Abstract
Over the past few years the field of innate immunity has undergone a revolution with the discovery of pattern recognition molecules (PRM) and their role in microbe detection. Among these molecules, the Nod‐like receptors (NLRs) have emerged as key microbial sensors that participate in the global immune responses to pathogens and contribute to the resolution of infections. This growing group of proteins is divided into subfamilies with basis in their different signaling domains. Prominent among them are Nod1, Nod2, Nalp3, Ipaf, and Naip that have been shown to play important roles against intracellular bacteria. Furthermore, mutations in the genes that encode these proteins have been associated with complex inflammatory disorders including Crohn's disease, asthma, familial cold urticaria, Muckle‐Wells syndrome, and Blau syndrome. In this review we will present the current knowledge on the role of these proteins in immunity and inflammatory diseases.
Introduction
Early control of microbial replication and successful eradication of an infection depend on rapid detection of the invading microbe and are essential for the survival of multicellular organisms Citation1. Because it does not require priming or the existence of cells with memory of earlier encounters with pathogens, the innate immune system provides the host with a rapid response that serves multiple functions such as pathogen detection, antimicrobial defense, and instruction of adaptive immune response. Such immediate activation is only possible because, in the innate immune system, highly conserved molecules expressed by microorganisms are recognized by a limited number of germ‐line‐encoded receptors, termed pattern recognition molecules (PRMs), which do not require somatic gene rearrangements, as is the case in the adaptive immune response with the generation of epitope‐specific antibodies. The microbial molecules detected by the PRMs are termed pathogen‐associated molecular patterns (PAMPs), and are usually vital for microbial survival and, therefore, unlikely to vary in their structures. Such molecules include parts of the bacterial cell envelope, such as lipopolysaccharide (LPS) and peptidoglycan (PG), and flagellin and microbial nucleic acids (viral RNA or bacterial DNA) Citation2.
The first family of proteins identified as PRMs was the Toll‐like receptors (TLRs) family Citation3. It has been estimated that most mammalian species have between 10 and 15 types of TLRs. Thirteen TLRs (named simply TLR1 to TLR13) have been identified in humans and mice together, and equivalent forms of many of these have been found in other mammalian species. However, equivalents of certain TLRs found in humans are not present in all mammals. For example, TLR10 is found in humans but not in mice. On the other hand, mice express TLRs 11, 12, and 13, none of which are present in humans Citation4. These proteins are transmembrane receptors that recognize PAMPs through leucine‐rich repeats (LRRs) present in their extracellular domains. The cytosolic domain of TLRs contains a Toll/interleukin‐1 receptor (TIR) interaction domain required for interaction with the TIR‐containing adaptor molecules MyD88, MyD88‐adaptor‐like (MAL (also known as TIR domain‐containing adaptor protein—TIRAP), TIR‐domain‐containing adaptor protein inducing IFNβ (TRIF), and TRIF‐related adaptor molecule TRAM. Once activated, TLRs signal through different intracellular signaling cascades, generally resulting in the activation of nuclear factor κB (NF‐κB) and activator protein 1 (AP‐1) in MyD88‐dependent pathways and/or type I interferons (IFNs) in TRIF‐dependent antiviral pathways Citation5.
Innate immunity is not only capable of detecting extracellular pathogens through TLRs and other membrane‐bound PRMs, but can also sense microbes in the cytoplasm with great efficiency. Indeed, having distinct possible locations of microbial sensing allows the host cell to cope with a broad array of infectious strategies. The largest known family of intracellular PRMs comprises the Nod‐like receptors (NLRs) Citation6. These proteins have high structural and functional homology to a subfamily of plant‐disease‐resistance proteins (R proteins) that recognize distinct effector molecules from pathogenic bacteria and elicit a defense response against pathogens, known as hypersensitivity response, that eventually leads to cell death Citation7. The NLRs share a characteristic tripartite structure: 1) a C‐terminus leucine‐rich repeat (LRR) domain that is assumed to be the sensing motif able to recognize conserved microbial patterns or other ligands; 2) a centrally located NACHT (Naip (neuronal apoptosis inhibitory protein), CIITA (MHC class II transcription activator), HET‐E (incompatibility locus protein from Podospora anserina) and TP1 (telomerase‐associated protein)) domain that mediates self‐oligomerization and is essential for activation of the NLRs; and 3) an N‐terminal effector domain, which can be a caspase recruitment domain (CARD, in the case of Nods and Ipaf), a pyrin domain (PYD, in the case of Nalps), or a baculovirus inhibitor of apoptosis protein repeat domain (BIR, in the case of Naip) and is responsible for homophilic interactions with other proteins containing domains of the same class, which results in triggering signal transduction (Figure ) Citation6.
Figure 1 NLR proteins are characterized by a tripartite structure: 1) a C‐terminal leucine‐rich repeat (LRR) domain that is involved in the sensing of their cognate ligands; 2) a centrally located NACHT domain, which mediates self‐oligomerization; and 3) at least one N‐terminal protein‐protein interaction domain that transduces signal upon NLR activation. Nalp1–14 display a pyrin domain (PYD), while Nod1 and 2 and Ipaf contain caspase recruitment and activation domains (CARD), and Naip presents three baculovirus inhibitor of apoptosis protein repeat domains (BIR). Notably, these proteins display a remarkable structural and functional homology to a subfamily of plant‐disease‐resistance proteins (R proteins), such as RPS2 from Arabidopsis and N protein from tobacco plants, demonstrating the high phylogenic conservations of NLRs in organisms from different kingdoms. NLR: Nod‐like receptors; NACHT: [Naip: neuronal apoptosis inhibitory protein; CIITA: MHC class II transcription activator; HET‐E: incompatibility locus protein from Podospora anserina; TP1: telomerase associated protein]; TIR: Toll/interleukin‐1 receptor; LRR: leucin‐rich repeat; RPS2 and LZ: no definition.
![Figure 1 NLR proteins are characterized by a tripartite structure: 1) a C‐terminal leucine‐rich repeat (LRR) domain that is involved in the sensing of their cognate ligands; 2) a centrally located NACHT domain, which mediates self‐oligomerization; and 3) at least one N‐terminal protein‐protein interaction domain that transduces signal upon NLR activation. Nalp1–14 display a pyrin domain (PYD), while Nod1 and 2 and Ipaf contain caspase recruitment and activation domains (CARD), and Naip presents three baculovirus inhibitor of apoptosis protein repeat domains (BIR). Notably, these proteins display a remarkable structural and functional homology to a subfamily of plant‐disease‐resistance proteins (R proteins), such as RPS2 from Arabidopsis and N protein from tobacco plants, demonstrating the high phylogenic conservations of NLRs in organisms from different kingdoms. NLR: Nod‐like receptors; NACHT: [Naip: neuronal apoptosis inhibitory protein; CIITA: MHC class II transcription activator; HET‐E: incompatibility locus protein from Podospora anserina; TP1: telomerase associated protein]; TIR: Toll/interleukin‐1 receptor; LRR: leucin‐rich repeat; RPS2 and LZ: no definition.](/cms/asset/5f998e0c-d251-495f-95a5-ce7bf2f3538f/iann_a_257472_f0001_b.jpg)
This variety of intracellular PRMs indicates that the mammalian cytosol is a rich environment for host‐pathogen interactions and is a site of active immune surveillance. Indeed, an intracellular detection system can fulfill various roles in the innate immune response: 1) in certain tissues, such as the intestinal epithelium, where expression of TLRs appears to be downregulated and/or cells expressing TLRs are compartmentalized Citation8, NLRs are crucial for initiating the innate immune response against invasive bacteria Citation1; 2) it serves as a backup system in the defense against certain pathogens that are capable of evading or minimizing extracellular innate immunity Citation9–11; and, finally, 3) NLRs can cooperate with other PRMs, such as the TLRs, to generate appropriate tailored responses for maximum effectiveness in resolving infection with different pathogens and, thus, providing substantial specificity to the innate immune response Citation12–15.
