Abstract
Recent findings of mitochondrial abnormalities in brains from subjects with neurological disorders have led to a renewed search for mitochondrial abnormalities in psychiatric disorders. A growing body of evidence suggests that there is mitochondrial dysfunction in schizophrenia, bipolar disorder, and major depressive disorder, including evidence from electron microscopy, imaging, gene expression, genotyping, and sequencing studies. Specific evidence of dysfunction such as increased common deletion and decreased gene expression in mitochondria in psychiatric illnesses suggests that direct examination of mitochondrial DNA from postmortem brain cells may provide further details of mitochondrial alterations in psychiatric disorders.
Introduction
The mitochondrion is a subcellular organelle located in the cytoplasm of all cells and contains copies of mitochondrial DNA (mtDNA). MtDNA encodes two ribosomal RNAs, 22 transfer RNAs, and 13 protein subunits of the electron transport chain and the displacement loop (D-loop), which is necessary for replication and transcription of the mitochondrial genome Citation1. In addition to the small number of genes encoded within the mitochondrion, it is estimated that at least 1000 nuclear-encoded proteins are also translocated to the mitochondria. The interactions between both mitochondrial proteins and translocated proteins are required for normal cellular function. The purpose of this review is to describe the current understanding of variations in mtDNA or nuclear DNA that impact on mitochondrial-related gene expression reported in schizophrenia (SZ), bipolar disorder (BPD), and major depressive disorder (MDD).
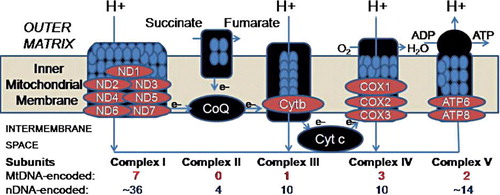
MtDNA is 16,569 base pair circular DNA with a guanine-rich heavy (H) strand and a cytosine-rich light (L) strand. The remaining subunits of the electron transport chain (see ) are encoded in the nuclear genome, which is physically separate from the cytoplasmically located mitochondrial genome. Each cell contains multiple mitochondria, and there are multiple copies of mtDNA in each mitochondrion Citation2. Muscle, brain, and liver often contain the highest number of mitochondria due to high energy utilization. Mitochondria provide energy for cellular processes by converting metabolites to adenosine triphosphate (ATP) via oxidative phosphorylation (OXPHOS). Mitochondria contain a highly permeable outer mitochondrial and a virtually impermeable inner membrane Citation3. The electron transport chain produces a pH gradient across the inner mitochondrial membrane that is important for ATP production and metabolite transport Citation4. OXPHOS is carried out by the five protein-lipid enzyme complexes of the respiratory chain, which are located in the inner membrane: complexes I (NADH: ubiquinone oxidoreductase), II (succinate: ubiquinone oxidoreductase), III (ubiquinol: ferrocytochrome c oxidoreductase), IV (ferrocytochrome c: oxygen oxidoreductase or cytochrome c oxidase), and V (ATP synthase) Citation5–8 (See ).
Key message
There is accumulating evidence of mitochondrial involvement in the pathophysiology of psychiatric disorders such as schizophrenia, bipolar disorder, and major depressive disorder.
Abbreviations
Figure 1. Diagram adapted from Lee et al. Citation106 showing five complexes involved in oxidative phosphorylation. We found decreased expression in brain for key mitochondrial DNA-encoded transcripts in neuropsychiatric disorders as shown in .
Mitochondria play a role in amino acid, lipid, and steroid metabolism, apoptosis, and act as calcium buffers and sources of free radicals Citation5–7, Citation9, Citation10, which can damage mtDNA resulting in production of more free radicals Citation11. Thus, complementary functions of mitochondria are cell death, cell bioenergetics, and production of amino acids. Related to psychiatry, monoamine oxidase A and B enzymes have been shown to be localized in the outer membrane of mitochondria Citation12, Citation13, and peripheral benzodiazepine receptors have also been shown to localize to the mitochondrial membrane Citation14.
MtDNA is inherited maternally and mtDNA is present at a much lower concentration in spermatozoa (approximately 1200 copies in a single spermatozoon), compared to oocytes (approximately 100,000 copies in a single oocyte). Species-specific sperm mitochondria are believed to be eliminated via ubiquination, which tags them for breakdown through proteolysis Citation15. Interestingly, there is some evidence for high maternal transmission of SZ Citation16–19, as well as BPD Citation20, which supports the hypothesis that increased risk for these disorders is related to mitochondrial dysfunction. However, it is also possible that this relationship may be due to environmental factors Citation21. Failure to find a pattern of maternal inheritance as well as evidence of paternal inheritance have also been reported in SZ and BPD Citation22, Citation23.
Although mtDNA mutations may be maternally transmitted, they are often sporadic. MtDNA is more susceptible to somatic deletions than nuclear DNA because mtDNA is not protected by histones and also has a poor DNA repair system. Thus mtDNA has been suggested as a potential ‘weak point’ of the genome Citation2, Citation24. Examples of clinical syndromes related to mtDNA mutations include Leber hereditary optic neuropathy (LHON), which is associated with a number of ND1 (see list of abbreviations) gene mutations Citation25, and myopathy and diabetes mellitus which are associated with point mutations in mitochondrial tRNA Citation26. Nuclear DNA mutations, such as the polymerase DNA-directed gamma (POLG) mutation Citation27, can sometimes predispose mtDNA deletions, as in the case of the mitochondrial chronic progressive external ophthalmoplegia (CPEO) disease which is associated with mutations of mitochondrial tRNA mutations as well as a ND5 T-C polymorphism. Another example of nuclear DNA mutations that affect mtDNA is the mitochondrial neurogastrointestinal encephalomyopathy (MNGIE) Citation28, which is related to a heteroplasmic tRNA mutation. The leucine-rich pentatricopeptide repeat cassette (LRPPRC) nuclear gene interacts with the mitochondrial genome, and a LRPPRC mutation results in reduced translation of mitochondrial genes cytochrome c oxidase 1 (MT-CO1) and possibly MT-CO3 Citation29. Although any organ can be affected by mitochondrial defects, the brain, skeletal muscle, and cardiac muscle are most commonly affected due to their high aerobic activity and higher mitochondrial content Citation7.
The evidence for involvement of mitochondria in neurodegenerative disorders such as Alzheimer's, Parkinson's, multiple sclerosis, and amyotrophic lateral sclerosis has been recently reviewed Citation30, Citation31 and is not the focus of this review. We survey the evidence for mitochondrial dysfunction in neuropsychiatric disorders including mitochondrial gene expression and mtDNA common deletion that are associated with SZ, BPD, and MDD.
Evidence for mitochondrial dysfunction in psychiatric disorders
Psychiatric symptoms of mitochondrial disease
Psychiatric symptoms have been documented in subjects with mitochondrial disease. In a review by Fattal et al. 2006 Citation10, the authors identified 19 confirmed case reports of mitochondrial disease with comorbid psychiatric problems, including MDD, psychosis, BPD, anxiety disorders, and personality change. Depression is a common symptom of the mitochondrial disease CPEO Citation32. A high comorbidity of mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) in SZ and bipolar affective disorder has also been documented Citation33–35. In addition, Grover et al. 2006 Citation36 reported a case of MELAS that was preceded by mania prior to diagnosis, further supporting the role of mtDNA mutations in the etiology of psychiatric disorders. Transgenic mice with mitochondrial DNA deletions related to a mutation in the nuclear-encoded POLG gene also displayed abnormal behaviors including decreased cued and contextual fear conditioning, increased startle response, and decreased and distorted day-night rhythm of wheel-running activity compared to normal mice Citation37 reminiscent of manic-like behaviors.
