Abstract
While the function of dystrophin in muscle disease has been thoroughly investigated, dystrophin and associated proteins also have important roles in the central nervous system. Many patients with Duchenne and Becker muscular dystrophies (D/BMD) have cognitive impairment, learning disability, and an increased incidence of some neuropsychiatric disorders. Accordingly, dystrophin and members of the dystrophin-associated glycoprotein complex (DGC) are found in the brain where they participate in macromolecular assemblies that anchor receptors to specialized sites within the membrane. In neurons, dystrophin and the DGC participate in the postsynaptic clustering and stabilization of some inhibitory GABAergic synapses. During development, α-dystroglycan functions as an extracellular matrix receptor controlling, amongst other things, neuronal migration in the developing cortex and cerebellum. Several types of congenital muscular dystrophy caused by impaired α-dystroglycan glycosylation cause neuronal migration abnormalities and mental retardation. In glial cells, the DGC is involved in the organization of protein complexes that target water-channels to the plasma membrane. Finally, mutations in the gene encoding ε-sarcoglycan cause the neurogenic movement disorder, myoclonus-dystonia syndrome implicating ε–sarcoglycan in dopaminergic neurotransmission. In this review we describe the recent progress in defining the role of the DGC and associated proteins in the brain.
Introduction
In the past two decades staggering progress has been made in elucidating the genetic basis for the muscular dystrophies. The genes for the most common forms of muscular dystrophy have been identified and their functions well defined Citation[1]. The DMD gene, mutated in Duchenne and Becker muscular dystrophies (D/BMD), is the largest gene in the human genome, spanning approximately 2.5 Mbp of the X chromosome and encodes several different proteins, including the giant actin-binding protein dystrophin. Dystrophin is expressed at high levels in striated muscle and to a lesser extent in neurons. In both cell types, dystrophin is found at the cytoplasmic face of the plasma membrane, in multiprotein complexes that can interact with both the cytoskeleton and extracellular matrix (ECM).
In common with many important proteins, dystrophin is part of multiprotein complex known as the dystrophin-associated glycoprotein complex (DGC). The DGC is a large, membrane-spanning protein complex that is thought to protect the muscle plasma membrane (sarcolemma) from mechanical damage (A). Biochemically, the core DGC can be divided into three subcomplexes: the sarcoglycan subcomplex, the dystroglycan subcomplex, and the cytoplasmic subcomplex composed of dystrophin, the dystrobrevins, and the syntrophins Citation[2]. Several forms of muscular dystrophy are caused by defects in the function and assembly of the DGC Citation[3]. Dystrophin can act as a macromolecular scaffold and is critically required for the assembly and stability of the DGC at the sarcolemma. In patients with D/BMD, the absence of dystrophin or a mutation that affects its function results in disassembly of the DGC at the membrane, effectively severing one of the links between the subsarcolemmal cytoskeleton and the ECM. Thus, the absence of dystrophin results in a secondary deficiency of other components of the DGC at the sarcolemma. In addition to the core DGC, many accessory proteins have been found to interact with this complex in muscle and non-muscle tissues. In this review, we have used a manually annotated protein interaction network to depict the conceptual organization of the DGC and associated proteins ().
Key messages
Duchenne and Becker muscular dystrophies are complex disorders that are often associated with cognitive impairment, behavioural, and psychiatric abnormalities.
Dystrophin and components of the dystrophin-associated glycoprotein complex (DGC) are associated with inhibitory GABAergic synapses in the hippocampus and cerebellum.
Congenital muscular dystrophies caused by defects in α-dystroglycan glycosylation are frequently associated with neuronal migration abnormalities and mental retardation.
DGC-like complexes anchor membrane-associated proteins, such as the water-channel aquaporin-4, to specialized sites in neurons and glia.
Mutations in the SGCE gene that encodes ε-sarcoglycan cause the movement disorder myoclonus-dystonia syndrome.
