Abstract
RET (REarranged during Transfection) is a transmembrane receptor tyrosine kinase that is activated by a complex consisting of a soluble glial cell line‐derived neurotrophic factor (GDNF) family ligand (GFL) and a glycosyl phosphatidylinositol‐anchored co‐receptor, GDNF family receptors α (GFRα). RET signalling is crucial for the development of the enteric nervous system. RET also regulates the development of sympathetic, parasympathetic, motor, and sensory neurons, and is necessary for the postnatal maintenance of dopaminergic neurons. The effect of GFLs on sensory, motor, and dopaminergic neurons has raised clinical interest towards these ligands. Outside the nervous system, RET is crucial for development of the kidney and plays a key role in spermatogenesis. Inactivating mutations in RET cause the Hirschsprung's disease characterized by megacolon aganglionosis. In contrast, activating mutations give rise to different types of cancer, multiple endocrine neoplasia type 2A and type 2B, familial medullary thyroid carcinoma, and papillary thyroid carcinoma. The multiple disease phenotypes correlate with differences in the molecular and cell biological functions of different oncogenic RET proteins. In this review we summarize how the different domains of the RET protein contribute to its normal function and how mutations in these domains affect the function of the receptor.
Introduction
RET (REarranged during Transfection) is a transmembrane receptor tyrosine kinase that can function as a growth factor receptor or as an oncogenic protein. Normally it is activated by a complex consisting of a soluble glial cell line‐derived neurotrophic factor (GDNF) family ligand (GFL) and a glycosyl phosphatidylinositol‐anchored co‐receptor, GDNF family receptors α (GFRα). Four different GFLs, GDNF, neurturin (NRTN), artemin (ARTN) and persephin (PSPN), can bind to and specifically activate RET through their cognate co‐receptors GFRα1–4, respectively. As a signal transducer of four different ligand/co‐receptor complexes, RET has many functions in different tissues Citation1. It is necessary for the development of the enteric nervous system and also regulates the development of sympathetic, parasympathetic, motor, and sensory neurons. The potential role of RET in dopamine neurons is still under debate. RET does not seem to be required for the embryonic development of dopaminergic neurons Citation2; however, new data have recently indicated that RET is required for the postnatal maintenance of dopamine neurons Citation3, Citation4. As a result of better understanding of how GFLs affect sensory, motor, and dopaminergic neurons, there has been a growing clinical interest in these ligands. GDNF and NRTN are in clinical trials for the treatment of the Parkinson's disease, while ARTN is under consideration for neuropathic pain clinical trials Citation5. Beyond the nervous system, RET is also known to play a crucial role in kidney development, spermatogenesis and regulation of thyroid function Citation6.
Although RET is widely expressed in developing and adult mammalian tissues, malfunction of this receptor results in diseases and conditions restricted to specific tissues and organs Citation7. For example, patients with Hirschsprung's (HSCR) disease suffer from a variable lack of neurons in the distal segments of the enteric nervous system Citation8. An impaired RET‐GDNF signalling might be involved in the pathogenesis of congenital central hypoventilation syndrome (Ondine's curse) Citation9. Multiple endocrine neoplasia type 2 (MEN 2) syndrome is characterized by medullary thyroid carcinoma (MTC), affecting the calcitonin‐producing C cells. Based on the presence and the type of additional symptoms, MEN 2 is classified into three subgroups Citation10: MEN 2A with MTC, phaeochromocytoma of adrenal medulla and hyperparathyroidism; MEN 2B with MTC, phaeochromocytoma, marfanoid habitus, thickened corneal nerve, ganglioneuromatosis of buccal membranes and gastrointestinal tract; and the familial medullary thyroid carcinoma (FMTC) characterized by MTC only. Papillary thyroid carcinoma (PTC) is frequently associated with genomic rearrangements in the RET gene Citation11. The reason why mutations in RET affect only some of the RET‐expressing tissues and give rise to limited symptomatology remains to be elucidated. It is also not clear why different types of mutations in different domains of the protein give rise to a wide variety of disease phenotypes.
Key messages
RET (REarranged during Transfection) is a transmembrane receptor tyrosine kinase that is activated by a complex consisting of a soluble glial cell line‐derived neurotrophic factor (GDNF) family ligand (GFL) and a glycosyl phosphatidylinositol‐anchored co‐receptor, GDNF family receptors α (GFRα). Four different GFLs, GDNF, neurturin (NRTN), artemin (ARTN), and persephin (PSPN), can bind to and specifically activate RET through their cognate co‐receptors GFRα1–4, respectively.
An auto‐activation of RET has been linked to the multiple endocrine neoplasia type 2 (MEN 2) syndrome which is characterized by medullary thyroid carcinoma (MTC), affecting the calcitonin‐producing C cells. Based on the presence and the type of additional symptoms, MEN 2 is classified into three subgroups: MEN 2A with MTC, phaeochromocytoma of adrenal medulla and hyperparathyroidism; MEN 2B with MTC, phaeochromocytoma, marfanoid habitus, thickened corneal nerve, ganglioneuro matosis of buccal membranes and gastrointestinal tract; and the familial medullary thyroid carcinoma (FMTC) characterized by MTC only. Papillary thyroid carcinoma (PTC) is frequently associated with genomic rearrangements in the RET gene.