Ultrastructure studies of mitochondrial abnormalities in psychiatric illness
Electron microscopy studies have revealed mitochondrial abnormalities in SZ. Uranova et al. (2001) studied mitochondrial volume density in oligodendrocytes in the prefrontal cortex and caudate nucleus in postmortem brains of subjects with SZ and observed a significant decrease in mitochondria number and density in both regions in subjects with SZ compared to controls Citation38. Morphological changes in mitochondria have been noted as well. Kung et al. (1999) observed a significant reduction in mitochondrial cross-sectional profiles in caudate and putamen sections from subjects with SZ compared to controls Citation39. They observed a lower number of mitochondria in patients who were not taking antipsychotic medications compared to those taking antipsychotics or controls, suggesting drug treatment normalized mitochondria number. Subjects with SZ were also observed to have monocyte and lymphocyte mitochondria that appeared swollen compared to those of controls Citation40. The same authors observed a significant increase in mitochondria density in lymphocytes and monocytes of neuroleptic-treated subjects with SZ compared to controls Citation40, which might be related to cellular shrinking or mitochondria swelling. There have been no studies to date examining morphological or morphometric abnormalities in BPD or MDD lymphocytes. Interestingly, two strong schizophrenia candidate genes, Disrupted in Schizophrenia 1 (DISC1) and G72, have splicing isoforms that are targeted to mitochondria although the exact functions of these proteins in the mitochondria remain unknown Citation41, Citation42.
Research suggests antipsychotic treatment may be related to increased antiapoptotic effects mediated by mitochondrial signals Citation43. Expression of antiapoptosis genes Bcl-2 and Bcl-xL is increased by haloperidol treatment Citation44–46, and both prevent the release of proapoptotic substances such as cytochrome c Citation44. It is possible that these antiapoptotic effects may be compensatory to the effects of antipsychotics on mitochondrial respiratory chain complex expression Citation47.
Altered metabolic activity
Adenosine triphosphate (ATP) is produced via mitochondrial respiratory chain complexes I–IV. Brain energy metabolism in psychiatric disorders has been studied using 31phosphorus magnetic resonance spectroscopy (31P-MRS) for measurement of cerebral membrane phospholipids and high-energy phosphates. Using 31P-MRS, Volz et al. (1997) found a significant decrease in phosphodiester percent values in the frontal cortex of subjects with SZ compared to controls and an increased ratio of phosphomonoesters/phosphodiesters Citation48. These authors also observed increased levels of phosphocreatine percent and phosphocreatine/ATP in the dorsolateral prefrontal cortex of subjects with SZ compared to controls, and a correlation of β-adenosine triphosphate and phosphocreatine/ATP with the CPZ-equivalent dose Citation48. Decreased levels of ATP in basal ganglia and temporal lobes of subjects with SZ were also observed by 31P-MRS Citation49.
Decreased intracellular pH and intracellular phosphocreatine in the frontal lobes as well as an altered response of phosphocreatine response to photic stimulation have been observed in patients with BPD Citation50, Citation51. Also, mRNA of two isoforms of creatine kinase, the enzyme that is involved in synthesis and metabolism of phosphocreatine, has been shown to be downregulated in the dorsolateral prefrontal cortex in subjects with BPD, although significant downregulation of these isoforms was not seen between subjects with SZ and controls Citation52.
Positron emission tomography (PET) scans with [18F]-fluoro-deoxy-glucose (FDG) have revealed altered metabolic decreases in SZ, BPD, and MDD. Hypofrontality, increased left and decreased right lentiform nucleus metabolic rate, and decreased temporal lobe metabolic rate have been reported in subjects with SZ compared to controls (for review, see Citation54). In major depressive disorder, reduced metabolic rate has been observed in the prefrontal cortex, anterior cingulate cortex, and caudate nucleus in depressed patients (for review, see Citation55). Similarly, depressed subjects with BPD display reduced whole-brain metabolism compared to manic subjects with BPD. Depressed subjects with BPD have also been shown to exhibit decreased total frontal lobe metabolism and medial temporal lobe metabolism. Increases in metabolism in the frontal lobe and medial temporal lobe have been reported during mania in BPD. Thalamus and occipital lobe metabolism has also been shown to be altered in subjects with BPD, although again these findings are also state-specific (for review, see Citation56).
N-acetylaspartate (NAA) is synthesized and exported from mitochondria Citation57. NAA synthesis correlated with mitochondrial metabolism, and NAA concentration has been used as an indicator of neuronal density and mitochondrial function Citation58. [1H]-MRS studies of NAA have shown deficits in SZ (for meta-analysis, see Citation59) and BPD Citation60.
In sum, the literature suggests energy metabolism abnormalities are widespread in the brains of subjects with SZ, BPD, and MDD. A better understanding of the origins of this dysfunction may give us insight into the disease processes in these psychiatric disorders.
Altered mitochondrial gene expression in psychiatric disorders
A broad mitochondrial dysfunction has been reported in neuropsychiatric disorders using microarray technology to survey nuclear-encoded gene expression Citation61–66. One study of BPD found mitochondrial gene expression was predominantly decreased in BPD compared to controls in the dorsolateral prefrontal cortex (DLPFC) when using Stanley samples with pH above 6.5 Citation62. In a select subgroup of medication-free BPD patients there were mitochondrial genes that showed increased expression Citation62. The authors suggested that medication may influence the direction of gene expression change and the precise genes altered in BPD patients; however, this subgroup of medication-free patients showed an increased brain pH as compared to controls. Since the contrast was not balanced between groups for pH, the interpretation that medications were driving the gene expression in the opposite direction can be explained equally by the strong effect of pH. Another microarray and proteomic study using a set of 105 DLPFC samples from the Stanley Array Collection found mitochondrial dysfunction in SZ Citation64. Subsets of genes in the oxidative phosphorylation pathway and proteasome family were found to be decreased in the hippocampus by microarray and also in the prefrontal cortex by Q-PCR in BPD but not SZ Citation63.
In a convincing microarray study of nuclear-encoded mitochondrial-related gene expression, Altar et al. (2005) Citation61 reported decreased expression of mitochondrial gene expression in hippocampal dentate granule neurons sampled with laser capture microscopy in SZ. Vawter et al. (2006) Citation67 found that after correction for pH, the nuclear-encoded mitochondrial-related dysregulation was not above a chance level of significance in the anterior cingulate cortex, dorsolateral prefrontal cortex (DLPFC), and cerebellum for BPD or MDD.
Two independent groups of researchers demonstrated in controls there were mitochondrial gene expression differences based upon mean group pH differences Citation62, Citation67. Iwamoto et al. (2005) reported a control group analysis, in which there were 144 mitochondrial genes (219 probe sets) dysregulated between low-pH control and high-pH control groups. The ratio of downregulated:upregulated was 3:1 comparing low-pH to high-pH controls. Agreement in fold change consistency between Vawter et al. (2006) Citation67 analysis of agonal-pH state in controls and the Iwamoto et al. (2005) Citation62 analysis was high (95.8%, 138/144 genes agreed in direction). Iwamoto et al. (2005) Citation62 reported 31% of genes Citation45 were altered in the control pH analysis and were codirectional with significantly dysregulated genes in either BPD or SZ. In a third study, Mexal et al. (2006) showed that hippocampal gene expression was highly dependent upon pH, especially for functional groups related to mitochondrial energy metabolism Citation68. Taken together, at least three independent microarray studies from different brain banks strongly support that pH differences in postmortem brain affects microarray gene expression for nuclear-encoded transcripts related to mitochondrial functions. pH is a surrogate for agonal hypoxia due to lactic acid accumulation in brain that commences with oxygen deprivation. A more difficult question to address technically is whether differences in mitochondria physiology associated with agonal-pH differences influence the same genes implicated with psychiatric disorders. Recently, Weis et al. (2007) Citation69 demonstrated that pH was decreased in BPD (n=113) and SZ (n=120) postmortem brain tissue compared to controls (n=120). This interesting study also compared control subjects with obvious histopathological abnormalities and cerebellar granular cell layer necrosis (CGLN) to controls subjects without CGLN and found decreased pH of 0.4 in CGLN + group. However, when comparing CGLN+ to CGLN- SZ groups the pH was reduced only 0.04 pH units, which was not significant. This diagnosis group×CGLN interaction for pH is one example of how the field is grappling with how to control for strong associations with postmortem gene expression overall. The Weis et al. 2007 Citation69 study did not rule out a relationship of agonal factors to pH which could have influenced the group differences. Linear regression models would be useful to predict pH using the data available at the Stanley Foundation-557 collection Citation69 to determine the strongest predictors of pH. Beyond the usual predictors of age, PMI, agonal factors and tissue necrosis, it would be interesting to see if the lifetime fluphenazine daily equivalents, smoking, and other variables such as mtDNA SNPs (see below) were strongly predictive.