Here we will review the recent advances in our understanding on the role of NLRs in sensing intracellular pathogens and how this impacts the innate immune response to infection. In particular, we will focus on those NLRs whose involvement in microbial motifs detection has already been established.
Key messages
Nod‐like receptors (NLRs) are involved in microbial sensing and initiation of the immune response against various pathogens.
The activation of the NLRs result in NF‐κB activation or the assembly of the inflammasome.
Polymorphisms or mutations in human or mouse NLRs are associated with susceptibility to inflammatory disorders.
NLRs sense diverse microbial molecules
To date, only few among the more than 20 members of the NLR family have known ligands and, like the TLRs, these NLRs sense characteristic microbial products. However, as for TLRs, the mechanism involved in PAMP recognition by NLRs has not yet been characterized and, to date, no direct interaction has been demonstrated. It is therefore possible that the recognition is indirect and involves other proteins.
Nod1 and Nod2 were the first NLRs identified as sensors of microbial products Citation16–19. Both proteins detect muropeptide monomers released from bacterial PG. PG is the major constituent of the cell wall of Gram‐positive bacteria, while in Gram‐negative bacteria, it is found as a thin layer in the periplasmic space. PG is responsible for providing shape and mechanical rigidity to bacteria and is composed of glycan chains that contain alternating N‐acetylglucosamine (GlcNAc) and N‐acetylmuramic acid (MurNAc) sugars cross‐linked to each other by short peptides. Apart from the thickness and the degree of stem peptide cross‐linking, an important difference between Gram‐positive and Gram‐negative PG resides in the nature of the third amino acid of the stem peptides. In Gram‐positive bacteria, the amino acid is commonly a lysine, whereas a diaminopimelic acid (DAP) is found in most Gram‐negative bacteria. During the bacterial life cycle, PG is constantly degraded by specific hydrolases, and newly synthesized subunits are integrated into the polymeric structure, allowing biological processes such as cell division. PG degradation leads to the formation of muropeptides that have strong immunomodulatory properties Citation20, Citation21.
The recognition of PG by PRMs has been shown to be an important feature in the innate immunity of flies and mammals. In mammals, TLR2 was the first PRM implicated in PG recognition Citation21. However, it has been recently demonstrated that TLR2 does not detect PG but likely detects lipoteichoic acid or lipoprotein contaminants found in commercial PG preparations, which would make Nod proteins the only known receptors for PG products in mammals Citation22. This matter, however, is not yet completely resolved since subsequent studies from Dziarski and colleagues showed that purified PG from Staphylococcus aureus induces NF‐κB through TLR2. The authors showed that, although insoluble PG and disaccharide‐peptide PG fragments generated by muramidase digestion are inactive or have greatly reduced activity, the polymeric PG or the solubilized PG does activate the cell in a TLR2‐dependent manner Citation23.
Nod1 and Nod2, however, recognize different moieties of bacterial PG. The muropeptides GlcNAc‐MurNAc‐L‐Ala‐D‐Glu‐meso‐DAP (GM‐triDAP) and GlcNAc‐MurNAc‐L‐Ala‐D‐Glu‐meso‐DAP‐D‐Ala (GM‐tetraDAP) have been identified as the naturally occurring products sensed by human and mouse Nod1, respectively, and the minimal structure detected by Nod1 is the dipeptide D‐Glu‐meso‐DAP Citation16, Citation17, Citation24. In contrast with these pioneer studies, Uehara and colleagues have recently shown that meso‐DAP itself can activate human epithelial cells through Nod1 to secrete antibacterial factors and cytokines Citation25. The reasons for such discrepancy are yet to be determined. Interestingly, the recognition of bacterial PG by Nod1 is dependent on the presence of the meso‐DAP, an amino acid absent from eukaryotes and that, therefore, represents a very simple signature of bacterial origin. The presence of meso‐DAP is characteristic of most Gram‐negative bacteria plus some Gram‐positive bacteria, such as Bacillus spp. Citation26. In contrast, Nod2 has been implicated as a general sensor for both Gram‐positive and Gram‐negative PGs, since biochemical and functional analyses have identified the muramyl dipeptide MurNAc‐L‐Ala‐D‐isoGln (MDP), the minimal motif in all PGs, as the essential structure recognized by Nod2 Citation18, Citation19. Again, the MDP motif is generally not found as a naturally occurring bacterial product, but the corresponding muropeptide, GlcNAc‐MurNAc‐L‐Ala‐D‐isoGln (GM‐Di), does exist.
Nalp3 seems to respond to multiple ligands, including bacterial RNA and imidazoquinolone compounds Citation27, dsRNA from viruses Citation28, uric acid Citation29, and K+ cellular efflux Citation30, which can arise from the insertion of a bacterial toxin through the plasma membrane Citation31. The fact that Nalp3 seems to be activated in response to uric acid, a host molecule released into the extracellular milieu by necrotic cells and by potassium efflux, suggests that this NLR may trigger the detection of danger signals, in addition to microbe‐derived stimuli. Exactly how Nalp3 is able either to sense these structurally disparate signals directly or to integrate signals from widely differing pathways is yet to be determined.
Finally, several groups have recently demonstrated that both Naip5 and Ipaf sense bacterial flagellin in a TLR5‐independent manner Citation32–34. It is still unclear if either Ipaf or Naip5 are the primary sensors for surveillance of cytosolic flagellin, but these two proteins coprecipitate suggesting that they might act in complex Citation35.
NLRs signaling: NF‐κB activation and inflammasome assembly
The NLRs are normally present in the cytoplasm in an inactive self‐repressed form. The current model suggests that the LRR domains fold back onto the NACHT domain, thereby inhibiting spontaneous oligomerization and activation of the NLR protein. Following direct or indirect binding of the ligand to the LRR, the molecule undergoes a conformational rearrangement, exposing the NACHT domain and thereby allowing oligomerization that eventually results in the exposure of the effector N‐terminal domain. The CARD and PYD domains of Nods and Nalps, respectively, normally link these proteins to downstream adaptor or effector proteins, such as Rip2 or inflammatory caspases, through homotypic interactions connecting CARD‐CARD or PYD‐PYD, similar to the manner in which the TIR domain links the TLRs to downstream TIR‐containing adaptors Citation36. Once activated, NLRs mediate proinflammatory signaling through two major pathways: activation of nuclear transcription factor (NF‐κB) and of the caspase‐1 inflammasome.
The consequences of the activation of Nod proteins seem to be largely dependent on NF‐κB activation. NF‐κB, a heterodimeric transcription factor, is a crucial component of the inflammatory response and is involved in the regulation of genes encoding proinflammatory cytokines, adhesion molecules, chemokines, growth factors, and inducible enzymes such as cyclooxygenase 2 (COX2) and inducible nitric oxide synthase (iNOS) Citation37. Recently, through the analysis of global gene expression changes using cDNA microarray, Masumoto et al. demonstrated that a great number of genes induced following Nod1 activation are downstream targets of NF‐κB. Such genes included molecules involved in innate immune responses (CXCL1, MBL2, and CD83), signaling molecules of TLR and interleukin‐1 (IL‐1) pathways (MAP3K7/TAK1 and MAIL/Iκ‐Bζ), components of the ubiquitin/proteasome pathway (UBE2M, UBE2D3, RYBP, and WSB1), and negative feedback regulators of inflammatory and apoptotic pathways (NFKBIA/IκBα, ATF3, TNFAIP3/A20, CFLAR/CLARP, and BIRC3/c‐IAP2) Citation38.