An impaired RET‐GDNF signalling has been linked to the Hirschsprung's (HSCR) disease. Patients with this disease suffer from a variable lack of neurons in the distal segments of the enteric nervous system.
The reason why mutations in RET affect only some of the RET‐expressing tissues and give rise to limited symptomatology remains to be elucidated. RET activity is crucially important in this process, and the signalling capacity of the different disease causing RET‐variants may depend on: 1) the subcellular localization, 2) the substrate specificity, 3) the turn‐over rate, 4) the proportion of activated RET—low in the case of abnormal folding and dimerization, or high in the case of a constitutively active kinase domain present in all receptors, 5) the genetic background—influencing the efficiency of the folding machinery in the endoplasmic reticulum, or signal‐transducing pathways.
The normal life cycle of RET
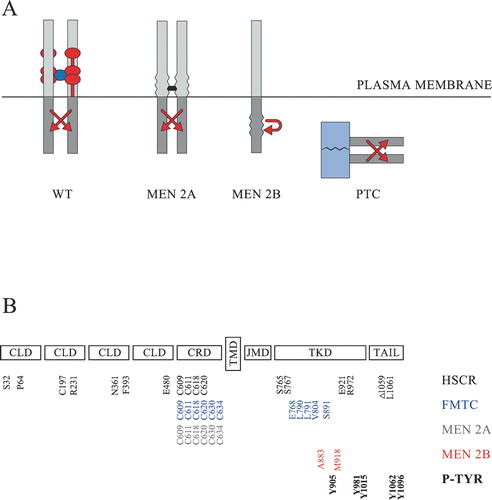
The first translated N‐terminal amino acids of human RET encode a signal sequence (residues 1–28) which guides the growing protein into the endoplasmic reticulum (ER). The signal sequence, which is cleaved off co‐translationally at the ribosomes, is followed by four cadherin‐like domains (residues 29–516), a cysteine‐rich domain (residues 517–635), a transmembrane domain (residues 636–657), a juxtamembrane domain (residues 658–723), a kinase domain (residues 724–1016), and a C‐terminal tail (Figure ). There are several splice variants of RET, and two of them (RET51 and RET9) are well characterized. These two isoforms share 1063 N‐terminal residues and differ from each other only in their very C‐termini. RET51 and RET9 have unique C‐terminal tails of 51 and 9 amino acid residues, respectively. The cadherin‐like domains and the cysteine‐rich domain are inserted into the ER‐lumen and become glycosylated during the transport of RET through the ER and Golgi apparatus to the plasma membrane. The eventual location of these domains is at the extracellular side of the plasma membrane, separated from the intracellular domains by the transmembrane domain (Figure ).
Figure 1 Schematic representation of RET and its oncogenic variants. A: Normally wild‐type RET (WT) is activated at the cell surface in the presence of a tetrameric GFL/GFRα complex. The dimeric GFL is shown in dark blue. The GPI‐anchored GFRα co‐receptors are normally comprised of three cysteine‐rich domains and are here shown in red. The most common multiple endocrine neoplasia type 2A (MEN 2A) variants are activated through the formation of abnormal covalent S‐S bridges (shown in black) between the extracellular domains of two RET molecules. The MEN 2B variants harbour activating mutations in the intracellular kinase domain and may activate signalling cascades either as monomers or as non‐covalently associated dimers. The papillary thyroid carcinoma (PTC) variants are not membrane‐bound, and the activation of their tyrosine kinase domains (shown in grey) is driven by the dimerizing nature of an N‐terminally fused protein (shown in light blue). B: The extracellular part of RET consists of four cadherin‐like domains (CLD) and a cysteine‐rich domain (CRD). The intracellular part consists of a juxtamembrane domain (JMD), a tyrosine kinase domain (TKD) and a C‐terminally located tail. The transmembrane domain (TMD) is located in the mid‐part of the protein. Part of the mutated amino acids which have been shown to be associated with Hirschsprung's (HSCR) disease (S32, P64, C197, R321, N361, F393, E480, C609, C611, C618, C620, S765, S767, E921, R972, Δ1059, L1061 in black), familial medullary thyroid carcinoma (FMTC) (C609, C611, C618, C620, C630, C634, E768, L790, L791, V804, S891 in blue), MEN 2A (C609, C611, C618, C620, C630, C634 in grey), or MEN 2B (A883, M918 in red) are shown at the bottom, together with the main phosphorylated tyrosines (P‐TYR) (Y905, Y981, Y1015, Y1062, Y1096 in bold) having an impact on normal RET‐signalling.