Since lowered intracellular pH has been demonstrated in BPD using whole-head 31P-MRS spectra Citation70, this seems to indicate that the mitochondrial pathophysiology preexists terminal conditions surrounding death and produce differences in physiological responses relevant to cellular bioenergetics. However, the reduced pH in vivo in BPD has not yet been replicated.
The evidence for nuclear-encoded mitochondrial gene expression alterations as part of the pathophysiology of SZ and BPD is not unequivocal, notwithstanding some evidence that has been found for psychiatric comorbidity in classical mitochondrial disorders and for brain imaging relevant to mitochondrial function such as hydrogen ion alterations in BPD Citation71. Some microarray studies have not reported a broad mitochondrial gene expression pattern in BPD Citation72, Citation73 or SZ Citation62, Citation74, Citation75. A few microarray studies in MDD have not reported that mitochondrial genes were differentially expressed in cortex Citation76–78. Further it was demonstrated that alcohol disorders and drug abuse comorbidity may play a factor in alterations involving mitochondrial-related genes Citation66. Taken together, a broad picture emerges from the nuclear gene expression studies, which suggests that mitochondrial dysfunction is modest when examined in view of strong agonal-pH background effects on nuclear-encoded transcripts and mtDNA copy number. The dilemma is caused by the agonal-pH effects that are almost always codirectional with the mitochondrial gene expression change in psychiatric disorder when the groups are unbalanced for agonal-pH factors. After parcellation of this strong effect of agonal-pH state on gene expression, the differential diagnostic effects on gene expression are significantly reduced Citation67. One caveat in performing human postmortem brain experiments at one time point prevents ruling out genes that have strong agonal-pH effects on gene expression as being part of the pathophysiology of the disorder. One method is to develop animal and cellular models that have profiles similar to BPD and SZ gene expression profiles and to show that the pathophysiology involves mitochondrial-related gene expression. The other method is to perform rigorously controlled studies of covariates and gene expression Citation62, Citation69, Citation79–81 which we further comment upon below.
Common and novel mitochondrial DNA SNPs associated with psychiatric disorders
A small body of literature has implicated sequence variations in the mitochondrial genome in the etiology of psychiatric disorders. Several studies have approached this question by resequencing the entire mitochondrial genomes of subjects with psychiatric disorders. These initial studies have been performed with small numbers of subjects and have not been validated with brain tissue. It has been shown that heteroplasmic mutations can increase with aging and neurodegeneration, thus more sensitive adjunct approaches to traditional sequence-based approaches will be needed to screen for variations in mitochondrial DNA. Traditional PCR-based approaches such as high-resolution thermal melt analysis and electrophoresis techniques will allow screening for private sequence variations. Microarray resequencing of the entire human mitochondrial genome is now possible using the Affymetrix platform (Santa Clara, CA).
Martorell et al. (2006) Citation82 resequenced the entire mitochondrial genome in leukocytes from six subjects with SZ with apparent maternal transmission and their schizophrenic mothers. Because multiple copies of mtDNA exist per cell, it is possible for a cell to contain mutant mtDNA and normal mtDNA in the same cell. This is known as heteroplasmy Citation28. The six subjects with SZ had 50 variants compared to the revised Cambridge reference sequence Citation82. The variants did not accumulate in a specific region of the mtDNA genome. Of the 50 variants, 6 were novel, and 3 of these were missense mutations. The heteroplasmic missense variant MT-ND4 12096T > A was found in 5 of the 6 mother/offspring schizophrenic pairs; however, none of the 3 missense variants were present in the 95 controls individuals. Two missense variants associated with SZ in complex I (MT-ND5 C12403T and MT-ND5 Asn105Thr) Citation82 were previously described variants associated with BPD Citation28.
Similarly, Munakata et al. (2007) Citation83 resequenced the entire mitochondrial genome from leukocytes in one proband from a family with suspected maternal inheritance of BPD, MDD, suicide, or other psychotic disorders. The proband was diagnosed with MDD and epilepsy. The authors compared the sequence of the probands to the human standard sequence of mtDNA and observed 34 base substitutions: 11 in the region of the D-loop, which is necessary for replication and transcription of the mitochondrial genome, 3 in rRNA, 13 synonymous substitutions, and 7 non-synonymous substitutions. All were registered polymorphisms in the mitochondrial database MITOMAP, except for two SNPs—a non-synonymous SNP in complex I (MT-ND1 T3394C) that has been previously implicated in two mitochondrial diseases, and a non-synonymous SNP in complex V (MT-ATP6 A9115G).
Kazuno et al. (2005) resequenced mitochondrial DNA from leukocytes of seven patients with atypical psychosis. A total of 67 SNPs were found in comparison to the revised Cambridge reference sequence Citation84. Two subjects shared 25 polymorphisms and these subjects belonged to the sub-haplogroup F1ba of the macro-haplogroup N, leading the authors to speculate that this haplotype may be a risk factor for atypical psychosis. However, Kirk et al. (1999) resequenced the mitochondrial genome from 25 subjects with bipolar affective disorder who had family histories suggestive of inheritance from the maternal side, but they did not find a relationship between haplotype frequency in subjects with bipolar affective disorder compared to controls Citation85.
In addition to whole mitochondrial genome resequencing studies, another approach has been to examine the relationship between specific polymorphisms and risk for psychiatric disorders. Marchbanks et al. (2003) examined the ratio of the heteroplasmic variant 12027T > C to the total 12027T, a non-synonymous SNP in the ND4 subunit of complex I Citation86. There was a significant increase in the variant-rich fraction in leukocytes from subjects with SZ (n=181) compared to controls (n=184); moreover, although there was not a significant difference in the variant-rich fraction between males and females, there was a significant increase in the variant-rich fraction in control females (36%) compared to control males (18%), raising the possibility that the variant increased risk for SZ more in males than females Citation86.
Munakata et al. (2005) Citation87 examined the mtDNA tRNALeu(UUR) 3243A > G mutation in the prefrontal cortex and liver in subjects with MDD, BPD, SZ, and controls. This mutation is associated with the mitochondrial diseases MELAS, diabetes mellitus, CPEO and mitochondrial myopathy. In a total of 57 subjects, with roughly equal numbers of subjects per group, the authors identified 2 subjects with BPD and 1 subject with SZ carrying this heteroplasmic mutation, while no subjects with MDD or controls were observed to carry this mutation. A mtDNA 3644T > C homoplasmic mutation in the NADH-ubiquinone dehydrogenase subunit 1 (MT-ND1) gene was observed in subjects with BPD Citation88.
Mitochondrial function may affect personality traits, as well as risk for psychiatric disorders. Kato et al. (2004) Citation89 examined two mtDNA polymorphisms, C5178A and A10398G, which are located in coding regions of complex I NADH dehydrogenase subunit 2 (MT-ND2) and NADH dehydrogenase subunit 3 (MT-ND3) genes. They reported a significant association between the C5178A polymorphism and extraversion in healthy controls (n=238). A separate study suggested the mtDNA A10398G polymorphism of the MT-ND3 genes may be related to lithium response in BPD Citation90.