Nod1 and Nod2 have been shown to interact with and use a common downstream molecule, the CARD‐containing adaptor protein Rip2 (also known as RICK or CARDIAK), to activate what appears to be identical signaling pathways. PG sensing is followed by oligomerization of Nod proteins, which in turn is sufficient to recruit Rip2 Citation39. Kobayashi et al. demonstrated that Nod1‐ or Nod2‐dependent NF‐κB activation is abolished in Rip2‐deficient mouse embryo fibroblasts but restored after exogenous addition of Rip2, supporting the role of Rip2 in the signaling pathways downstream of Nod1 and Nod2 Citation40. Rip2 then interacts with IKKγ (also known as NEMO), the regulatory subunit of the IκB kinase (IKK) complex, thereby linking these proteins to the IKK complex (Figure ). Recently, Silva‐Correia et al. proposed that Rip2 is also required for Nod1‐dependent caspase‐8‐mediated apoptosis in a breast cancer cell line Citation41.
Figure 2 Activation of NLRs by microbial products and/or host‐derived motifs. Upon stimulation with meso‐diaminopimelic acid‐containing muropeptides or muramyl‐dipeptide, Nod1 or Nod2 recruit the serine‐threonine kinase Rip2 that in turn leads to NF‐κB activation. The Nod proteins are also involved in the immune response to Shigella flexneri, Helicobacter pylori, Pseudomonas aeruginosa, Escherichia coli, Chlamydia pneumoniae, Listeria monocytogenes, Streptococcus pneumoniae, Salmonella typhimurium, and Mycobacterium tuberculosis. The activation of Nod2 can also result in the release of IL‐1β. The Nalp3 inflammasome is activated when cells are stimulated with MDP, bacterial RNA, or uric acid, or in response to K+ efflux induced by adenosine triphosphate (ATP) or bacterial toxins. Ipaf and Naip also form inflammasomes in response to flagellin. The inflammasome assembly comprises the interaction of the pyrin domains from Nalp3 and ASC and the CARD domain from the latter with the caspase‐1 CARD domain. Ipaf can interact directly with caspase‐1 through its CARD domain. Several pathogens have been shown to activate the inflammasome by different means: Salmonella typhimurium and Legionella pneumophila require Ipaf and/or Naip, while Listeria monocytogenes and Staphylococcus aureus need Nalp3. Finally, Francisella tularensis was shown to require ASC to activate the inflammasome, but the primary sensor is still unknown. NLR: Nod‐like receptors; NF‐κB: nuclear factor‐κB; IL‐1β: interleukin‐1β; MDP: muramyl dipeptide; ASC: apoptosis‐associated speck‐like protein; CARD: caspase activation and recruitment domain; TLR: Toll‐like receptors.
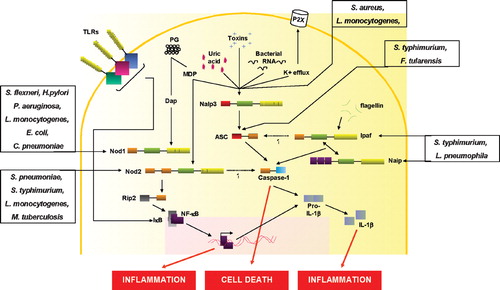
In addition to NF‐κB, Nod1 has been reported to activate c‐Jun N‐terminal kinase in response to bacteria, although the mechanism involved awaits further investigation Citation39.
A novel CARD‐containing protein, CARD6, has been identified as a modulator of Nod1‐ and Rip2‐dependent activation of NF‐κB. Even though a preliminary study suggested that CARD6 may downregulate Nod1‐dependent NF‐κB activation Citation42, a more recent study, using the full‐length CARD6 molecule, has demonstrated that the interaction between CARD6 and Nod1 and Rip2 actually positively modulates NF‐κB activation Citation43.
More recently, CARD9 has also been shown to associate with both Nod2 and Rip2 and to play a critical role in Nod2‐dependent activation of mitogen‐activated protein kinases (MAPKs). In addition, CARD9 has been shown to induce NF‐κB activation signaling through Bcl10 and Malt‐1 Citation44, Citation45.
Erbin, a member of the leucine‐rich repeat‐ and PSD‐95/Discs‐large/ZO‐1 (PDZ) domain‐containing (LAP) family, has also been shown to physically interact with Nod2 and to regulate negatively Nod2‐mediated NF‐κB Citation46, Citation47. During infection with Shigella flexneri, Nod2 and Erbin were shown to colocalize at the entry foci of the bacteria, and maximal affinity between the two proteins was observed after 30–40 minutes of infection Citation46. Taken together, these results suggest an important role for Erbin in manipulating Nod2 signaling during bacterial infection Citation46, Citation47.
Nalps, Ipaf, and Naip initiate inflammatory responses through a mechanism distinct from Nod proteins; indeed, these proteins participate in the formation of caspase‐1‐activating complexes, termed inflammasomes, which result in the secretion of the proinflammatory cytokine IL‐1β. IL‐1β is a major mediator of inflammation and, in general, initiates and/or amplifies a wide variety of effects associated with innate immunity and host responses to microbial invasion and tissue injury Citation48.
Although only a few Nalps among the 14 identified in human genome are well characterized, a common feature of the members of this NLR subfamily is their ability to recruit the adaptor protein ASC (apoptosis‐associated speck‐like protein, also known as TMS1) through homophilic PYD‐PYD interactions. ASC contains an N‐terminal PYD and a C‐terminal CARD that allows the recruitment of caspase‐1 to the platform through CARD‐CARD interactions. The oligomerization of Nalps is believed to bring inflammatory caspases into close proximity, leading to their activation within the inflammasome (Figure ). The primary function of the activated caspase‐1 is to process the inflammatory cytokines pro‐IL‐18 and IL‐1β to their mature and active forms, resulting in inflammation and cell death. IL‐33, a cytokine that is involved in generating a T helper 2 (Th2)‐cell response, is also cleaved by caspase‐1 Citation48.
In the case of Ipaf, its CARD domain can associate either with the adaptor protein ASC or directly with the CARD of pro‐caspase‐1 through homotypic interactions to form the inflammasome and activate caspase‐1 Citation6 (Figure ).
Naip is unique among the NLRs because it harbors three N‐terminal BIR domains to transduce signals. The function of the BIR domains in Naip is currently unclear and remains to be determined. It has been proposed, however, that its murine form, Naip5, interacts with Ipaf, indicating that it may be part of the same caspase‐1 activating complex Citation1.
NLRs in bacterial infection: ready to cope with different microbial strategies
Even before the identification of the microbial motifs sensed by different NLRs, a role for these proteins on the innate immune response to certain bacterial infections had been suggested by several studies. Recognition of live bacteria by NLRs upregulates host cell synthesis of antimicrobial defense mechanisms, such as antimicrobial peptides, and proinflammatory factors, such as chemokines (IL‐8, MIP‐2, and KC) and cytokines (IL‐1β, IL‐6 and IL‐18). Those factors are thought to mediate the recruitment of inflammatory cell populations needed for bacterial clearance Citation6.
The first evidence of a role for Nod proteins in innate immunity against a bacterial pathogen was provided by studies showing that NF‐κB activation and IL‐8 secretion by epithelial cells following infection with the Gram‐negative invasive pathogen Shigella flexneri were dependent on Nod1 Citation39. Several subsequent studies using cell culture and animal models corroborated the role of Nod proteins in initiating inflammation upon bacterial infection. In line with their different ligand‐specificities, Nod1 has been shown to be involved mainly in the host defense to Gram‐negative bacterial pathogens such as enteroinvasive Escherichia coliCitation9, Chlamydia pneumoniaeCitation49, Citation50, and Pseudomonas aeruginosaCitation11, and some Gram‐positive bacteria that possess DAP‐containing PG like Listeria monocytogenesCitation51. In contrast, Nod2 is a general bacterial sensor that participates in the innate immunity against Gram‐positive (Streptococcus pneumoniaeCitation52 and Listeria monocytogenesCitation13), Gram‐negative (Salmonella typhimuriumCitation53), and mycobacteria (Mycobacterium tuberculosisCitation54). Interestingly, despite its cytosolic localization, Nod1 has been reported to respond to certain bacterial pathogens in an invasion‐independent way. For example, Helicobacter pylori is able to deliver muropeptides within epithelial cells through its type IV secretion apparatus encoded by the cag pathogenicity island and initiate Nod1‐dependent signaling pathways Citation10. Other microbial pathogens, such as Salmonella typhimurium and Legionella pneumophila, have since similarly been shown to use their bacterial secretion systems to initiate NLR signaling in host cells Citation32–35.