RET is the only known receptor tyrosine kinase that does not directly bind its ligands and requires co‐receptors for activation. The current view is that RET is synthesized as an inactive monomer, which can get into contact with its ligands and co‐receptors only at the cell surface. The dimerization and subsequent transphosphorylation of RET at the cell surface are thought to be triggered by the homodimeric ligands. The interaction between the dimeric GDNF and two GFRα1 co‐receptors has been modelled Citation12, and dimeric ARTN was recently shown by X‐ray crystallography to bind two GFRα3 receptors Citation13. How these tetrameric GFL/GFRα complexes interact with RET, and whether RET participates in some type of preformed complex (RET‐RET, RET‐GFRα, or RET/RET‐GFRα/GFRα) in the absence of the ligand remains unclear. It is thus also unclear if the ligand triggers a dimerization of GFRα1 and thereby also of RET, or whether the ligand triggers a conformational change of an already preformed complex Citation5. GDNF and ARTN have been shown to bind to heparin Citation14, Citation15, but it is also unknown whether heparin is involved in the formation and stabilization of the ligand‐receptor complex. Although the details of the formation of the full ligand‐receptor complex still remains unclear, the dimerization of the RET extracellular domain is thought to trigger the dimerization‐activation of its intracellular tyrosine kinase domains Citation16, Citation17. The kinase domains then transphosphorylate each other on crucial tyrosine residues and thereby trigger the activation of several intracellular signalling cascades Citation18.
GFRα receptors are GPI‐anchored proteins which are thought to be located in special membrane microdomains (lipid rafts) at the cell surface. Optimal signal transduction has been suggested to depend on the efficient GDNF‐induced recruitment of RET into these lipid rafts Citation19, but it has also been shown that PSPN‐activated RET can stimulate both neurite outgrowth and neuronal survival without a significant association with the raft fraction Citation20. Therefore it remains unclear how important lipid rafts are for RET‐related signalling.
After ligand‐induced activation, RET associates with the ubiquitin ligase Cbl, becomes ubiquitinated, internalised, and degraded Citation21, Citation22. Several tyrosine phosphatases (LAR, SHP‐1, SHP‐2, and PTPRJ) have been shown to interact with RET Citation23–25, which after internalization has been co‐localized with RAB5a, a marker of clathrin‐coated vesicles and early endosomes Citation26. The signal transduction capacity of RET has furthermore been shown to be modulated by the Sprouty2 protein Citation27, Citation28, which can interfere with protein complexes involved in intracellular signalling cascades. Taken together, several components which have been linked to the down‐regulation of other receptor tyrosine kinases have also been found to be involved in the regulation of RET, but the RET‐specific inactivating mechanisms require further characterization. Unexpectedly, it has been suggested that RET in the absence of a ligand in some cell types triggers apoptosis before the inactive form of the receptor is degraded Citation29.
Cadherin‐like domains for cell adhesion?
Little is known about the function of the N‐terminally located four cadherin‐like domains of RET Citation30, but based on their homology to cadherins it is tempting to speculate that these domains could be important for cell adhesion. It remains to be studied if these domains influence such RET‐regulated functions as cell migration and neurite outgrowth. Between the second and third cadherin‐like domain there is a Ca2+‐binding site, and Ca2+ is indeed needed for the folding, secretion, as well as signal transducing capacity of RET Citation30–32. Therefore, the binding of Ca2+ probably has a direct stabilizing effect on the structure of RET. Efforts have been made to map the domains which are important for the interaction of RET with its co‐receptor GFRα, but the results are so far contradictory Citation33, Citation34. Crystallographic data would be needed to clarify if more than one domain of RET is important for an optimal RET‐GFRα‐GDNF interaction.
The folding of this extracellular part of RET is apparently delicate, and its extensive glycosylation might somehow contribute to the stability of the mature protein. The cadherin‐like domains of RET harbour 11 out of the 12 putative N‐linked glycosylation sites in RET. N‐linked core glycosylation takes place in the ER, and the molecular weight of RET thereby increases from approximately 120 kDa to 150 kDa Citation35. During secretion through the Golgi apparatus, the molecular weight of RET further increases to 170 kDa, which indicates that it is further modified in an yet unknown way. Therefore, it is clear that mutations in this part of the protein easily affect its folding. Database analysis shows that the vast majority of the mutations in this region of RET are linked to HSCR disease. Some of these RET variants (such as S32, F393, R231) have been characterized in detail Citation36, Citation37 and have indeed been shown to be synthesized as misfolded proteins which become degraded instead of being secreted to the cell surface.
A cysteine‐rich domain for receptor complex formation?
The cysteine‐rich domain of RET is, as indicated by its name, rich in cysteine residues (16 cysteines out of 117 residues). As described above it is unclear if RET stays as an inactive monomer, forms inactive RET‐RET dimers, or forms inactive RET‐GFRα1 dimers/tetramers Citation5. The cysteine‐rich domain of RET could be involved in the formation of either inactive dimers or in the formation of ligand‐triggered active dimers, but this domain may also be involved in the interaction with the GFRα co‐receptors.