Figure 1. The molecular organization of the DGC and ‘DGC-like’ complexes in muscle and brain. Conceptual organization of the core DGC, ‘DGC-like’ complexes and associated proteins in skeletal muscle (A), neurons (B) and glia (C). The composition of the core DGC in muscle was determined by the direct biochemical purification of the complex from tissue. By contrast, the complexes shown in panels B and C have been inferred from different studies (yeast two-hybrid interactions, co-localization and co-immunoprecipitation) and do not represent a biochemically defined entity. For the purposes of clarity, only a subset of syntrophin-associated proteins, such as the Na+ channels, nNOS and aquaporin-4 have been included in each complex. A comprehensive list of syntrophin-associated proteins is presented in . In each panel, the dystrophin/β-dystroglycan binding region is reduced in scale in comparison with the similar region in dystrobrevin. Question marks denote imputed interactions that have not been demonstrated in vivo. (Abbreviations: DGC = dystrophin-associated glycoprotein complex; SBD = syntrophin-binding domain; nNOS = neuronal nitric oxide synthase; SAST = syntrophin-associated serine/threonine kinase; SSPN = sarcospan; ABD = actin-binding domain; PH = plecstrin homology domain; PDZ = PSD-95, (postsynaptic density protein 95), discs–large and ZO-1 (zonula occludens-1); SU = syntrophin-unique region; Kir 4.1 = inwardly rectifying K+ channel 4.1; WW = WW domain ; EFI/EFII = EF hand domains ; ZZ = zinc finger domain; H1/H2 = helical domains.)
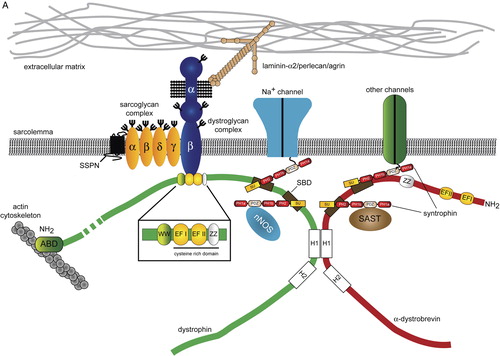
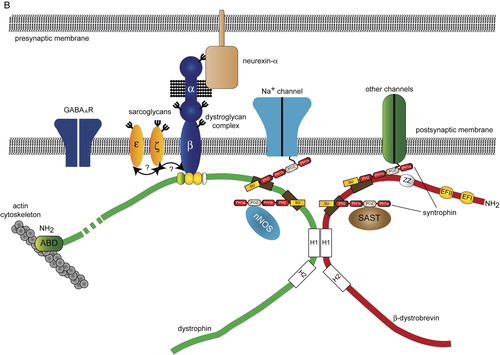
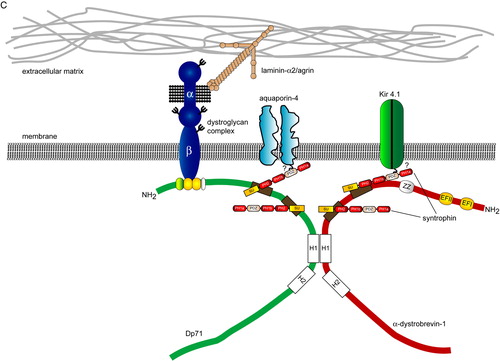
Figure 2. Protein interaction network for the dystrophin-associated glycoprotein complex (DGC). A systems view of the molecular interactions between the core DGC and associated proteins. The map was created using the Human GRID (General Repository of Interaction Datasets, http://www.thebiogrid.org/) database and was assembled using the Osprey network visualization tool Citation[106]. Gene names: LAMA2 = laminin-α2; AGRN = agrin; HSPG2 = perlecan (heparan sulphate proteoglycan-2); NRXN = neurexin; DAG1 = dystroglycan; CAV3 = caveolin-3; SGCA = α-sarcoglycan; SGCB = β-sarcoglycan; SGCG = γ-sarcoglycan; SGCD = δ-sarcoglycan; SSPN = sarcospan; FLNC = filamin C; DMD = dystrophin; ACTG1 = γ-(cytoplasmic) actin; PGM5 = phosphoglucomutase 5; SNTA1 = α-syntrophin; SNTB1 = β1-syntrophin; SNTB2 = β2-syntrophin; DNTA = α-dystrobrevin; SYNC1 = syncoilin; DMN = desmuslin; DTNBP1 = dystrobrevin-binding protein-1; SCN5A = Nav1.5; SCN4A = Nav1.4; NOS = neuronal nitric oxide synthase; DGKZ = diacylglycerol kinase, zeta; CALM1 = calmodulin 1; GRB2 = growth factor receptor bound protein 2; MAST = microtubule associated serine/threonine kinase 1; KCNJ4 = potassium inwardly-rectifying channel J4 (Kir2.3); KCNJ12 = potassium inwardly-rectifying channel, subfamily J12 (Kir2.2). Colour coding: red = core DGC components; green = cytoskeletal proteins; yellow = signalling proteins; dark blue = transmembrane proteins; light blue = extracellular matrix proteins; grey = trafficking proteins.