A direct evidence for Nod1‐mediated host defense in vivo came from a study showing that Nod1‐deficient mice were more susceptible to infection by H. pylori strains displaying functional type IV secretion systems Citation10. This finding was consistent with subsequent in vitro studies demonstrating Nod1‐dependent regulation of antimicrobial peptide (β‐defensin) production in epithelial cells stimulated with H. pylori strains expressing type IV secretion systems Citation55.
Similarly, Hisamatsu et al. proposed a direct antibacterial role for Nod2 in epithelial intestinal cells after the demonstration that the number of viable internalized Salmonella typhimurium in Caco‐2 cells stably transfected with a Nod2 expression plasmid was lower than in vector‐transfected cells Citation53. The mechanism by which Nod2 can be directly antibacterial was not investigated in this study, but a more recent report suggested that Nod2 may regulate the antimicrobial peptides cryptidins synthesis as part of host defense strategies against Listeria monocytogenes infection in vivo. Consistent with that function, Nod2‐deficient mice are more sensitive than wild‐type mice to oral challenge with Listeria monocytogenes. Interestingly, the authors reported no difference between Nod2‐deficient and wild‐type mice after intravenous infection with Listeria monocytogenes, suggesting that the predominant function of Nod2 in antibacterial immunity appears to occur at the intestinal level, not systemically, thus strengthening the idea that the route of infection and the types of host cells infected influence the array of PRMs involved in host defense Citation13.
Recent reports showed that CARD9 is required for the innate immune response against fungal, bacterial, and viral intracellular pathogens. CARD9‐deficient mice failed to clear infection and showed altered cytokine production after challenge with the bacterial pathogen Listeria monocytogenes and with the fungal pathogen Candida albicansCitation44, Citation45.
Nalp1 has been implicated in caspase‐1 activation and necrosis in response to anthrax lethal toxin. The virulence of Bacillus anthracis (the causative agent of anthrax) depends on the secretion of factors that form functional toxins such as edema toxin and lethal toxin. Nalp1 is highly polymorphic in mice, and five alleles have been identified in a total of 18 mouse strains. Mice carrying an allele of Nalp1 encoding the protein Nalp1b are susceptible to the effects of lethal toxin, and it has been suggested that Nalp1b‐dependent caspase‐1 activation might be linked to macrophage susceptibility to lethal toxin, even though the molecular basis for the susceptibility remains uncharacterized Citation56.
Both Nalp3 and the adaptor protein ASC are required for caspase‐1 activation by the Gram‐positive bacteria Staphylococcus aureus and Listeria monocytogenesCitation27, Citation30. Nalp3 or ASC deficiency in macrophages causes a specific defect in caspase‐1 activation and secretion of IL‐1β and IL‐18 when cultured with Staphylococcus aureus or Listeria monocytogenes. Macrophages that were cultured with Listeria monocytogenes deficient in listeriolysin O, a pore‐forming toxin that is required for rapid phagosome lysis, did not secrete IL‐1β, which indicates that the escape of this pathogen into the cytosol is crucial for the detection and engagement of the inflammasome by pathogen‐mediated effector molecules or by PAMPs that are released into the cytosol Citation27, Citation30.
However, ASC, but not Nalp3, is required for activation of caspase‐1 by Salmonella typhimurium and Francisella tularensisCitation30, Citation57–59. Caspase‐1 plays an important role in innate immunity against Salmonella typhimurium, as macrophages and dendritic cells infected with Salmonella typhimurium undergo caspase‐1‐mediated cell death. ASC‐deficient macrophages are unable to activate caspase‐1 following Salmonella typhimurium infection Citation57, Citation58. Similarly, F. tularensis requires ASC to activate caspase‐1 and to trigger a competent immune response following internalization and escape from the phagosome Citation59. In contrast to ASC‐deficient mice, Nalp3‐deficient mice have no apparent defect in caspase‐1 activation by F. tularensis or Salmonella typhimuriumCitation30, Citation57–59.
ASC does not have an apparent sensor domain and it is unlikely that it would act as the primary sensor in a bacterial infection. In the case of Salmonella typhimurium, cytosolic detection can occur through flagellin recognition by Ipaf, that also uses ASC as a downstream adaptor molecule, resulting in caspase‐1 activation and subsequent macrophage cell death and IL‐1β secretion. When Ipaf‐deficient macrophages were infected with Salmonella typhimurium, caspase‐1 processing and IL‐1β release, as well as host cell death, were abrogated. Importantly, the flagellin‐dependent activation of caspase‐1 requires the type three secretion system (TTSS) encoded on the Salmonella pathogenicity island 1 (SPI1) Citation33, Citation34. Ordinarily, flagellin is secreted by a specialized flagellum secretion apparatus to the bacterial cell surface for the assembly of the flagellum. The Salmonella typhimurium virulence‐associated SPI1 apparatus is evolutionarily related to the flagellum secretion system. In fact, it has been shown that effector proteins that are delivered to the host cell by the SPI1 apparatus can be secreted from the bacterium by the flagellum secretion apparatus Citation1. So, it is possible that the activation of caspase‐1 that is mediated by Salmonella typhimurium might occur by a mechanism that involves the accidental delivery of flagellin into the macrophage cytosol.
Polymorphisms in the murine Naip5 (also known as Birc1e) locus, found in the A/J mouse strain, result in enhanced susceptibility to infection by the Gram‐negative pathogen Legionella pneumophila, the causative agent of Legionnaire's disease Citation32, Citation60. In this infection model, detection of bacterial flagellin by Naip5 correlates directly with the survival of Legionella pneumophila bacteria both in macrophages in vitro and in the lungs of experimentally infected mice. Similarly to what is observed for Salmonella typhimurium, Legionella pneumophila‐induced cell death was shown to be dependent upon a functional bacterial type IV secretion system and flagellin expression Citation32. Interestingly, although Ipaf has been shown to be crucial for Legionella pneumophila growth restriction in vitro, increased susceptibility to bacterial infection is only observed in mice that have mutant Naip5 alleles (A/J), not in mice deficient in Ipaf Citation61. In addition, Naip5 also responds to flagellin from Salmonella typhimurium, indicating that its sensing is not specific to Legionella flagellin Citation62.
The innate immune response: a cooperative effort
In the context of an infection, bacteria likely activate the immune response through several PRMs simultaneously. A major effort has been made to identify the molecules responsible for the initiation of inflammation following infection with different pathogens. Even though some PRMs may play a more relevant role than others depending on the nature of the pathogen, it is likely that none of these PRMs function individually as the only component responsible for microbial‐induced immune response in vivo. As discussed along this review, the different NLRs sense different PAMPs (and possibly danger signals) to activate diverse signaling pathways that result in a number of outcomes. The immune response to an infection, however, does not seem to be the simple addition of these outcomes but the result of a complex interaction between various signaling pathways. Thus, it is becoming clear now that NLRs can cooperate among each other and with other PRMs, such as the TLRs, to induce appropriate immune responses.
Nods and TLRs pathways were proposed to be linked by the adaptor molecule Rip2 Citation40. Rip2‐deficient mice were shown to display reduced cytokine production upon stimulation of TLRs with LPS, PG, and double‐stranded RNA, indicating that Nods could also be downstream of TLR4, TLR2, and TLR3, respectively. In addition, Rip2 was also implicated in LPS signaling in macrophages Citation13. However, a recent report showed that backcrossed Rip‐deficient mice displayed unimpaired responses to synthetic and highly purified TLR agonists, clearly demonstrating that Rip2 mediates responses induced through Nod1 and Nod2 but not TLRs Citation63.