Most probably, part of the extracellular cysteine residues of RET normally form intramolecular cysteine bridges. Therefore, mutations in this part of the protein may easily affect its folding. Most of the disease‐associated point mutations in this region of RET indeed affect six specific cysteine residues. The mutations in the cysteine‐rich domain can be divided into two groups. In the first group, the mutations (C630, C634) are related to the MEN 2A and FMTC phenotypes. These mutations lead to partially misfolded proteins, in which abnormal cysteine bridges are formed between two RET molecules. The abnormal S‐S bridges on one hand stabilize the structure of the receptor, but on the other hand also dimerize and thereby activate the RET receptor even in the absence of a ligand. As folding of the extracellular part of secretory proteins takes place in the ER, it is very likely that the MEN 2A and FMTC variants of RET form covalent dimers and thereby also are activated already during their synthesis in the ER, before they reach the cell surface. Surprisingly, in the second group, the mutations (C609, C611, C618, C620) are associated not only with the MEN 2A and FMTC phenotypes, but also with the HSCR phenotype. Pedigree analysis shows that the MEN 2A/FMTC and HSCR phenotypes even can co‐segregate in some families (Citation38 and references therein). Members of such families can be asymptomatic carriers of the mutation, be diagnosed as HSCR patients, develop MEN 2A/FMTC syndromes, or suffer from a combination of both HSCR and MEN 2A/FMTC. Apparently the same point mutation can give rise to both a severe misfolding of the receptor leading to its degradation and/or a partial misfolding of the receptor leading to its dimerization and activation Citation39, Citation40. However, it remains unclear whether differential folding of RET in different tissues, or differential folding due to genetically determined differences of the folding machinery in the ER can explain this enigma.
A transmembrane domain to connect the outside to the inside
The transmembrane domain of RET has traditionally been regarded as a passive link between the extracellular ligand‐sensing part and the intracellular signal‐conducting part. However, new data show that also the transmembrane domain might play an active role during the activation of RET. The transmembrane domain may drive a self‐association of RET which may stimulate the formation of covalent cysteine‐bridges close to the plasma membrane in the extracellular domain of mutant forms of RET (C630 and C634). This could provide an explanation for how some MEN 2A variants of RET are activated Citation41. Interestingly, mutations in three residues (A639, A640, A641) which reside in the transmembrane domain have been identified in patients with MTC or MEN 2.
A juxtamembrane domain for the modulation of the extracellular signal?
The juxtamembrane domain is a forgotten domain of RET. It is not part of the tyrosine kinase domain, and it harbours only two tyrosine residues (Y660 and Y687). Although Y687 has been shown to become autophosphorylated in an in vitro kinase assay Citation42, neither of the tyrosine residues has been found to take part in any ligand‐dependent signalling. However, it was recently shown that the ligand‐dependent tyrosine kinase activity of RET is modulated by the intracellular level of cyclic adenosine‐3′,5′‐monophosphate (cAMP). Most interestingly, cAMP was shown to regulate the activity of RET through a protein kinase A site (S696) which is located in the juxtamembrane domain. When this site is mutated the RET‐dependent formation of lamellipodia is impaired Citation43, and the migration of enteric neural crest cells is affected Citation44. Therefore, it is possible that the juxtamembrane domain has a key role in mediating a cross‐talk between RET and signalling pathways of other receptors.
A few point mutations in this domain have also been reported to be involved in the development of diseases. A single point mutation (G691S) in the juxtamembrane domain has been linked to sporadic MTC Citation45, whereas a deletion/insertion mutation (K666N with an addition of an extra serine residue, S667) has recently been shown to be associated with FMTC Citation46. The molecular mechanisms underlying these activating mutations remain to be solved.
A kinase domain to trigger intracellular signalling
The activation of the kinase domain of RET is an obviously central function of the receptor. The kinase domain is homologous to other tyrosine kinases Citation16, Citation17, and its activation is believed to depend on a transphosphorylation reaction between two adjacent RET molecules. A number of tyrosine residues (Y905, Y981, Y1015, Y1062, Y1096) inside the kinase domain and the C‐terminal tail are phosphorylated, and several downstream signalling pathways (MAPK, PI3K/AKT, JUN, Src, PLCγ) have been mapped to depend on the phosphorylation of specific tyrosine residues. However, it remains unsolved why RET activation triggers cell migration in some cells, while it triggers survival, proliferation, or differentiation in others Citation18. It has been suggested that RET, depending on its localization in the cell could trigger different signalling pathways. RET located on the cell surface outside lipid rafts would have the capacity to activate SHC (Src Homology 2 domain‐Containing), while RET still on the cell surface but inside lipid rafts would activate fibroblast growth factor receptor substrate 2 (FRS2) Citation47. It has also recently been suggested that RET could activate AKT from the cell surface, but ERK (extracellular signal‐regulated kinase) only after it has become internalized Citation26.
Database analysis shows that HSCR‐related mutations are scattered all over the kinase domain of RET. A few of them (such as K907, E921, R972) have been studied in detail Citation36, Citation37 and shown to give rise to enzymatically inactive receptors. Other mutations residing in this region change the properties of the kinase domain and underlie MEN 2B and FMTC. Depending on the affected codons, the disease phenotype is more or less severe. The most aggressive oncogenic phenotype, MEN 2B has been linked to only few codons (encoding V804/Y806, A883, and M918). Activation of at least the M918 variant of MEN 2B takes place already during its synthesis in the ER Citation48. Interestingly, mutations in some codons (encoding L790, Y791) have been linked to both a mild MEN 2A phenotype and a FMTC phenotype. Different types of mutations underlie the PTC disease. Patients with PTC mostly harbour a chromosomal rearrangement in which the kinase domain of RET is fused at its N‐terminus to a soluble protein which spontaneously forms cytoplasmic dimers and thereby induces activation of the RET kinase domain Citation11. Therefore, the PTC variants of RET differ from the transmembrane MEN 2 variants of RET in their subcellular localization. Interestingly, the membrane‐bound receptor‐type tyrosine phosphatase PTPRJ has been shown to interact with the wild‐type and RET MEN 2A variants, but not with the MEN 2B or PTC1 variants of RET Citation25, while another membrane‐bound phosphatase (LAR) reduces the kinase activity of MEN 2A, but not that of MEN 2B Citation23. Furthermore, it has been suggested that the properties or substrate specificity of different types of oncogenic RET are altered. Tyrosine 1062 has been shown to be more phosphorylated in the MEN 2B variant than in the MEN 2A variant Citation49, and the M918T mutation (MEN 2B) has been shown to increase the ATP‐binding affinity of RET Citation50. Moreover, it is possible that the MEN 2A, MEN 2B, FMTC and PTC variants of RET activate the transcription factor STAT3 (Signal Transducer and Activator of Transcription 3) differently (Citation51–54 and references therein). Taken together, an altered subcellular localization as well as a modified activity of the different types of oncogenic RET variants probably underlie the wide variety of phenotypic traits found in patients. However, the picture is further complicated by the finding that some rare kindreds, carrying point mutations in RET, are affected by both PTC and MTC Citation55.