The majority of the studies to date have failed to implicate any particular variant in association with psychiatric disorders, although these studies involved resequencing or genotyping of leukocyte DNA. It is possible that heteroplasmic mutations that accumulate with age in the brain may be involved in psychiatric disorders and will go undetected when DNA from peripheral blood, rather than brain tissue, is sequenced. Another reason for this failure to replicate the many mitochondrial findings may be the relatively small sample sizes generally seen in these studies. It is also possible that the differences in results are due to haplogroup variation, as well as private mutations occurring within families and individuals. It is possible that dysfunction of the electron transport chain can be caused by combinations of mtDNA genotypes derived from haplogroup, which could increase risk for SZ or BPD, in conjunction with nuclear DNA variations in mitochondrial-related genes.
Copy number variations or deletions/insertions associated with psychiatric disorders
The most frequent mitochondrial deletion is a large 4977 base pair ‘common deletion’, and several groups have investigated this deletion in psychiatric disorders. As the original observation of an age-related increase in the common deletion, Wallace et al. (1987) showed that the common deletion increased between ages 45 and 55 Citation91. Depending on the sensitivity of the method to detect the common deletion, it might be missed entirely in younger subjects. Kato et al. (1997) Citation92 reported an increased ratio of the common deletion in the cerebral cortex of subjects with BPD (n=7) compared to age-matched control subjects (n=8). However, no abnormalities were reported in a study Citation93 of prefrontal cortex common deletion levels from subjects with SZ (n=30) or BPD (n=25) compared to controls (n=29). Cavalier et al. (1995) Citation94 found significantly reduced cytochrome c oxidase enzyme activity in the frontal cortex and the caudate nucleus in subjects with SZ compared to controls, but, again, no change in common deletion levels was seen in subjects with SZ compared to controls. However, these authors also failed to observe age-related accumulation of the common deletion in SZ Citation94, which is a feature of normal control brains Citation95. Kakiuchi et al. (2005) Citation96 also failed to see an increase in the common deletion in subjects with BPD or SZ, or age-related increases in levels of the deletion. However, the lack of an increase in age-related deletions may be related to the overall younger age of the subjects, as age-matched controls apparently did not display age-related increases in deletion Citation96, or perhaps the methodology. Sabunciyan et al. (2007) Citation93 reported that antidepressant and alcohol use in bipolar patients and first-generation typical antipsychotics in SZ were correlated to the level of common deletion.
Several groups have examined mtDNA copy number in psychiatric disorders. Vawter et al. (2006) Citation67 reported a strong association of prolonged death (presumably longer intervals of hypoxia) with increased mtDNA copy number in DLPFC. After controlling for the strong effects of agonal duration, there was a trend towards decreased mtDNA copy number in BPD compared to controls Citation67.The copy number of mtDNA can be induced in hypoxic states Citation97 resulting in mitochondrial replication and/or mtDNA replication. As mentioned earlier, multiple copies of mtDNA are found within single mitochondria. Vawter et al. (2006) Citation67 reported about 50–500 copies of mtDNA per copy of nuclear DNA in DLPFC homogenate depending on agonal-pH state of individual subjects. Others have reported a 10-fold higher range of mtDNA copy number of 1259–4617 varying widely among brain regions, agonal factors, and methodology Citation98.
Investigation of the relationship between mitochondrial gene expression and common deletion in psychiatry disorders
The reviewed studies provided the motivation to investigate both mitochondrial gene expression and common deletion in DLPFC in a cohort of subjects with SZ, MDD, and BPD compared to matched controls.
Materials and methods
Subjects
The DLPFC (area 9, plus 46) was collected at the University of California, Irvine Brain Repository Citation67. A total of 78 subjects (12 with BPD, 14 with SZ, 15 with MDD, and 37 controls) were studied. Age, gender and pH of each subject/sample are summarized in . The subjects all had rapid deaths, as a prolonged death was previously shown to increase the mtDNA copy number Citation67.
Table I. Demographics of four groups of subjects from which DLPFC was obtained. All subjects had a rapid death and absence of prolonged hypoxia prior to death.
RNA and DNA extraction
RNA and DNA were extracted using Trizol Reagent (Invitrogen). RNA was cleaned with RNeasy Plus Mini Kits and treated with RNase Free-DNase Set (Qiagen, Valencia, CA, USA). The protocol included passage through a membrane-based Qiagen column (QIAamp DNA Mini Kit, Qiagen Sciences, MD). Total RNA quality, indicated by the ratio of 28S/18S ribosomal peaks, was analyzed on an Agilent Bioanalyzer 2100 (Agilent, Palo Alto, CA, USA). The cleaned RNA was reverse-transcribed with Taqman Reverse Transcript Reagent (N808-0234, Applied Biosystems).
Real-time PCR
Real-time PCR with both SybrGreen and Taqman was performed on a 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA) with SybrGreen PCR Master Mix (Applied Biosystems, Foster City, CA) or Taqman Universal PCR Master Mix (Applied Biosystems, Foster City, CA), respectively. For quantitative real-time PCR, the relative quantization method was used. A serial dilution of a control sample from the same brain region was used for a standard curve in every assay, and Ct was converted to a relative concentration from the standard curve on each plate.
Primers
Expression of the 13 mitochondrial-encoded genes and D-loop were chosen to compare expression between diagnosis groups and controls. The primers for targeting conservative or deletion regions were based on MITOMAP (A Human Mitochondrial Genome Database: http://www.mitomap.org, 2006) using the revised Cambridge Reference sequence. All primers were designed with Primer Express (Applied Biosystems) and synthesized at ABI (sequences available upon request from authors). The average of three nuclear genes (GAPDH, BEXL1, and CFL1) was used as a reference for normalization. D-loop primers and probes were designed in a non-variable region.
MtDNA common deletion
The amount of the mtDNA common deletion was screened by two methods. We tested coamplification in a linear range and direct visualization of the common deletion band on Agilent gel-based quantification (PCR amplicon of deleted band is 350 bp compared to 5000 bp for non-deleted mtDNA band). We also used a TaqMan PCR method to compare the amplification of the relative level of deleted mtDNA to wild-type DNA by amplifying a region of the mitochondrial genome that is commonly deleted (in the MT-ND4 gene) and a region that is not commonly deleted (DLOOP). This allows for an estimation of ratio of non-deleted mtDNA (MT-ND4) to copy number (DLOOP). We hypothesized that the ratio of non-deleted mtDNA to copy number would be higher in controls than in experimental groups. Both screening methods showed an increase in BPD for indirect measures of the common deletion.
Data analysis
Both t test and ANCOVA (Partek, St Louis, MO) were used for significance test and covariance corrections. The copy number of the subjects was assessed in homogenized frontal cortex tissue by measuring nuclear DNA, mtDNA, and forming a ratio using a method previously reported in dissected single neurons from the substantia nigra Citation99.
MtDNA-encoded gene expression
After adjusting for age, gender, pH, and ASR covariates Citation100, there were significantly decreased expressions of 10/13 and 6/13 mitochondrial-encoded transcripts for SZ and MDD, respectively, compared to controls (). Lithium treatment in BPD was associated with a decrease in expression of all 13 transcripts by 19%–37% compared to non-lithium-treated subjects with BPD, although this decrease was not significant for individual mtDNA transcripts. After normalizing data using the D-loop as a reference gene, two significantly increased mitochondrial-expressed genes were noted in BPD (MT-ND1 and MT-ND5). Overall, the expression results suggest decreased mitochondrial transcription in SZ and MDD. However, when mtDNA transcripts were internally normalized to D-loop, there were no expression differences. This is likely because the nascent message RNA is without introns. Further examination of the D-loop region for variants is required as well as the nuclear DNA genes that impact mtDNA transcript expression.