Even though the proposed involvement of Rip2 in TLR‐mediated pathways may be due, at least in part, to contamination of the PAMPs preparations, it is nonetheless clear that Nods and TLRs cooperate to induce full activation of signaling pathways. A growing body of evidence shows that both Nod1 and Nod2 act in synergy with TLRs to induce maximal cytokine production Citation12–15. The positive cross‐talk between these pathways allows 1) the generation of more potent responses to microbes, and 2) a more controlled response since the maximal effect would necessitate to trigger simultaneously the two arms of the defense pathway. In the future, it will be interesting to determine whether this synergistic response allows only for a more intense response, or if the repertoire of genes modulated by the combined action of the microbial motifs is wider than the sum of the genes induced separately by each microbial pattern alone Citation64.
In many cases, the control of infection does not require adaptive immune responses; nevertheless, when necessary, the innate immune system is also very efficient at instructing the cellular mediators of adaptive immunity. By using human as well as murine dendritic cells (DCs), several reports have demonstrated that Nod1 and Nod2 agonists and TLR‐activating molecules synergistically induce maturation of DCs and the production of cytokines that can tip the balance between Th1 and Th2 adaptive immune responses Citation13–15.
More recently, Fritz and colleagues showed that Nod1 is key for priming antigen‐specific T‐cell immunity and subsequent full‐blown antibody responses in vivo. The authors showed a critical role for Nod1, acting in synergy with TLRs, in priming Th1 as well as Th17 immune pathways in vivo. In contrast, when acting alone, Nod1 agonist elicited priming of antigen‐specific T‐ and B‐cell immunity with a predominant Th2 polarization profile. These results were further highlighted by the observation that the antigen‐specific response to H. pylori, a bacterium that was shown to trigger Nod1 signaling in gastric epithelial cells, is diminished in Nod1‐deficient animals. Finally, the authors showed that, even though dendritic cells (DCs) are thought to be key players in integrating microbial and antigen signals to instruct adaptive immune responses, DCs and hematopoietic cells in general could not compensate for Nod1 deficiency leading to the idea that innate immune priming by Nod1 is carried by cells other than those in the myeloid compartment Citation12.
In contrast, Watanabe et al. proposed an unanticipated role for Nod2 in negatively regulating the TLR2‐mediated induction of Th1 type cytokines IL‐12 and IFNγ in CD11b+ splenocytes Citation65, Citation66. Although subsequent studies have challenged these conclusions, it is an interesting hypothesis that, at least for certain stimuli and in certain cell types, Nods and TLRs could antagonize each other. In the case of the study by Watanabe et al., the possibility that Nod2 influences the balance Th1/Th2 and thereby modulates the adaptive immune response to bacteria could have a significant impact in the understanding of an inflammatory bowel disease, Crohn's disease, which is associated with mutations in Nod2 (see below).
The Nalps seem to cooperate with TLRs as well. It is now generally accepted that activation and release of IL‐1β requires two distinct signals. What constitutes these signals in vivo either during an infection or during autoinflammatory responses is not known definitively. However, many studies demonstrated that the first signal can be provided by TLRs, resulting in the NF‐κB‐dependent synthesis of pro‐IL‐1β. A second signal is needed to activate caspase‐1 for cleavage and secretion of mature IL‐1β. The inflammasome has been shown to fulfill this role of second signal since it results in caspase‐1 activation Citation48. Consistent with this, it has been demonstrated that the secretion of IL‐1β and IL‐18 is fully abrogated in macrophages from ASC‐deficient mice in response to various TLR agonists Citation57. The requirement of a second signal for IL‐1β production might constitute a mechanism that ensures that the activation of potent inflammatory responses occurs only in the presence of a bona fide stimulus, such as the presence of invasive pathogens (not commensal bacteria alone) and/or tissue injury. In autoinflammatory disorders, this tight regulation seems to be deregulated, thereby leading to the inappropriate activation of IL‐1β in the absence of infection (see below).
As mentioned above, the primary function of the activated caspase‐1 is to process the inflammatory cytokines pro‐IL‐18 and IL‐1β to their mature and active forms. These two cytokines are able to bind to and activate the IL‐1 receptor (IL‐1R) and IL‐18 receptor (IL‐18R) complexes, respectively. These two receptors, similar to the TLRs, have a cytoplasmic TIR domain that, once activated, recruits TIR‐containing adaptors such as MyD88 and induce NF‐κB activation and other signaling cascades. In this sense, Nalps can be considered to be upstream in the IL‐1R and IL‐18R signaling cascades, directly linking intracellular pathogen sensing to signaling pathways shared with TLRs Citation48.
Interestingly, Nods agonists (MDP and Mur‐triDAP) have been shown to induce IL‐1β secretion in macrophages, suggesting that Nods could also be able to directly stimulate the inflammasome Citation67. Since Martinon et al. identified Nalp3 as an intermediate factor in MDP‐induced activation of the inflammasome Citation68, it is now unclear if Nod2 and Nalp3 are two independent systems of MDP detection, or if Nalp3 acts downstream of Nod2 in the specific pathways leading to the inflammasome activation. It is possible that activation of Nod2 by MDP then leads to the activation and/or recruitment of the inflammasome, which in turn activates caspase‐1.
Recently, CARD9 has been shown to be critical for Dectin‐1‐dependent sensing of zymosan, a major yeast cell wall component. Dectin‐1 represents the prototype of non‐TLR transmembrane PRMs and is known to mediate antifungal responses. Dectin‐1‐dependent activation of NF‐κB following stimulation with zymosan or infection with Candida albicans is completely abolished in CARD9‐deficient mice Citation44, Citation45.
In summary, it is likely that immune response is a result of coordinated outputs of activation of multiple PRMs, leading to development of innate immunity and generating signals for the adaptive immune response.
NLRs in inflammatory human diseases
Polymorphisms or mutations in human or mouse NLRs are associated with susceptibility to inflammatory disorders, which further strengthens the idea that these molecules are important in inflammation and immunity.
Nod2 has gained notoriety, since mutations in the gene encoding Nod2 (CARD15) have been linked to increased susceptibility to the chronic inflammatory disease of the intestine, Crohn's disease, and to Blau Syndrome. Crohn's disease is an inflammatory bowel disease characterized by granulomatous inflammation of the distal ileum, associated with increased production of IL‐1β, IL‐6, IL‐12, and TNFα, and activation of NF‐κB, in immune cells of the lamina propria Citation69. Almost 50% of familial Crohn's disease cases in Western populations are linked to three CARD15 mutant alleles: R702W, G908R, and L1007fsinsC. Homozygosity for either allele increases the risk of Crohn's disease by up to 40‐fold, but many patients who are homozygotes for these mutations remain healthy, and even those who develop the disease are commonly asymptomatic for the first 10–15 years of their life Citation70. Thus, it is likely that additional factors, both genetic and environmental, act in conjunction with Nod2 mutations. Two of the mutant alleles—R702W and G908R—specify single amino acid substitutions within the LRR, whereas the third—L1007fsinsC—results in a frameshift mutation that removes the last 33 amino acids of the Nod2 polypeptide Citation70. The L1007fsinsC mutation is the most prevalent in Crohn's disease patients and has been shown to abrogate PG and MDP sensing Citation71. Monocytes with the L1007fsinsC mutation are also impaired in IL‐1β secretion upon stimulation with PG or MDP. In addition, impaired anti‐inflammatory IL‐10 secretion in L1007fsinsC human monocytes stimulated by TLR2 ligands or whole bacteria has been reported Citation64. However, these results, which suggest that Crohn's‐disease‐linked Nod2 mutations are loss‐of‐function mutations, are not entirely consistent with the common clinical observations of elevated proinflammatory cytokines and presence of activated NF‐κB in the lamina propria of Crohn's disease patients. Indeed, the analysis of dendritic cells derived from such patients revealed increased expression of certain inflammatory marker genes including IL‐1β upon incubation with muramyl dipeptide Citation72.