Different C‐terminal tails for an optimal signal transmission?
Several of the functionally important tyrosine residues in RET lie within its kinase domain. However, the C‐terminal tails in the two well characterized splice variants RET9 and RET51 harbour one (Y1062), and respectively two (Y1062, Y1096) important tyrosine residues. Interestingly it has been shown that the two RET isoforms form only homodimeric complexes and do not transactivate each other Citation56, but contradictory results have emerged based on monoisoformic mouse models. According to these results, RET51 may, or may not, be biologically less active than RET9 Citation57, Citation58. In any case it seems clear that the function of Y1062 is vitally important for the signalling capacity of RET9 Citation57, Citation59, and that its function somehow can be compensated by Y1096 in RET51 Citation57, Citation60. Moreover, two cytoskeleton‐associated proteins (Enigma and Shank3) have been shown to interact with RET9 more strongly than with RET51 Citation61, Citation62, while RET51 is internalized and degraded more quickly than RET9 from the cell surface Citation21.
At least three mutations (Δ1059, L1061, Y1062) close to Y1062 have been found in HSCR patients Citation63, Citation64, further strengthening the idea that Y1062 has a crucial role in the function of RET. Surprisingly, some MEN 2B‐ or FMTC‐related mutations (E768, V804, A883, S891, M918, A919, E768/A919) that lie within the kinase domain display a much stronger transforming activity when introduced into RET51, than the corresponding mutations in RET9 Citation65–67. Therefore, it is possible that the two isoforms fulfil different functions Citation57, Citation68.
Concluding remarks
As described above, RET‐related mutations give rise to a complicated spectrum of clinical symptoms and disease phenotypes. Based on the normal function of RET it is possible to highlight several possible reasons for the varying phenotypes. The signalling capacity of the different RET variants may depend on: 1) the subcellular localization, 2) the substrate specificity, 3) the turn‐over rate, 4) the proportion of activated RET—low in the case of abnormal folding and dimerization, or high in the case of a constitutively active kinase domain present in all receptors, 5) the genetic background—influencing the efficiency of the folding machinery in the ER, or signal transducing pathways. Therefore different types of clinical symptoms related to RET may need to be cured with different types of drugs targeted to specific domains of RET. Ideally, such drugs could improve the folding of RET in the ER, hinder the formation of abnormal S‐S bridges during the secretion of RET, affect its degradation, or modify its intracellular kinase activity.
Acknowledgements
We thank Urmas Arumäe, Jukka Kallijärvi, Anu Planken, and Heidi Virtanen for useful comments to the manuscript.
References
- Airaksinen M. S., Saarma M. The GDNF family: signalling, biological functions and therapeutic value. Nat Rev Neurosci 2002; 3: 383–94
- Jain S., Golden J. P., Wozniak D., Pehek E., Johnson E. M Jr., Milbrandt J. RET is dispensable for maintenance of midbrain dopaminergic neurons in adult mice. J Neurosci 2006; 26: 11230–8
- Kramer E. R., Aron L., Ramakers G. M. J., Seitz S., Zhuang X., Beyer K., et al. Absence of Ret signaling in mice causes progressive and late degeneration of the nigrostriatal system PLoS Biol. 2007; 5: e39
- Mijatovic J., Airavaara M., Planken A., Auvinen P., Raasmaja A., Piepponen T. P., et al. Constitutive Ret activity in knock‐in MEN2B mice induces profound elevation of brain dopamine concentration via enhanced synthesis and increases number of TH‐positive cells in substantia nigra. J Neurosci 2007; 27: 4799–809
- Bespalov M. M., Saarma M. GDNF family receptor complexes are emerging drug targets. Trends Pharmacol Sci 2007; 28: 68–74
- Saarma M. GFL neurotrophic factors: Physiology and pharmacology. The New Encyclopedia of Neuroscience. Elsevier Press 2007, In press
- Plaza‐Menacho I., Burzynski G. M., de Groot J. W., Eggen B. J. L., Hofstra R. M. W. Current concepts in RET‐related genetics, signaling and therapeutics. Trends Genet 2006; 22: 627–36
- Lantieri F., Griseri P., Ceccherini I. Molecular mechanisms of RET‐induced Hirschsprung pathogenesis. Ann Med 2006; 38: 11–9