Table II. Gene expression results of 13 mitochondrial genes and D-loop. The t test and ANCOVA values are shown.
The estimated ratio of deletion by PCR analysis was detected at significantly higher levels in BPD compared to controls (2.12-fold change, ) after appropriate age and gender adjustment. Further, MDD and SZ showed trends toward increase (). We verified that the ratio of common deletion was highly correlated at two different genomic DNA concentrations (r=0.98 after removing two outliers), and normalized directly by the amount of mtDNA in the same reaction tube to increase the measurement reliability. Consistent with previous reports, there was a significant positive relationship between age and the amount of estimated common deletion in brain (; r=0.40). Overall, this study suggests mitochondrial dysfunction and transcript alterations in the brain (, ) after age adjustment.
Figure 2. The mitochondrial common deletion is increased in psychiatric disorders. The individual subjects are plotted by age and common deletion (x-axis ratio of 350/5000 bp). Subjects with BPD died at a younger age in this study; there was a significant increase in common deletion after age adjustment. The blue circle indicates the 95% confidence limits around the control subjects. The increase in the common deletion for nine psychiatric subjects is shown outside of the blue circle. Group abbreviations are shown in .
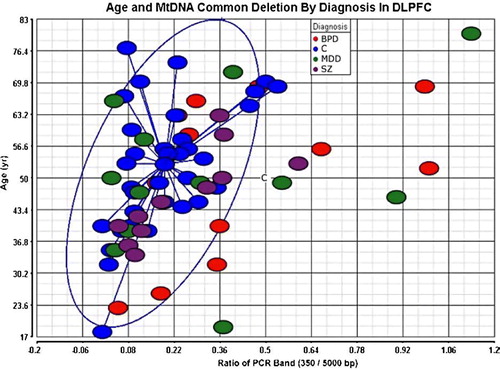
Figure 3. Genotyping for large common deletion. Subjects were genotyped in duplicate at two concentrations. Arrows indicate presence of deletion relative to 5000 bp upper non-deleted band. Group abbreviations are shown in .
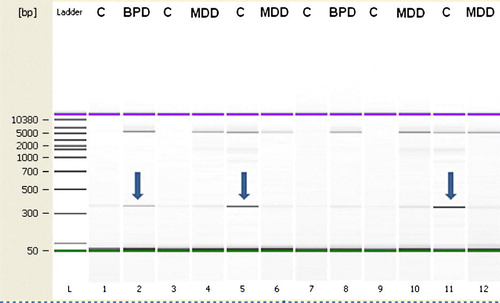
Table III. Mitochondrial common deletion (ratio of 350 bp/5000 bp) is increased in BPD. PCR amplification around the deleted fragment (350 bp) and the non-deleted fragment (5000 bp) was measured in the same PCR reaction and Agilent gel lane (see ). This assay was linear in the range of 10–20 ng of DNA (r=0.98), and duplicate values were averaged for each sample.
Conclusions
Multiple lines of evidence suggest mitochondrial dysfunction may play a role in psychiatric disorders. The small but growing body of evidence, including morphological, genetic, comorbidity with mitochondrial disorders, and imaging data, supports the role of mitochondrial dysfunction in SZ, BPD, and MDD. A high comorbidity between mitochondrial disease and psychiatric disorder has been noted Citation10, Citation32, Citation101. Further, mitochondrial dysfunction as evidenced by abnormal mitochondrial morphology and morphometry has been noted Citation38–40, and altered metabolic activity has been observed in subjects with psychiatric disorders Citation48, Citation49, Citation53–56.
Altered mitochondrial genome expression has also been suggested, particularly of genes encoding complex I Citation102. Searching for candidate mtDNA SNPs associated with SZ, BPD and MDD has yielded a number of positive findings, although these results tend not to be replicated, possibly due to low sample sizes or an association of multiple nuclear and mitochondrial genomic variants in combination that lead to respiratory chain dysfunction and predisposition to these disorders. Nevertheless, the results of these studies suggest a role of mitochondrial dysfunction in the etiology of psychiatric disorders.
This is the first examination of the 13 coding transcripts and levels of common deletion in brain tissue from three psychiatric disorders. There were significant alterations in expression of 10/13 and 6/13 mitochondrial transcripts for SZ and MDD, respectively. In BPD an increased mtDNA common deletion was found and non-significant increases were seen for SZ or MDD. It is important to note that originally this method for quantification of the common deletion was reported in single cells from the substantia nigra Citation99 while the present study is the first to adapt this method to quantify the deletion in homogenized frontal cortex tissue. A shortcoming of this method when used on DNA from homogenized brain tissue is that it approximates the ratio of total deleted to non-deleted mtDNA and therefore may result in an underestimation of common deletion when a relatively low proportion of cells have the heteroplasmic common deletion. This may explain the lack of a significant increase seen in SZ and MDD. However, despite this limitation, we were still able to detect an increase in mtDNA common deletion in BPD.
The findings of alterations in mitochondrial transcript expression appeared to be independent of the common deletion as there were no significant correlations between mitochondrial gene expressions for the 13 transcripts with the levels of common deletion. However, the direction in the data suggests that an increased common deletion is associated with decreased mitochondrial gene expression. This observation requires confirmation with a larger cohort and suggests that accumulation of the common deletion and decrease in transcription overall may be susceptibility factors for neuropsychiatric disorders Citation103, Citation104. The effect of medications cannot be excluded, especially as some medications, such as haloperidol, have been suggested to have mitochondrial effects Citation105.
Future directions
The further assessment of mtDNA variations to correlate with transcript abnormalities and accumulation of common deletions is warranted in brain. Examination of the entire mitochondrial DNA sequence is feasible to determine detailed sub-haplogroupings, novel mutations, and common SNPs previously reported. MtDNA resequencing can determine private mutations, especially if homoplasmic, which accounts for a substantial proportion of mtDNA variation between individuals.
Further, resequencing the D-loop coding region may help elucidate whether variations in this region may be associated with decreased transcription. SNP variations in the hypervariable D-loop region might account for differential mtDNA copy number, transcript, and deletions Citation106. While information from homogenized brain tissue does not yield cellular specificity of the mtDNA alterations that distinguishes controls from neuropsychiatric groups, the use of single-cell expression quantification may lead to a better understanding of the role of mitochondrial pathology in psychiatric disorders.
Acknowledgements
We appreciate the assistance of David Walsh, PsyD, Preston Cartagena, PsyD, and Richard Stein, PhD, for their contributions to postmortem collections and clinical characterization of subjects. We acknowledge Kathleen Burke as well as Jacque Berndt and the investigators and medical examiners at the Orange County Coroner's Office for procurement of brain tissue. F. Warren Lovell, MD, performed a neuropathological evaluation of the postmortem brains. Tissue specimens were processed and stored at the University of California, Davis and the Human Brain and Spinal Fluid Resource Center, Veteran's Medical Center, Los Angeles under the direction of Wallace W. Tourtellotte, MD, PhD. This project is supported by the NIMH Conte Center Grant P50 MH60398, Pritzker Family Philanthropic Fund, William Lion Penzner Foundation (UCI), Della Martin Foundation (UCI), NIMH Grant #MH54844 (EGJ), WM Keck Foundation (EGJ), and the NIMH Program Project MH42251 (SJW and HA). The authors are members of a Conte Center supported by the NIMH and members of the Pritzker Neuropsychiatric Disorders Research Consortium, which is supported by the Pritzker Family Philanthropic Fund. A shared intellectual property agreement exists between the Pritzker Family Philanthropic Fund and all the universities involved, in order to encourage the development of appropriate findings for research and clinical applications. The academic and philanthropic entities involved in this Consortium are jointly filing patent applications related to the present findings.