Using murine models, it has been proposed that Nod2 signaling could limit TLR2‐driven activation of NF‐κB and IL‐12 expression while, conversely, Nod2 deficiency or expression of the human L1007fsinsC mutation increased TLR2‐mediated activation of NF‐κB and secretion of IL‐12 and IFNγ. As a result, the authors of this study concluded that Nod2 deficiency leads to exacerbated Th1 responses, which is a hallmark of Crohn's disease. Citation66. Moreover, another study from the same group reported that Nod2 deficiency leads to deregulated TLR2 signaling and induction of antigen‐specific colitis Citation67.
Using a mouse model in which the human frameshift mutation has been introduced by knock‐in, Maeda et al. reported the unexpected observation that macrophages harboring such a mutation exhibited a gain‐of‐function phenotype with increased NF‐κB and more efficient IL‐1β secretion upon MDP stimulation Citation73. Although these data would fit with the global increased inflammation observed in Crohn's patients, they are at odds with those obtained with human monocytes harboring the L1007fsinsC mutation or with macrophages from Nod2‐deficient mice that have been shown to be impaired in MDP sensing and synergy with TLRs Citation71. The function of Nod2 in the biology in Crohn's disease is apparently complex and further investigation is required for the understanding of these striking discrepancies.
Blau syndrome is a rare autosomal dominant genetic disorder not related directly to Crohn's disease, and which is characterized by granulomatous arthritis, uveitis, and skin lesions Citation36. All the missense point mutations in the Nod2 gene associated with this disease are localized in the NACHT domain. These mutations might therefore confer a gain of function to Nod2 rather than disrupting the folding of this domain. This concept is supported by increased basal MDP‐independent NF‐κB activation and is consistent with the heterozygosity pattern that is found in affected patients. Therefore, in this case, inflammation might be due to an aberrant activation of Nod2 in the absence of pathogenic stimuli Citation74.
Mutations in Nod1 are associated with increased susceptibility to asthma and inflammatory bowel disease Citation75, Citation76. In both cases, the mutation has been found to be an insertion‐deletion polymorphism within intron nine of Card4. Both studies suggest that this mutation may contribute to differences in expression levels of naturally occurring splice variants of Nod1. In normal tissue, isoforms of Nod1 are readily detected that lack either the 7th, 7th to 8th, or 7th to 9th repeats within the LRR domain. In vitro, it has been shown that although these isoforms maintained the ability to activate NF‐κB in overexpression studies, none of the truncated isoforms were able to sense TriDAP compared with the activity of full‐length Nod1 Citation77. These findings suggest that at physiological levels, Nod1 splicing variants may contribute to shutting down the NF‐κB pathway triggered by the full‐length molecule. In diseases such as asthma or inflammatory bowel disease, the expression of these isoforms appears to be altered which could lead to abnormal inflammation. In the future, it will be interesting to examine the expression of these isoforms in different disease states and how they then may impact on the development of disease.
Mutations in Nalp3 (encoded by the Cias1 gene) are involved in the autosomal dominant diseases familial cold autoinflammatory syndrome, Muckle‐Wells syndrome, and chronic infantile neurological cutaneous and articular syndrome/neonatal onset multisystemic inflammatory disease (CINCA). All three disorders are closely related autoinflammatory syndromes characterized by periodic fever, skin rashes, amyloidosis, and, in the case of CINCA, the eventual development of neurological complications Citation48. The approximately 40 known disease‐associated mutations in Nalp3 are localized in the NACHT domain and resemble the Nod2 mutations found in the Blau syndrome. For example, the Nalp3 R260W mutation corresponds structurally to the R334W mutation in the NACHT of Nod2 found in Blau syndrome patients. Specifically, this amino acid change in the NACHT of Nalp3 confers a gain of function to the protein, resulting in a constitutively active Nalp3, overactivation of caspase‐1, and aberrant maturation and release of IL‐1β from peripheral blood leukocytes and/or monocytes. In addition, macrophages from these patients are hyperresponsive to MDP Citation78.
More recently, the Nalp3 inflammasome and subsequent aberrant caspase‐1 activation were linked to gout and pseudogout, two other autoinflammatory syndromes Citation29. In these cases, the acute and chronic inflammatory response is associated with the deposition of monosodium urate or calcium pyrophosphate dihydrate crystals, respectively, in joints and periarticular tissues. Monosodium urate and calcium pyrophosphate dihydrate crystals stimulate the caspase‐1‐activating Nalp3 inflammasome to produce active IL‐1β. Mice deficient in various components of the inflammasome, such as caspase‐1, ASC, and Nalp3, are defective in crystal‐induced IL‐1β activation.
Finally, some mutations in Cias1 have also been identified in people with no inflammatory diseases and in patients with undefined or other inflammatory disorders, such as rheumatoid arthritis or systemic lupus erythematosus Citation6.
Mutations in Naip have been associated with the development of spinal muscular atrophy, a neurodegenerative disorder. However, spinal muscular atrophy appears to be dependent on a mutation of spinal motor neuron 1 (SMN1), a neighboring gene. In this context, Naip deletion might contribute to the severity of the disease Citation79.
Concluding remarks
In the last few years the innate immunity field has observed a huge expansion in the understanding of how microbes are sensed and how this leads to a response that (under normal situations) is very efficient in eliminating them. Several questions remain to be elucidated about the sensing of bacterial products through NLR proteins. First, it is not known how NLR‐activating motifs are delivered to the cytosol. Second, it is not clear whether NLRs interact directly with ligands, or if other molecules are required. Another point that needs to be studied in more detail is the participation of NLRs in cell death and the pathways that are triggered in such events. On the other hand, although it is suspected that NLRs may play an important role in intracellular pathogen killing, very little is known about the mechanisms employed by these proteins to eliminate intracellular pathogens. Further studies, including analysis of NLR‐deficient mice, are required to answer these questions and thus provide a complete understanding of how they contribute to fight infections.
Acknowledgements
Research in the laboratory of S. E. Girardin is supported by grants from the Canadian Institutes for Health Research and the Crohn's and Colitis Foundation of Canada.