- Sasaki A., Kanai M., Kijima K., Akaba K., Hashimoto M., Hasegawa H., et al. Molecular analysis of congenital central hypoventilation syndrome. Hum Genet 2003; 114: 22–6
- Hansford J. R., Mulligan L. M. Multiple endocrine neoplasia type 2 and RET: from neoplasia to neurogenesis. J Med Genet 2000; 37: 817–27
- Santoro M., Melillo R. M., Fusco A. RET/PTC activation in papillary thyroid carcinoma: European journal of endocrinology prize lecture. Eur J Endocrinol 2006; 155: 645–53
- Leppänen V‐M., Bespalov M. M., Runeberg‐Roos P., Puurand Ü., Merits A., Saarma M., et al. The structure of GFRα1 domain 3 reveals new insights into GDNF binding and RET activation. EMBO J 2004; 23: 1452–62
- Wang X., Baloh R. H., Milbrandt J., Garcia K. C. Structure of artemin complexed with its receptor GFRα3: convergent recognition of glial cell line‐derived neurotrophic factors. Structure 2006; 14: 1083–92
- Rickard S. M., Mummery R. S., Mulloy B., Rider C. C. The binding of human glial cell line‐derived neurotrophic factor to heparin and heparan sulfate: importance of 2‐O‐sulfate groups and effect on its interaction with its receptor, GFRα1. Glycobiology 2003; 13: 419–26
- Silvian L., Jin P., Carmillo P., Boriack‐Sjodin P. A., Pelletier C., Rushe M., et al. Artemin crystal structure reveals insights into heparan sulfate binding. Biochemistry 2006; 45: 6801–12
- Knowles P. P., Murray‐Rust J., Kjaer S., Scott R. P., Hanrahan S., Santoro M., et al. Structure and chemical inhibition of the RET tyrosine kinase domain. J Biol Chem 2006; 281: 33577–87
- Tuccinardi T., Manetti F., Schenone S., Martinelli A., Botta M. Construction and Validation of a RET TK Catalytic Domain by Homology Modeling. J Chem Inf Model 2007; 47: 644–55
- Kodama Y., Asai N., Kawai K., Jijiwa M., Murakumo Y., Ichihara M., et al. The RET proto‐oncogene: A molecular therapeutic target in thyroid cancer. Cancer Sci 2005; 96: 143–8
- Tansey M. G., Baloh R. H., Milbrandt J., Johnson E. M Jr. GFRα‐mediated localization of RET to lipid rafts is required for effective downstream signaling, differentiation, and neuronal survival. Neuron 2000; 25: 611–23
- Yang J., Lindahl M., Lindholm P., Virtanen H., Coffey E., Runeberg‐Roos P., et al. PSPN/GFR(4 has a significantly weaker capacity than GDNF/GFR(1 to recruit RET to rafts, but promotes neuronal survival and neurite outgrowth. FEBS Lett 2004; 569: 267–71
- Scott R. P., Eketjäll S., Aineskog H., Ibáňez C. F. Distinct turnover of alternatively spliced isoforms of the RET kinase receptor mediated by differential recruitment of the Cbl ubiquitin ligase. J Biol Chem 2005; 280: 13442–9
- Pierchala B. A., Milbrandt J., Johnson E. M Jr. Glial cell line‐derived neurotrophic factor‐dependent recruitment of Ret into lipid rafts enhances signaling by partitioning Ret from proteasome‐dependent degradation. J Neurosci 2006; 26: 2777–87
- Qiao S., Iwashita T., Furukawa T., Yamamoto M., Sobue G., Takahashi M. Differential effects of leukocyte common antigen‐related protein on biochemical and biological activities of RET‐MEN2A and RET‐MEN2B mutant proteins. J Biol Chem 2001; 276: 9460–7
- Incoronato M., D'Alessio A., Paladino S., Zurzolo C., Carlomagno M. S., Cerchia L., et al. The Shp‐1 and Shp‐2, tyrosine phosphatases, are recruited on cell membrane in two distinct molecular complexes including Ret oncogenes. Cell Signal 2004; 16: 847–56
- Iervolino A., Iuliano R., Trapasso F., Viglietto G., Melillo R. M., Carlomagno F., et al. The receptor‐type protein tyrosine phosphatase J antagonizes the biochemical and biological effects of RET‐derived oncoproteins. Cancer Res 2006; 66: 6280–7
- Richardson D. S., Lai A. Z., Mulligan L. M. RET ligand‐induced internalization and its consequences for downstream signaling. Oncogene 2006; 25: 3206–11
- Chi L., Zhang S., Lin Y., Prunskaite‐Hyyryläinen R., Vuolteenaho R., Itäranta P., et al. Sprouty proteins regulate ureteric branching by coordinating reciprocal epithelial Wnt11, mesenchymal Gdnf and stromal Fgf7 signalling during kidney development. Development 2004; 131: 3345–56
- Ishida M., Ichihara M., Mii S., Jijiwa M., Asai N., Enomoto A., et al. Sprouty2 regulates growth and differentiation of human neuroblastoma cells through RET tyrosine kinase. Cancer Sci 2007; 98: 815–21
- Bordeaux M‐C., Forcet C., Granger L., Corset V., Bidaud C., Billaud M., et al. The RET proto‐oncogene induces apoptosis: a novel mechanism for Hirschsprung disease. EMBO J 2000; 19: 4056–63