![Figure 2. Protein interaction network for the dystrophin-associated glycoprotein complex (DGC). A systems view of the molecular interactions between the core DGC and associated proteins. The map was created using the Human GRID (General Repository of Interaction Datasets, http://www.thebiogrid.org/) database and was assembled using the Osprey network visualization tool Citation[106]. Gene names: LAMA2 = laminin-α2; AGRN = agrin; HSPG2 = perlecan (heparan sulphate proteoglycan-2); NRXN = neurexin; DAG1 = dystroglycan; CAV3 = caveolin-3; SGCA = α-sarcoglycan; SGCB = β-sarcoglycan; SGCG = γ-sarcoglycan; SGCD = δ-sarcoglycan; SSPN = sarcospan; FLNC = filamin C; DMD = dystrophin; ACTG1 = γ-(cytoplasmic) actin; PGM5 = phosphoglucomutase 5; SNTA1 = α-syntrophin; SNTB1 = β1-syntrophin; SNTB2 = β2-syntrophin; DNTA = α-dystrobrevin; SYNC1 = syncoilin; DMN = desmuslin; DTNBP1 = dystrobrevin-binding protein-1; SCN5A = Nav1.5; SCN4A = Nav1.4; NOS = neuronal nitric oxide synthase; DGKZ = diacylglycerol kinase, zeta; CALM1 = calmodulin 1; GRB2 = growth factor receptor bound protein 2; MAST = microtubule associated serine/threonine kinase 1; KCNJ4 = potassium inwardly-rectifying channel J4 (Kir2.3); KCNJ12 = potassium inwardly-rectifying channel, subfamily J12 (Kir2.2). Colour coding: red = core DGC components; green = cytoskeletal proteins; yellow = signalling proteins; dark blue = transmembrane proteins; light blue = extracellular matrix proteins; grey = trafficking proteins.](/cms/asset/946fff60-0269-4dc9-9947-e03756493ac1/iann_a_367022_f0002_b.jpg)
In addition to its mechanical role in muscle, dystrophin and the DGC are also involved in intracellular Ca2 + homeostasis. In dystrophin-deficient muscle, raised intracellular [Ca2 +] has been shown to affect the function of some Ca2 +-permeable channels and activate Ca2 +-dependent proteases Citation[4]. It has also been suggested that the DGC has a regulatory function, participating in signal transduction between the ECM and the cytoskeleton Citation[5]. Furthermore, central nervous system (CNS) involvement in some D/BMD patients supports the notion that dystrophin and the DGC have pleiotropic roles in excitable cells. Several recent reviews have focused on the role of dystrophin and DGC in muscle Citation[5–7]. In this review, we will discuss the role of the DGC in the CNS.
Abbreviations
The cognitive and neuropsychiatric spectrum in D/BMD
Duchenne muscular dystrophy (DMD) is a complex, multisystemic disorder that can affect the CNS. It is now well established that a significant proportion of boys with DMD have mild cognitive impairment Citation[8]. Although normally distributed, full-scale intelligence quotient (IQ) scores are shifted downwards by one standard deviation from the population mean of 100, such that the mean IQ for DMD boys is 80.2 (see Citation[9] for meta-analysis). Furthermore, approximately 35% of DMD boys, compared to only 2% within the general population, have full IQ scores of 70 or below Citation[9]. DMD boys also have an increased incidence of three types of learning disabilities: dyslexia, dyscalculia, and dysgraphia (disorder of written communication) Citation[10]. Although there is little correlation between the site of the mutation in the DMD gene and cognitive impairment, mutations in the 3’-end of the gene are frequently associated with lower IQ scores Citation[11]. Although generally rarer, truncating mutations in the 3′-end of the DMD gene are often associated with moderate to severe mental retardation and have the propensity to disrupt the assembly of the DGC by deleting the β-dystroglycan binding site at the C-terminus of dystrophin Citation[12], Citation[13].
In addition to cognitive impairment, D/BMD patients have been reported to have an increased incidence of neuropsychiatric and behavioural disorders including autism spectrum disorder and attention deficit hyperactivity disorder (ADHD) (). A recent questionnaire-based study by Hendriksen and Vles found that 11.7% of males with DMD had a co-morbid diagnosis of ADHD Citation[14]. Similarly, Hinton and colleagues found that 1 in 4 boys with DMD had significant problems with attention Citation[15]. These studies point to a complex pattern of neuropsychiatric and neurocognitive deficits in a significant proportion of D/BMD patients. Educational provision is clearly an important aspect for the treatment and management of D/BMD Citation[16].
Table I. Neurocognitive and neuropsychiatric deficits associated with Duchenne and Becker muscular dystrophies (D/BMD). The Table summarizes some of the findings in research articles reporting cognitive, behavioural, and psychiatric abnormalities described in some boys with DMD and BMD. Key references are given.