References
- Delbridge L. M., O'Riordan M. X. The innate recognition of intracellular bacteria. Curr Opin Immunol 2006; 19: 1–7
- Akira S., Uematsu S., Takeuchi O. Pathogen recognition and innate immunity. Cell 2006; 124: 783–801
- Pasare C., Medzhitov R. Toll‐like receptors: linking innate and adaptive immunity. Adv Exp Med Biol 2005; 560: 11–18
- Uematsu S., Akira S. Toll‐like receptors and innate immunity. J Mol Med 2006; 84: 712–24
- Creagh E. M., O'Neill L. A. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co‐operate in innate immunity. Trends Immunol 2006; 27: 352–7
- Fritz J. H., Ferrero R. L., Philpott D. J., Girardin S. E. Nod‐like proteins in immunity, inflammation and disease. Nat Immunol 2006; 7: 1250–7
- DeYoung B. J., Innes R. W. Plant NBS‐LRR proteins in pathogen sensing and host defense. Nat Immunol 2006; 7: 1243–49
- Harris G., KuoLee R., Chen W. Role of Toll‐like receptors in health and diseases of gastrointestinal tract. World J Gastroenterol 2006; 12: 2149–60
- Kim J. G., Lee S. J., Kagnoff M. F. Nod1 is an essential signal transducer in intestinal epithelial cells infected infected with bacteria that avoid recognition by Toll‐like receptors. Infect Immun 2004; 72: 1487–95
- Viala J., Chaput C., Boneca I. G., Cardona A., Girardin S. E., Moran A. P., et al. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 2004; 5: 1166–74
- Travassos L. H., Carneiro L. A., Girardin S. E., Boneca I. G., Lemos R., Bozza M. T., et al. Nod1 participates in the innate immune response to Pseudomonas aeruginosa. J Biol Chem 2005; 280: 36714–18
- Fritz J. H., LeBourhis L., Sellge G., Magalhaes J. G., Fsihi H., Kufer T. A., et al. Nod1‐mediated innate immune recognition of peptidoglycan contributes to the onset of adaptive immunity. Immunity 2007; 26: 445–59
- Kobayashi K. S., Chamaillard M., Ogura Y., Henegariu O., Inohara N., Nunez G., et al. Nod2‐dependent regulation of innate and adaptive immunity in the intestinal tract. Science 2005; 307: 731–4
- van Heel D. A., Hunt K. A., Ghosh S., Herve M., Playford R. J. Normal responses to specific Nod1‐activating peptidoglycan agonists in the presence of the Nod2 frameshift and other mutations in Crohn's disease. Eur J Immunol 2006; 36: 1629–35
- Traub S., Kubasch N., Morath S., Kresse M., Hartung T., Schmidt R. R., et al. Structural requirements of synthetic muropeptides to synergize with lipopolysaccharide in cytokine induction. J Biol Chem 2004; 279: 8694–700
- Girardin S. E., Boneca I. G., Carneiro L. A., Antignac A., Jehanno M., Viala J., et al. Nod1 detects a unique muropeptide from gram‐negative bacterial peptidoglycan. Science 2003; 300: 1584–7
- Chamaillard M., Hashimoto M., Horie Y., Masumoto J., Qiu S., Saab L., et al. An essential role for Nod1 in host recognition of bacterial peptidoglycan containing diaminopimelic acid. Nat Immunol 2003; 4: 702–7
- Girardin S. E., Boneca I. G., Viala J., Chamaillard M., Labigne A., Thomas G., et al. Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J Biol Chem 2003; 278: 8869–72
- Inohara N., Ogura Y., Fontalba A., Gutierrez O., Pons F., Crespo J., et al. Host recognition of bacterial muramyl dipeptide mediated through Nod2. Implications for Crohn's disease. J Biol Chem 2003; 278: 5509–12
- Girardin S. E., Travassos L. H., Herve M., Blanot D., Boneca I. G., Philpott D. J., et al. Peptidoglycan molecular requirements allowing detection by Nod1 and Nod2. J Biol Chem 2003; 278: 41702–08
- Boneca I. G. The role of peptidoglycan in pathogenesis. Curr Opin Microbiol 2005; 8: 46–53
- Travassos L. H., Girardin S. E., Philpott D. J., Blanot D., Nahori M. A., Werts C., et al. Toll‐like receptor 2‐dependent bacterial sensing does not occur via peptidoglycan recognition. EMBO Rep 2004; 5: 1000–6
- Dziarski R., Gupta D. Staphylococcus aureus peptidoglycan is a toll‐like receptor 2 activator: a reevaluation. Infect Immun 2005; 73: 5212–16
- Magalhaes J. G., Philpott D. J., Nahori M. A., Jehanno M., Fritz J., Bourhis L. L., et al. Murine Nod1 but not its human orthologue mediates innate immune detection of tracheal cytotoxin. EMBO Rep 2005; 6: 1201–7
- Uehara A., Fujimoto Y., Kawasaki A., Kusumoto S., Fukase K., Takada H. Meso‐diaminopimelic acid and meso‐lanthionine, amino acids specific to bacterial peptidoglycans, activate human epithelial cells through Nod1. J Immunol 2006; 177: 1796–804
- Carneiro L. A., Travassos L. H., Philpott D. J. Innate immune recognition of microbes through Nod1 and Nod2: implications for disease. Microbes Infect 2004; 6: 609–16
- Kanneganti T. D., Ozoren N., Body‐Malapel M., Amer A., Park J. H., Franchi L., et al. Bacterial RNA and small antiviral compounds activate caspase‐1 through cryopyrin/Nalp3. Nature 2006; 440: 233–6
- Kanneganti T. D., Body‐Malapel M., Amer A., Park J. H., Whitfield J., Franchi L., et al. Critical role for Cryopyrin/Nalp3 in activation of caspase‐1 in response to viral infection and double‐stranded RNA. J Biol Chem 2006; 281: 36560–8
- Martinon F., Petrilli V., Mayor A., Tardivel A., Tschopp J. Gout‐associated uric acid crystals activate the Nalp3 inflammasome. Nature 2006; 440: 237–41
- Mariathasan S., Weiss D. S., Newton K., McBride J., O'Rourke K., Roose‐Girma M., et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006; 440: 228–32
- Gurcel L., Abrami L., Girardin S., Tschopp J., van der Goot F. G. Caspase‐1 activation of lipid metabolic pathways in response to bacterial pore‐forming toxins promotes cell survival. Cell 2006; 126: 1135–45
- Molofsky A. B., Byrne B. G., Whitfield N. N., Madigan C. A., Fuse E. T., Tateda K., et al. Cytosolic recognition of flagellin by mouse macrophages restricts Legionella pneumophila infection. J Exp Med 2006; 203: 1093–104
- Franchi L., Amer A., Body‐Malapel M., Kanneganti T. D., Ozoren N., Jagirdar R., et al. Cytosolic flagellin requires Ipaf for activation of caspase‐1 and interleukin 1beta in salmonella‐infected macrophages. Nat Immunol 2006; 7: 576–82
- Miao E. A., Alpuche‐Aranda C. M., Dors M., Clark A. E., Bader M. W., Miller S. I., et al. Cytoplasmic flagellin activates caspase‐1 and secretion of interleukin 1beta via Ipaf. Nat Immunol 2006; 7: 569–75
- Zamboni D. S., Kobayashi K. S., Kohlsdorf T., Ogura Y., Long E. M., Vance R. E., et al. The Birc1e cytosolic pattern‐recognition receptor contributes to the detection and control of Legionella pneumophila infection. Nat Immunol 2006; 7: 318–25
- Martinon F., Tschopp J. NLRs join TLRs as innate sensors of pathogens. Trends Immunol 2005; 26: 447–54
- Baldwin Jr A. S. The transcription factor NF‐kB and human disease. J Clin Invest 2001; 107: 3–6
- Masumoto J., Yang K., Varambally S., Hasegawa M., Tomlins S. A., Qiu S., et al. Nod1 acts as an intracellular receptor to stimulate chemokine production and neutrophil recruitment in vivo. J Exp Med 2006; 203: 203–13
- Girardin S. E., Tournebize R., Mavris M., Page A. L., Li X., Stark G. R., et al. CARD4/Nod1 mediates NF‐kB and JNK activation by invasive Shigella flexneri EMBO Rep. 2001; 2: 736–42
- Kobayashi K., Inohara N., Hernandez L. D., Galan J. E., Nunez G., Janeway C. A., et al. RICK/Rip2/CARDIAK mediates signalling for receptors of the innate and adaptive immune systems. Nature 2002; 416: 194–9
- da Silva Correia J., Miranda Y., Austin‐Brown N., Hsu J., Mathison J., Xiang R., et al. Nod1‐dependent control of tumor growth. Proc Natl Acad Sci U S A 2006; 103: 1840–5