- Anders J., Kjaer S., Ibáňez C. F. Molecular modeling of the extracellular domain of the RET receptor tyrosine kinase reveals multiple cadherin‐like domains and a calcium‐binding site. J Biol Chem 2001; 276: 35808–17
- Nozaki C., Asai N., Murakami H., Iwashita T., Iwata Y., Horibe K., et al. Calcium‐dependent Ret activation by GDNF and neurturin. Oncogene 1998; 16: 293–9
- van Weering D. H. J., Moen T. C., Braakman I., Baas P. D., Bos J. L. Expression of the receptor tyrosine kinase Ret on the plasma membrane is dependent on calcium. J Biol Chem 1998; 273: 12077–81
- Kjaer S., Ibáňez C. F. Identification of a surface for binding to the GDNF‐GFRα1 complex in the first cadherin‐like domain of RET. J Biol Chem 2003; 278: 47898–904
- Amoresano A., Incoronato M., Monti G., Pucci P., de Franciscis V., Cerchia L. Direct interactions among Ret, GDNF and GFRα1 molecules reveal new insights into the assembly of a functional three‐protein complex. Cell Signal 2005; 17: 717–27
- Takahashi M., Buma Y., Taniguchi M. Identification of the ret proto‐oncogene products in neuroblastoma and leukemia cells. Oncogene 1991; 6: 297–301
- Carlomagno F., De Vita G., Berlingieri M. T., de Franciscis V., Melillo R. M., Colantuoni V., et al. Molecular heterogeneity of RET loss of function in Hirschsprung's disease. EMBO J 1996; 15: 2717–25
- Pelet A., Geneste O., Edery P., Pasini A., Chappuis S., Atti T., et al. Various mechanisms cause RET‐mediated signaling defects in Hirschsprung's disease. J Clin Invest 1998; 101: 1415–23
- Dvořáková S., Dvořáková K., Malíková M., Škába R., Vlček P., Bendlová B. A novel Czech kindred with familial medullary thyroid carcinoma and Hirschsprung's disease. J Pediatr Surg 2005; 40: e1–6
- Ito S., Iwashita T., Asai N., Murakami H., Iwata Y., Sobue G., et al. Biological properties of Ret with cysteine mutations correlate with multiple endocrine neoplasia type 2A, familial medullary thyroid carcinoma, and Hirschsprung's disease phenotype. Cancer Res 1997; 57: 2870–2
- Chappuis‐Flament S., Pasini A., De Vita G., Ségouffin‐Cariou C., Fusco A., Attié T., et al. Dual effect on the RET receptor of MEN 2 mutations affecting specific extracytoplasmic cysteines. Oncogene 1998; 17: 2851–61
- Kjaer S., Kurokawa K., Perrinjaquet M., Abrescia C., Ibáňez C. F. Self‐association of the transmembrane domain of RET underlies oncogenic activation by MEN2A mutations. Oncogene 2006; 25: 7086–95
- Liu X., Vega Q. C., Decker R. A., Pandey A., Worby C. A., Dixon J. E. Oncogenic RET receptors display different autophosphorylation sites and substrate binding specificities. J Biol Chem 1996; 271: 5309–12
- Fukuda T., Kiuchi K., Takahashi M. Novel mechanism of regulation of Rac activity and lamellipodia formation by RET tyrosine kinase. J Biol Chem 2002; 277: 19114–21
- Asai N., Fukuda T., Wu Z., Enomoto A., Pachnis V., Takahashi M., et al. Targeted mutation of serine 697 in the Ret tyrosine kinase causes migration defect of enteric neural crest cells. Development 2006; 133: 4507–16
- Elisei R., Cosci B., Romei C., Bottici V., Sculli M., Lari R., et al. RET exon 11 (G691S) polymorphism is significantly more frequent in sporadic medullary thyroid carcinoma than in the general population. J Clin Endocrinol Metab 2004; 89: 3579–84
- Cordella D., Muzza M., Alberti L., Colombo P., Travaglini P., Beck‐Peccoz P., et al. An in‐frame complex germline mutation in the juxtamembrane intracellular domain causing RET activation in familial medullary thyroid carcinoma. Endocr Relat Cancer 2006; 13: 945–53
- Paratcha G., Ledda F., Baars L., Coulpier M., Besset V., Anders J., et al. Released GFRα1 potentiates downstream signaling, neuronal survival, and differentiation via a novel mechanism of recruitment of c‐Ret to lipid rafts. Neuron 2001; 29: 171–84
- Runeberg‐Roos P., Virtanen H., Saarma M. RET(MEN 2B) is active in the endoplasmic reticulum before reaching the cell surface. Oncogene 2007 Jun 18, [Epub ahead of print]
- Salvatore D., Melillo R. M., Monaco C., Visconti R., Fenzi G., Vecchio G., et al. Increased in vivo phosphorylation of ret tyrosine 1062 is a potential pathogenetic mechanism of multiple endocrine neoplasia type 2B. Cancer Res 2001; 61: 1426–31
- Gujral T. S., Singh V. K., Jia Z., Mulligan L. M. Molecular mechanisms of RET receptor‐mediated oncogenesis in multiple endocrine neoplasia 2B. Cancer Res 2006; 66: 10741–9