Ling Shao and Maureen V. Martin made equal contributions to this article.
References
- Anderson S, Bankier AT, Barrel BG, de Bruijn MHL. Sequence and organization of the human mitochondrial genome. Nature. 1981; 290: 457–65
- Kato T. The other, forgotten genome: mitochondrial DNA and mental disorders. Mol Psychiatry. 2001; 6: 625–33
- Horton HR, Moran LA, Ochs RS, Rawn JD, Scrimgeour KG. Principles of Biochemistry. Neil Patterson Publishers, Englewood Cliffs, NJ 1993
- Moyes CD, Buck LT, Hochachka PW. Temperature effects on pH of mitochondria isolated from carp red muscle. Regulatory, integrative and comparative physiology. Am J Physiol. 1988; 254: 611–5
- Chinnery PF, Schon EA. Mitochondria. J Neurol Neurosurg Psychiatry. 2003; 74: 1188–99
- Shanske AL, Shanske S, DiMauro S. The other human genome. Arch Pediatr Adolesc Med. 2001; 155: 1210–6
- Orth M, Schapira AH. Mitochondria and degenerative disorders. Am J Med Genet. 2001; 106: 27–36
- Johns DR. The other human genome: mitochondrial DNA and disease. Nat Med. 1996; 2: 1065–8
- Shoffner JM, Wallace D. The metabolic and molecular bases of inherited disease7th ed. McGraw-Hill, New York 1995
- Fattal O, Budur K, Vaughan AJ, Franco K. Review of the literature on major mental disorders in adult patients with mitochondrial diseases. Psychosomatics. 2006; 47: 1–7
- Dröge W, Schipper HM. Oxidative stress and aberrant signaling in aging and cognitive decline. Aging Cell. 2007; 6: 361–70
- Schaitman C, Greenawalt JW. Enzymatic properties of the inner and outer membranes of rat liver mitochondria. J Cell Biol. 1968; 38: 158–75
- Smith D, Filipowicz C, McCauley R. Monoamine oxidase A and monoamine oxidase B activities are catalyzed by different proteins. Biochim Biophys Acta. 1985; 831: 1–7
- Krueger KE, Papdopoulos V. Mitochondrial benzodiazepine receptors and the regulation of steroid biosynthesis. Annu Rev Pharmacol Toxicol. 1992; 32: 211–37
- Schwartz M, Vissing J. New patterns of inheritance in mitochondrial disease. Annu Rev Pharmacol Toxicol. 2003; 310: 247–51
- Gottesman II, Bertelsen A. Confirming unexpressed genotypes for schizophrenia. Risks in the offspring of Fischer's Danish identical and fraternal discordant twins. Arch Gen Psychiatry. 1991; 46: 867–72
- Goldstein JM, Faraone SV, Chen WJ, Tsuang MT. Gender and the familial risk for schizophrenia. Disentangling confounding factors. Schizophr Res. 1992; 7: 135–40
- Wolyniec PS, Pulver AE, McGrath JA, Tam D. Schizophrenia: gender and familial risk. J Psychiatr Res. 1992; 26: 17–27
- Shimizu A, Kurachi M, Yamaguchi N, Torii H, Isaki K. Morbidity risk of schizophrenia to parents and siblings of schizophrenic patients. J Psychiatry Neurol. 1987; 41: 65–70
- McMahon FJ, Stine OC, Meyers DA, Simpson SG, DePaulo JR. Patterns of maternal transmission in bipolar affective disorder. Am J Hum Genet. 1995; 56: 1277–86
- Doi N, Hoshi Y. Persistence problem in schizophrenia and mitochondrial DNA. Am J Med Genet B Neuropsychiatr Genet. 2007; 144B: 1–4
- DeLisi LE, Razi K, Stewart J, Relja M, Shields G, Smith AB, et al. No evidence for a parent-of-origin effect detected in the pattern of inheritance of schizophrenia. Biol Psychiatry. 2000; 48: 706–9
- Kornberg JR, Brown JL, Sadovnick AD, Remick RA, Keck PE, McElroy SL, et al. Evaluating the parent-of-origin effect in bipolar affective disorder. Is a more penetrant subtype transmitted paternally?. J Affect Disord. 2000; 59: 183–92
- Wallace DC. Diseases of the mitochondrial DNA. Annu Rev Biochem. 1992; 61: 1175–212
- Hudson G, Carelli V, Spruijt L, Gerards M, Mowbray C, Achilli A, et al. Clinical expression of Leber hereditary optic neuropathy is affected by the mitochondrial DNA-haplogroup background. Am J Hum Genet. 2007; 81: 228–33
- Hao H, Bonilla E, Manfredi G, DiMauro S, Moraes CT. Segregation patterns of a novel mutation in the mitochondrial tRNA glutamic acid gene associated with myopathy and diabetes mellitus. Am J Hum Genet. 1995; 56: 1017–25
- Di Fonzo A, Bordoni A, Crimi M, Sara G, Del Bo R, Bresolin N, et al. POLG mutations in sporadic mitochondrial disorders with multiple mtDNA deletions. Hum Mutat. 2003; 22: 498–506
- Simon DK, Johns DR. Mitochondrial disorders: clinical and genetic features. Annu Rev Med. 1999; 50: 111–27
- Xu F, Morin C, Mitchell G, Ackerly A, Robinson BH. The role of the LRPPRC (leucine-rich pentatricopeptide repeat cassette) gene in cytochrome oxidase assembly: mutation causes lowered levels of COX (cytochrome c oxidase) I and COX III mRNA. Biochem J. 2004; 382: 331–6
- Petrozzi L, Ricci G, Giglioli NJ, Siciliano G, Mancuso M. Mitochondria and neurodegeneration. Biosci Rep. 2007; 27: 87–104
- Kalman B, Laitinen K, Komoly S. The involvement of mitochondria in the pathogenesis of multiple sclerosis. J Neuroimmunol. 2007; 188: 1–12
- DiMauro S, Moraes CT. Mitochondrial encephalomyopathies. Arch Neurol. 1993; 50: 1197–208
- Oexle K, Zwirner A. Advanced telomere shortening in respiratory chain disorders. Hum Mol Genet. 1997; 6: 905–8
- Prayson RA, Wang N. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes (MELAS) syndrome: an autopsy. Arch Pathol Lab Med. 1998; 122: 978–81
- Siciliano G, Tessa A, Petrini S, Mancuso M, Bruno C, Grieco GS, et al. Autosomal dominant external ophthalmoplegia and bipolar affective disorder associated with a mutation in theANT1 gene. Neuromuscul Disord. 2003; 13: 162–5
- Grover S, Padhy SK, Das CP, Vasishta RK, Sharan P, Chakrabarti S. Mania as a first presentation in mitochondrial myopathy. Psychiatry Clin Neurosci. 2006; 60: 774–5
- Kasahara T, Kubota M, Miyauchi T, Noda Y, Mouri A, Nabeshima T, et al. Mice with neuron-specific accumulation of mitochondrial DNA mutations show mood disorder-like phenotypes. Mol Psychiatry. 2006; 11: 577–93
- Uranova N, Orlovskaya D, Vikhreva O, Zimina I, Kolomeets N, Vostrikov V, et al. Electron microscopy of oligodendroglia in severe mental illness. Brain Res Bull. 2001; 55: 597–610
- Kung L, Roberts RC. Mitochondrial pathology in human schizophrenic striatum: a postmortem ultrastructural study. Synapse. 1999; 31: 67–75
- Inuwa IM, Peet M, Williams MA. QSAR modeling and transmission electron microscopy stereology of altered mitochondrial ultrastructure of white blood cells in patients diagnosed as schizophrenia and treated with antipsychotic drugs. Biotech Histochem. 2005; 80: 133–7
- James R, Adams RR, Christie S, Buchanan DJ, Porteous DJ, Millar JK. Disrupted in Schizophrenia 1 (DISC1) is multicompartmentalized protein that predominantly localizes to mitochondria. Mol Cell Neurosci. 2004; 26: 112–22
- Kvajo M, Dhilla A, Swor DE, Karayiorgou M, Gogos JA. Evidence implicating the candidate schizophrenia/bipolar disorder susceptibility gene G72 in mitochondrial function. Mol Psychiatry. 2007 Aug 7 [ Epub ahead of print].