- Stehlik C., Hayashi H., Pio F., Godzik A., Reed J. C. CARD6 is a modulator of NF‐kappa B activation by Nod1‐ and Cardiak‐mediated pathways. J Biol Chem 2003; 278: 31941–49
- Dufner A., Pownall S., Mak T. W. Caspase recruitment domain protein 6 is a microtubule‐interacting protein that positively modulates NF‐kappaB activation. Proc Natl Acad Sci U S A 2006; 103: 988–93
- Hsu Y. M., Zhang Y., You Y., Wang D., Li H., Duramad O., et al. The adaptor protein CARD9 is required for innate immune responses to intracellular pathogens. Nat Immunol 2007; 8: 198–205
- Gross O., Gewies A., Finger K., Schafer M., Sparwasser T., Peschel C., et al. Card9 controls a non‐TLR signalling pathway for innate anti‐fungal immunity. Nature 2006; 442: 651–6
- Kufer T. A., Kremmer E., Banks D. J., Philpott D. J. Role for erbin in bacterial activation of Nod2. Infect Immun 2006; 74: 3115–24
- McDonald C., Chen F. F., Ollendorff V., Ogura Y., Marchetto S., Lecine P., et al. A role for Erbin in the regulation of Nod2‐dependent NF‐kappaB signaling. J Biol Chem 2005; 280: 40301–9
- Martinon F., Tschopp J. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ 2007; 14: 10–22
- Opitz B., Forster S., Hocke A. C., Maass M., Schmeck B., Hippenstiel S., et al. Nod1‐mediated endothelial cell activation by Chlamydophila pneumonie. Circ Res 2005; 96: 319–26
- Welter‐Stahl L., Ojcius D. M., Viala J., Girardin S., Liu W., Delarbre C., et al. Stimulation of the cytosolic receptor for peptidoglycan, Nod1, by infection with Chlamydia trachomatis or Chlamydia muridarum. Cell Microbiol 2006; 8: 1047–57
- Opitz B., Puschel A., Beermann W., Hocke A. C., Forster S., Schmeck B., et al. Listeria monocytogenes activated p38 MAPK and induced IL‐8 secretion in a nucleotide‐binding oligomerization domain 1‐dependent manner in endothelial cells. J Immunol 2006; 176: 484–90
- Opitz B., Puschel A., Schmeck B., Hocke A. C., Rosseau S., Hammerschmidt S., et al. Nucleotide‐binding oligomerization domain proteins are innate immune receptors for internalized Streptococcus pneumoniae. J Biol Chem 2004; 279: 36426–32
- Hisamatsu T., Suzuki M., Reinecker H. C., Nadeau W. J., McCormick B. A., Podolsky D. K. CARD15/Nod2 functions as an antibacterial factor in human intestinal epithelial cells. Gastroenterology 2003; 124: 993–1000
- Ferwerda G., Girardin S. E., Kullberg B. J., Le Bourhis L., de Jong D. J., Langenberg D. M., et al. Nod2 and toll‐like receptors are nonredundant recognition systems of Mycobacterium tuberculosis. PLoS Pathog 2005; 1: 279–85
- Boughan P. K., Argent R. H., Body‐Malapel M., Park J. H., Ewings K. E., Bowie A. G., et al. Nucleotide‐binding oligomerization domain‐1 and epidermal growth factor receptor: critical regulators of beta‐defensins during Helicobacter pylori infection. J Biol Chem 2006; 281: 11637–48
- Boyden E. D., Dietrich W. F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat Genet 2006; 38: 137–8
- Mariathasan S., Newton K., Monack D. M., Vucic D., French D. M., Lee W. P., et al. Differential activation of the inflammasome by caspase‐1 adaptors ASC and Ipaf. Nature 2004; 430: 213–8
- Sutterwala F. S., Ogura Y., Szczepanik M., Lara‐Tejero M., Lichtenberger G. S., Grant E. P., et al. Critical role for Nalp3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase‐1. Immunity 2006; 24: 317–27
- Mariathasan S., Weiss D. S., Dixit V. M., Monack D. M. Innate immunity against Francisella tularensis is dependent on the ASC/caspase‐1 axis. J Exp Med 2005; 202: 1043–9
- Wright E. K., Goodart S. A., Growney J. D., Hadinoto V., Endrizzi M. G., Long E. M., et al. Naip5 affects host susceptibility to the intracellular pathogen Legionella pneumophila. Curr Biol 2003; 13: 27–36
- Amer A., Franchi L., Kanneganti T. D., Body‐Malapel M., Ozoren N., Brady G., et al. Regulation of Legionella phagosome maturation and infection through flagellin and host Ipaf. J Biol Chem 2006; 281: 35217–23
- Ren T., Zamboni D. S., Roy C. R., Dietrich W. F., Vance R. E. Flagellin‐deficient Legionella mutants evade caspase‐1‐ and Naip5‐mediated macrophage immunity. PLoS Pathog 2006; 2: e18
- Park J. H., Kim Y. G., McDonald C., Kanneganti T. D., Hasegawa M., Body‐Malapel M., et al. RICK/RIP2 mediates innate immune responses induced through Nod1 and Nod2 but not TLRs. J Immunol 2007; 178: 2380–6
- Werts C., Girardin S. E., Philpott D. J. TIR, CARD and PYRIN: three domains for an antimicrobial triad. Cell Death Differ 2006; 13: 798–815
- Watanabe T., Kitani A., Murray P. J., Strober W. Nod2 is a negative regulator of Toll‐like receptor 2‐mediated T helper type 1 responses. Nat Immunol 2004; 5: 800–8
- Watanabe T., Kitani A., Murray P. J., Wakatsuki Y., Fuss I. J., Strober W. Nucleotide binding oligomerization domain 2 deficiency leads to dysregulated TLR2 signaling and induction of antigen‐specific colitis. Immunity 2006; 25: 473–85
- Netea M. G., Azam T., Ferwerda G., Girardin S. E., Walsh M., Park J. S., et al. IL‐32 synergizes with nucleotide oligomerization domain (Nod) 1 and Nod2 ligands for IL‐1beta and IL‐6 production through a caspase 1‐dependent mechanism. Proc Natl Acad Sci U S A 2005; 102: 16309–14
- Martinon F., Agostini L., Meylan E., Tschopp J. Identification of bacterial muramyl dipeptide as activator of the Nalp3/cryopyrin inflammasome. Curr Biol 2004; 14: 1929–34
- Podolsky D. K. Inflammatory bowel disease. N Engl J Med 2002; 347: 417–29
- Hugot J. P., Chamaillard M., Zouali H., Lesage S., Cezard J. P., Belaiche J., et al. Association of Nod2 leucine‐rich repeat variants with susceptibility to Crohn's disease. Nature 2001; 411: 599–603
- Inohara N., Chamaillard M., McDonald C., Nunez G. Nod‐LRR proteins: role in host‐microbial interactions and inflammatory disease. Annu Rev Biochem 2005; 74: 355–83
- Braat H., Stokkers P., Hommes T., Cohn D., Vogels E., Pronk I., et al. Consequence of functional Nod2 and Tlr4 mutations on gene transcription in Crohn's disease patients. J Mol Med 2005; 83: 601–09
- Maeda S., Hsu L. C., Liu H., Bankston L. A., Iimura M., Kagnoff M. F., et al. Nod2 mutation in Crohn's disease potentiates NF‐kappaB activity and IL‐1beta processing. Science 2005; 307: 734–8
- Miceli‐Richard C., Lesage S., Rybojad M., Prieur A. M., Manouvrier‐Hanu S., Hafner R., et al. CARD15 mutations in Blau syndrome. Nat Genet 2001; 29: 19–20
- Hysi P., Kabesch M., Moffatt M. F., Schedel M., Carr D., Zhang Y., et al. Nod1 variation, immunoglobulin E and asthma. Hum Mol Genet 2005; 14: 935–41
- McGovern D. P., Hysi P., Ahmad T., van Heel D. A., Moffatt M. F., Carey A., et al. Association between a complex insertion/deletion polymorphism in Nod1 (CARD4) and susceptibility to inflammatory bowel disease. Hum Mol Genet 2005; 14: 1245–50
- Girardin S. E., Jehanno M., Mengin‐Lecreulx D., Sansonetti P. J., Alzari P. M., Philpott D. J. Identification of the critical residues involved in peptidoglycan detection by Nod1. J Biol Chem 2005; 280: 38648–56
- Agostini L., Martinon F., Burns K., McDermott M. F., Hawkins P. N., Tschopp J. Nalp3 forms an IL‐1beta‐processing inflammasome with increased activity in Muckle‐Wells autoinflammatory disorder. Immunity 2004; 20: 319–25
- Roy N., Mahadevan M. S., McLean M., Shutler G., Yaraghi Z., Farahani R., et al. The gene for neuronal apoptosis inhibitory protein is partially deleted in individuals with spinal muscular atrophy. Cell 1995; 80: 167–78