- Schuringa J. J., Wojtachnio K., Hagens W., Vellenga E., Buys C. H., Hofstra R., et al. MEN2A‐RET‐induced cellular transformation by activation of STAT3. Oncogene 2001; 20: 5350–8
- Hwang J. H., Kim D. W., Suh J. M., Kim H., Song J. H., Hwang E. S., et al. Activation of signal transducer and activator of transcription 3 by oncogenic RET/PTC (rearranged in transformation/ papillary thyroid carcinoma) tyrosine kinase: roles in specific gene regulation and cellular transformation. Mol Endocrinol 2003; 17: 1155–66
- Yuan Z‐L., Guan Y‐J., Wang L., Wei W., Kane A. B., Chin Y. E. Central role of the threonine residue within the p+1 loop of receptor tyrosine kinase in STAT3 constitutive phosphorylation in metastatic cancer cells. Mol Cell Biol 2004; 24: 9390–400
- Plaza‐Menacho I., van der Sluis T., Hollema H., Gimm O., Buys C. H. C. M., Magee A. I., et al. Ras/ERK1/2‐mediated STAT3 Ser727 phosphorylation by familial medullary thyroid carcinoma‐associated RET mutants induces full activation of STAT3 and is required for c‐fos promoter activation, cell mitogenicity, and transformation. J Biol Chem 2007; 282: 6415–24
- Melillo R. M., Cirafici A. M., De Falco V., Bellantoni M., Chiappetta G., Fusco A., et al. The oncogenic activity of RET point mutants for follicular thyroid cells may account for the occurrence of papillary thyroid carcinoma in patients affected by familial medullary thyroid carcinoma. Am J Pathol 2004; 165: 511–21
- Tsui‐Pierchala B. A., Ahrens R. C., Crowder R. J., Milbrandt J., Johnson E. M Jr. The long and short isoforms of Ret function as independent signaling complexes. J Biol Chem 2002; 277: 34618–25
- Jain S., Encinas M., Johnson E. M Jr., Milbrandt J. Critical and distinct roles for key RET tyrosine docking sites in renal development. Genes Dev 2006; 20: 321–33
- de Graaff E., Srinivas S., Kilkenny C., D'Agati V., Mankoo B. S., Costantini F., et al. Differential activities of the RET tyrosine kinase receptor isoforms during mammalian embryogenesis. Genes Dev 2001; 15: 2433–44
- Wong A., Bogni S., Kotka P., de Graaff E., D'Agati V., Costantini F., et al. Phosphotyrosine 1062 is critical for the in vivo activity of the Ret9 receptor tyrosine kinase isoform. Mol Cell Biol 2005; 25: 9661–73
- Degl'Innocenti D., Arighi E., Popsueva A., Sangregorio R., Alberti L., Rizzetti M. G., et al. Differential requirement of Tyr1062 multidocking site by RET isoforms to promote neural cell scattering and epithelial cell branching. Oncogene 2004; 23: 7297–309
- Borrello M. G., Mercalli E., Perego C., Degl'Innocenti D., Ghizzoni S., Arighi E., et al. Differential interaction of Enigma protein with the two RET isoforms. Biochem Biophys Res Commun 2002; 296: 515–22
- Schuetz G., Rosário M., Grimm J., Boeckers T. M., Gundelfinger E. D., Birchmeier W. The neuronal scaffold protein Shank3 mediates signaling and biological function of the receptor tyrosine kinase Ret in epithelial cells. J Cell Biol 2004; 167: 945–52
- Geneste O., Bidaud C., De Vita G., Hofstra R. M. W., Tartare‐Deckert S., Buys C. H. C. M., et al. Two distinct mutations of the RET receptor causing Hirschsprung's disease impair the binding of signalling effectors to a multifunctional docking site. Hum Mol Genet 1999; 8: 1989–99
- Wu T‐T., Tsai T‐W., Chu C‐T., Lee Z‐F., Hung C‐M., Su C‐C., et al. Low RET mutation frequency and polymorphism analysis of the RET and EDNRB genes in patients with Hirschsprung disease in Taiwan. J Hum Genet 2005; 50: 168–74
- Pasini A., Geneste O., Legrand P., Schlumberger M., Rossel M., Fournier L., et al. Oncogenic activation of RET by two distinct FMTC mutations affecting the tyrosine kinase domain. Oncogene 1997; 15: 393–402
- Rossel M., Pasini A., Chappuis S., Geneste O., Fournier L., Schuffenecker I., et al. Distinct biological properties of two RET isoforms activated by MEN 2A and MEN 2B mutations. Oncogene 1997; 14: 265–75
- Iwashita T., Kato M., Murakami H., Asai N., Ishiguro Y., Ito S., et al. Biological and biochemical properties of Ret with kinase domain mutations identified in multiple endocrine neoplasia type 2B and familial medullary thyroid carcinoma. Oncogene 1999; 18: 3919–22
- Lee D. C. W., Chan K. W., Chan S. Y. RET receptor tyrosine kinase isoforms in kidney function and disease. Oncogene 2002; 21: 5582–92