- Rodrigues T, Santos AC, Pigoso AA, Mingatto FE, Uyemura SA, Curti C. Thrioridazine interacts with the membrane of mitochondria acquiring antioxidant activity toward apoptosis-potentially implicated mechanisms. Br J Pharmacol. 2002; 136: 136–42
- Tsujimoto Y. Role of Bcl-2 family proteins in apoptosis: apoptosomes or mitochondria?. Genes Cells. 1998; 3: 697–707
- Saldoña M, Bonastre M, Aguilar E, Marin C. Differential nigral expression of Bcl-2 protein family in chronically haloperidol and clozapine-treated rats: role in neurotoxicity and stereotyped behavior. Exp Neurol. 2007; 203: 302–8
- Wei Z, Mousseau DD, Richardson JS, Dyck LE, Li XM. Atypical antipsychotics attenuate neurotoxicity of beta-amyloid (25–35) by modulating Bax and Bcl-X(1/s) expression and localization. J Neurosci Res. 2003; 74: 942–7
- Dean CE. Antipsychotic-associated neuronal changes in the brain: Toxic, therapeutic, or irrelevant to the long-term outcome of schizophrenia?. Prog Neuropsychopharmacol Biol Psychiatry. 2006; 30: 174–89
- Volz HP, Rzanny R, May S, Hegewald H, Preussler B, Hajek M, et al. 31P magnetic resonance spectroscopy in the dorsolateral prefrontal cortex of schizophrenics with a volume selective technique—preliminary findings. Biol Psychiatry. 1997; 41: 644–8
- Kegeles LS, Humaran TJ, Mann JJ. In vivo neurochemistry of the brain in schizophrenia as revealed by magnetic resonance spectroscopy. Biol Psychiatry. 1998; 44: 382–98
- Deicken RF, Fein G, Weiner MW. Abnormal frontal lobe phosphorous metabolism in bipolar disorder. Am J Psychiatry. 1995; 152: 915–8
- Hamakawa H, Kato T, Shioiri T, Inubushi T, Kato N. Quantitative proton magnetic resonance spectroscopy of the bilateral frontal lobes in patients with bipolar disorder. Psychol Med. 1999; 29: 639–44
- MacDonald ML, Naydenov A, Chu M, Matzilevich D, Konradi C. Decrease in creatine kinase messenger RNA expression in the hippocampus and dorsolateral prefrontal cortex in bipolar disorder. Bipolar Disord. 2006; 8: 255–64
- Prince JA, Harro J, Blennow K, Gottfries CG, Oreland L. Putamen mitochondrial energy metabolism is highly correlated to emotional and intellectual impairment in schizophrenics. Neuropsychopharmacology. 2000;22:284–92. doi: 10.1038/sj.mp.4002052.
- Buchsbaum MS, Hazlett EA. Positron emission tomography studies of abnormal glucose metabolism in schizophrenia. Schizophr Bull. 1998; 24: 343–64
- Videbech P. PET measurements of brain glucose metabolism and blood flow in major depressive disorder: a critical review. Acta Psychiatrica Scandinavica. 2000; 101: 11–20
- Strakowski SM, DelBello MP, Adler C, Cecil KM, Sax KW. Neuroimaging in bipolar disorder. Bipolar Disord. 2000; 2: 148–64
- Patel TB, Clark JB. Synthesis of N-acetyl-L-aspartate by rat brain mitochondria and its involvement in mitochondrial/cytosolic carbon transport. Biochem J. 1979; 184: 539–46
- Clark JB. N-acetyl aspartate: a marker for neuronal loss or mitochondrial dysfunction. Dev Neurosci. 1998; 20: 271–6
- Steen RG, Hamer RM, Lieberman JA. Measurement of brain metabolites by 1H magnetic resonance spectroscopy in patients with schizophrenia: a systematic review and meta-analysis. Neuropsychopharmacology. 2005; 30: 1949–62
- Yildiz-Yesiloglu A, Ankerst DP. Neurochemical alterations of the brain in bipolar disorder and their implications for pathophysicology: A systematic review of the in vivo proton magnetic resonance spectroscopy findings. Prog Neuropsychopharmacol Biol Psychiatry. 2006; 30: 969–95
- Altar CA, Jurata LW, Charles V, Lemire A, Liu P, Bukhman Y, et al. Deficient hippocampal neuron expression of proteasome, ubiquitin, and mitochondrial genes in multiple schizophrenia cohorts. Biol Psychiatry. 2005; 58: 85–96
- Iwamoto K, Bundo M, Kato T. Altered expression of mitochondria-related genes in postmortem brains of patients with bipolar disorder or schizophrenia, as revealed by large-scale DNA microarray analysis. Hum Mol Genet. 2005; 14: 241–53
- Konradi C, Eaton M, MacDonald ML, Walsh J, Benes FM, Heckers S. Molecular evidence for mitochondrial dysfunction in bipolar disorder. Arch Gen Psychiatry. 2004; 61: 300–8
- Prabakaran S, Swatton JE, Ryan MM, Huffaker SJ, Huang JT, Griffin JL, et al. Mitochondrial dysfunction in schizophrenia: evidence for compromised brain metabolism and oxidative stress. Mol Psychiatry. 2004; 9: 684–97
- Middleton FA, Mirnics K, Pierri JN, Lewis DA, Levitt P. Gene expression profiling reveals alterations of specific metabolic pathways in schizophrenia. J Neurosci. 2002; 22: 2718–29
- Solokov B, Jiang L, Trivedi N, Aston C. Transcription profiling reveals mitochondrial, ubiquitin and signaling systems abnormalities in postmortem brains from subjects with a history of alcohol abuse or dependence. J Neurosci Res. 2003; 72: 756–67
- Vawter MP, Tomita H, Meng F, Bolstad B, Li J, Evans S, et al. Mitochondrial-related gene expression changes are sensitive to agonal-pH state: implications for brain disorders. Mol Psychiatry. 2006; 11: 663–79
- Mexal S, Berger R, Adams C, Ross R, Freedman R, Leonard S. Brain pH has a significant impact on human postmortem hippocampal gene expression profiles. Brain Res. 2006; 1106: 1–11
- Weis S, Llenos IC, Dulay JR, Elashoff M, Martinez-Murillo F, Miller CL. Quality control for microarray analysis of human brain samples: The impact of postmortem factors, RNA characteristics, and histopathology. J Neurosci Methods. 2007; 165: 198–209
- Hamakawa H, Murashita J, Yamada N, Inubushi T, Kato N, Kato T. Reduced intracellular pH in the basal ganglia and whole brain measured by 31P-MRS in bipolar disorder. Psychiatry Clin Neurosci. 2004; 58: 82–8
- Kato T, Kato N. Mitochondrial dysfunction in bipolar disorder. Bipolar Disord. 2000; 2: 180–90
- Jurata LW, Bukhman YV, Charles V, Capriglione F, Bullard J, Lemire AL, et al. Comparison of microarray-based mRNA profiling technologies for identification of psychiatric disease and drug signatures. J Neurosci Methods. 2004; 138: 173–88
- Bezchlibnyk YB, Wang JF, McQueen GM, Young LT. Gene expression differences in bipolar disorder revealed by cDNA array analysis of post-mortem frontal cortex. J Neurochem. 2001; 79: 826–34
- Hakak Y, Walker JR, Li C, Wong WH, Davis KL, Buxbaum JD, et al. Genome-wide expression analysis reveals dysregulation of myelination-related genes in chronic schizophrenia. Proc Natl Acad Sci U S A. 2001; 98: 4746–51
