., et al. Depletion of intracellular Ca2+ stores enhances flow‐induced vascular dilatation in rat small mesenteric artery. Br J Pharmacol 2006; 147: 506–15
- Sokoya E. M., Burns A. R., Setiawan C. T., Coleman H. A., Parkington H. C., Tare M. Evidence for the involvement of myoendothelial gap junctions in EDHF‐mediated relaxation in the rat middle cerebral artery. Am J Physiol Heart Circ Physiol 2006; 291: H385–93
- McNeish A. J., Dora K. A., Garland C. J. Possible role for K+ in endothelium‐derived hyperpolarizing factor‐linked dilatation in rat middle cerebral artery. Stroke 2005; 36: 1526–32
- McSherry I. N., Sandow S. L., Campbell W. B., Falck J. R., Hill M. A., Dora K. A. A role for heterocellular coupling and EETs in dilation of rat cremaster arteries. Microcirculation 2006; 13: 119–30
- Pannirselvam M., Ding H., Anderson T. J., Triggle C. R. Pharmacological characteristics of endothelium‐derived hyperpolarizing factor‐mediated relaxation of small mesenteric arteries from db/db mice. Eur J Pharmacol 2006; 551: 98–107
- Morikawa K., Shimokawa H., Matoba T., Kubota H., Akaike T., Talukder M. A., et al. Pivotal role of Cu,Zn‐superoxide dismutase in endothelium‐dependent hyperpolarization. J Clin Invest 2003; 112: 1871–9
- Dong H., Waldron G. J., Galipeau D., Cole W. C., Triggle C. R. NO/PGI2‐independent vasorelaxation and the cytochrome P450 pathway in rabbit carotid artery. Br J Pharmacol 1997; 120: 695–701
- Bauersachs J., Popp R., Hecker M., Sauer E., Fleming I., Busse R. Nitric oxide attenuates the release of endothelium‐derived hyperpolarizing factor. Circulation 1996; 94: 3341–7
- Griffith T. M., Chaytor A. T., Taylor H. J., Giddings B. D., Edwards D. H. cAMP facilitates EDHF‐type relaxations in conduit arteries by enhancing electrotonic conduction via gap junctions. Proc Natl Acad Sci U S A 2002; 99: 6392–7
- Zhang D. X., Gauthier K. M., Chawengsub Y., Campbell W. B. Acetylcholine‐induced relaxations of rabbit small mesenteric arteries: role of arachidonic acid metabolites and K+. Am J Physiol Heart Circ Physiol 2007 Mar 2, [Epub ahead of print]
- Ujiie H., Chaytor A. T., Bakker L. M., Griffith T. M. Essential role of Gap junctions in NO‐ and prostanoid‐independent relaxations evoked by acetylcholine in rabbit intracerebral arteries. Stroke 2003; 34: 544–50
- Matoba T., Shimokawa H., Morikawa K., Kubota H., Kunihiro I., Urakami‐Harasawa L., et al. Electron spin resonance detection of hydrogen peroxide as an endothelium‐derived hyperpolarizing factor in porcine coronary microvessels. Arterioscler Thromb Vasc Biol 2003; 23: 1224–30
- Randriamboavonjy V., Kiss L., Falck J. R., Busse R., Fleming I. The synthesis of 20‐HETE in small porcine coronary arteries antagonizes EDHF‐mediated relaxation. Cardiovasc Res 2005; 65: 487–94
- Nishikawa Y., Stepp D. W., Chilian W. M. In vivo location and mechanism of EDHF‐mediated vasodilation in canine coronary microcirculation. Am J Physiol 1999; 277: H1252–9
- Tanaka M., Kanatsuka H., Ong B. H., Tanikawa T., Uruno A., Komaru T., et al. Cytochrome P‐450 metabolites but not NO, PGI2, and H2O2 contribute to ACh‐induced hyperpolarization of pressurized canine coronary microvessels. Am J Physiol Heart Circ Physiol 2003; 285: H1939–48
- Drummond G. R., Selemidis S., Cocks T. M. Apamin‐sensitive, non‐nitric oxide (NO) endothelium‐dependent relaxations to bradykinin in the bovine isolated coronary artery: no role for cytochrome P450 and K+. Br J Pharmacol 2000; 129: 811–9
- Kenny L. C., Baker P. N., Kendall D. A., Randall M. D., Dunn W. R. The role of gap junctions in mediating endothelium‐dependent responses to bradykinin in myometrial small arteries isolated from pregnant women. Br J Pharmacol 2002; 136: 1085–8