- Vawter MP, Crook JM, Hyde TM, Kleinman JE, Weinberger DR, Becker KG, et al. Microarray analysis of gene expression in the prefrontal cortex in schizophrenia: a preliminary study. Schizophr Res. 2002; 58: 11–20
- Aston C, Jiang L, Sokolov BP. Microarray analysis of postmortem temporal cortex from patients with schizophrenia. J Neurosci Res. 2004; 77: 858–66
- Iwamoto K, Kakiuchi C, Bundo M, Ikeda K, Kato T. Molecular characterization of bipolar disorder by comparing gene expression profiles of postmortem brains of major mental disorders. Mol Psychiatry. 2004; 9: 406–16
- Sibille E, Arango V, Galfalvy HC, Pavlidis P, Erraji-Benchekroun L, Ellis SP, et al. Gene expression profiling of depression and suicide in human prefrontal cortex. Neuropsychopharmacology. 2004; 29: 351–61
- Mexal S, Frank M, Berger R, Adams CE, Ross RG, Freedman R, et al. Differential modulation of gene expression in the NMDA postsynaptic density of schizophrenic and control smokers. Brain Res Mol Brain Res. 2005; 139: 317–32
- Arion D, Unger T, Lewis DA, Levitt P, Mirnics K. Molecular evidence for increased expression of genes related to immune and chaperone function in the prefrontal cortex in schizophrenia. Biol Psychiatry. 2007; 62: 711–21
- Atz M, Walsh D, Cartagena P, Li J, Evans S, Choudary P, et al. Methodological considerations for gene expression profiling of human brain. J Neurosci Methods. 2007; 163: 295–309
- Martorell L, Segues T, Folch G, Valero J, Joven J, Labad A, et al. New variants in the mitochondrial genomes of schizophrenic patients. Eur J Hum Genet. 2006; 14: 520–8
- Munakata K, Fujii K, Nanko S, Kunugi H, Kato T. Sequence and functional analyses of mtDNA in a maternally inherited family with bipolar disorder and depression. Mutat Res. 2007; 617: 119–24
- Kazuno AA, Munakata K, Mori K, Tanaka M, Nanko S, Kunugi H, et al. Mitochondrial DNA sequence analysis of patients with'atypical psychosis’. Psychiatry Clin Neurosci. 2005; 59: 497–503
- Kirk R, Furlong RA, Amos W, Cooper G, Rubinsztein JS, Walsh C, et al. Mitochondrial genetic analyses suggest selection against maternal lineages in bipolar affective disorder. Am J Hum Genet. 1999; 65: 508–18
- Marchbanks RM, Ryan M, Day IN, Owen M, McGuffin P, Whatley SA. A mitochondrial DNA sequence variant associated with schizophrenia and oxidative stress. Schizophr Res. 2003; 65: 33–8
- Munakata K, Iwamoto K, Bundo M, Kato T. Mitochondrial DNA 3243A > G mutation and increased expression of LARS2 gene in the brains of patients with bipolar disorder and schizophrenia. Biol Psychiatry. 2005; 57: 525–32
- Munakata K, Tanaka M, Mori K, Washizuka S, Yoneda M, Tajima O, et al. Mitochondrial DNA 3644T– > C mutation associated with bipolar disorder. Genomics. 2004; 84: 1041–50
- Kato C, Umekage T, Tochigi M, Otowa T, Hibino H, Ohtani T, et al. Mitochondrial DNA polymorphisms and extraversion. Am J Med Genet B Neuropsychiatr Genet. 2004; 128: 76–9
- Washizuka S, Ikeda A, Kato N, Kato T. Possible relationship between mitochondrial DNA polymorphisms and lithium response in bipolar disorder. Int J Neuropsychopharmacol. 2003; 6: 421–4
- Wallace DC, Ye JH, Neckelmann SN, Singh G, Webster KA, Greenberg BD. Sequence analysis of cDNAs for the human and bovine ATP synthase beta subunit: mitochondrial DNA genes sustain seventeen times more mutations. Curr Genet. 1987; 12: 81–90
- Kato T, Stine OC, McMahon FJ, Crowe RR. Increased levels of a mitochondrial DNA deletion in the brain of patients with bipolar disorder. Biol Psychiatry. 1997; 42: 871–5
- Sabunciyan S, Kirches E, Krause G, Bogerts B, Mawrin C, Llenos IC, et al. Quantification of total mitochondrial DNA and mitochondrial common deletion in the frontal cortex of patients with schizophrenia and bipolar disorder. J Neural Transm. 2007; 114: 665–74
- Cavelier L, Jazin EE, Eriksson I, Prince J, Bave U, Oreland L, et al. Decreased cytochrome-c oxidase activity and lack of age-related accumulation of mitochondrial DNA deletions in the brains of schizophrenics. Genomics. 1995; 29: 217–24
- Corral-Debrinski M, Horton T, Lott MT, Shoffner JM, Beal MF, Wallace DC. Mitochondrial DNA deletions in human brain: regional variability and increase with advancing age. Nat Genet. 1992; 2: 324–9
- Kakiuchi C, Ishiwata M, Kametani M, Nelson C, Iwamoto K, Kato T. Quantitative analysis of mitochondrial DNA deletions in the brains of patients with bipolar disorder and schizophrenia. Int J Neuropsychopharmacol. 2005; 8: 515–22
- Liu CS, Tsai CS, Kuo CL, Chen HW, Lii CK, Ma YS, et al. Oxidative stress-related alteration of the copy number of mitochondrial DNA in human leukocytes. Free Radic Res. 2003; 37: 1307–17
- Frahm T, Mohamed SA, Bruse P, Gemünd C, Oehmichen M, Meissner C. Lack of age-related increase of mitochondrial DNA amount in brain, skeletal muscle and human heart. Mech Ageing Dev. 2005; 126: 1192–200
- Kraytsberg Y, Kudryavtseva E, McKee AC, Geula C, Kowall NW, Khrapko K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat Genet. 2006; 38: 518–20
- Li JZ, Vawter MP, Walsh DM, Tomita H, Evans SJ, Choudary PV, et al. Systemic changes in gene expression in postmortem human brains associated with tissue pH and terminal medical conditions. Hum Mol Genet. 2004; 13: 609–16
- Fattal O, Link J, Quinn K, Cohen BH, Franco K. Psychiatric comorbidity in 36 adults with mitochondrial cytopathies. CNS Spectr. 2007; 12: 429–38
- Karry R, Klein E, Ben Shachar D. Mitochondrial complex I subunits expression is altered in schizophrenia: a postmortem study. Biol Psychiatry. 2004; 55: 676–84
- Dairaghi DJ, Shadel GS, Clayton DA. Human mitochondrial transcription factor A and promoter spacing integrity are required for transcription initiation. Biochim Biophys Acta. 1995; 1271: 127–34
- Bonawitz ND, Clayton DA, Shadel GS. Initiation and beyond: multiple functions of the human mitochondrial transcription machinery. Mol Cell. 2006; 24: 813–25
- Ukai W, Ozawa H, Tateno M, Hashimoto E, Saito T. Neurotoxic potential of haloperidol in comparison with risperidone: implication of Akt-mediated signal changes by haloperidol. J Neural Transm. 2004; 111: 667–81
- Lee HC, Li SH, Lin JC, Wu CC, Yeh DC, Wei YH. Somatic mutations in the D-loop and decrease in the copy number of mitochondrial DNA in human hepatocellular carcinoma. Mutat Res. 2004; 547: 71–8