There have been surprisingly few neuroimaging studies examining brain function in DMD patients. Positron emission tomography (PET) imaging has been used to study glucose metabolism in the brains of ten boys with DMD. This study found that region-specific glucose hypometabolism might be linked to cognitive and behavioural abnormalities in this cohort Citation[17]. Further metabolic defects have been reported in the brains of DMD patients using phosphorus-31 magnetic resonance spectroscopy. DMD patients were found to have higher ratios of inorganic phosphate to adenosine triphosphate (ATP), phosphocreatine and phosphomonoesters compared to normal controls Citation[18]. Thus, altered brain metabolism may contribute to the cognitive and behavioural deficits in DMD patients.
Dystrophin and its isoforms in the brain
There are a variety of reasons why the DGC in brain differs in composition and function from the core DGC in muscle. First, there are several distinct dystrophin isoforms that are present in the brain. Second, the DGC has not been purified from the brain using the biochemical techniques initially used to isolate the complex from striated muscle. Several distinct complexes have been shown to exist in neurons and glia, suggesting that there is considerable heterogeneity in the composition of the DGC in brain (B and 1C) Citation[19], Citation[20]. For the purposes of this review we have called these complexes ‘DGC-like’, differentiating them from the core DGC in muscle. Finally, with the exception of ε- and ζ-sarcoglycan, most of the sarcoglycans are expressed predominantly in striated and smooth muscle and are only weakly expressed in the CNS. Thus, if a sarcoglycan complex exists in the brain it is unlikely to resemble the cognate complex in muscle.
As mentioned previously, in addition to dystrophin, the DMD gene encodes several other proteins, most of which are found in the CNS and peripheral nervous system (for review see Citation[3]). Three forms of full-length dystrophin are encoded by transcripts originating from independent promoters in the 5′-end of the DMD gene. Specifically, the P-promoter produces dystrophin that is specifically expressed in cerebellar Purkinje cells whereas the C-promoter transcribes dystrophin in the cortex and subcortical regions including the hippocampus Citation[21]. The most abundant product of the DMD gene that is expressed in the CNS is Dp71 Citation[22]. The Dp71 mRNA is transcribed from a promoter located between exons 62 and 63. The Dp71 protein contains the binding sites for dystroglycan as well as the dystrobrevins and syntrophins but does not have an N-terminal actin-binding domain Citation[23]. In addition to these variants, Dp140 and Dp260 are also found in the CNS Citation[24]. Dp140 appears to be restricted to the microvasculature, whereas Dp260 is found almost exclusively in the retina. It is noteworthy that the dystrophin isoform Dp116 is found in Schwann cells of peripheral nerves but is not associated with myelinated structures in the adult CNS.
Full-length dystrophin is expressed almost exclusively in neurons Citation[25]. By contrast, many of the shorter isoforms, notably Dp71 and Dp140, are expressed in glial cells, notably perivascular astrocytes and Bergmann glia Citation[19], Citation[24]. This diversity is mirrored by the distribution of the brain-expressed components of the DGC and suggests that there are several distinct DGC-like complexes in the brain (see below). Dystrophin is located within membrane-associated puncta in cerebellar Purkinje cells, pyramidal neurons in the neocortex, and pyramidal neurons of the hippocampal formation Citation[25]. In these brain regions, dystrophin co-localizes with inhibitory GABAA receptor clusters Citation[26]. In the mdx mouse there is a significant reduction in the number of clusters that are immunoreactive for the α1 and α2 GABAA subunits Citation[26].
A role for dystrophin and the DGC at inhibitory synapses has gained increasing weight and may explain some of the behavioural and electrophysiological findings described in the preceding sections. In cultured hippocampal neurons, Brunig and colleagues found that dystrophin is extensively co-localized with the α2 subunit of the GABAA receptor Citation[27]. Dystrophin is also able to recruit other components of the DGC, including the syntrophins and β-dystroglycan, to nascent receptor clusters. This study also made the intriguing observation that selective signalling from GABAergic terminals contributes to postsynaptic clustering of dystrophin Citation[27]. The clustering of dystrophin at inhibitory GABAergic synapses is independent of postsynaptic GABAA receptors and gephyrin, a scaffolding protein that clusters inhibitory neurotransmitter receptors Citation[27]. As alluded to above, dystroglycan is also clustered at GABAergic synapses. GABAergic receptor clusters from mice lacking dystroglycan do not contain dystrophin, suggesting that dystroglycan is required for the assembly of DGC-like complexes at inhibitory synapses in the hippocampus Citation[28]. Conversely, dystroglycan remains clustered in neurons from both mdx and gephyrin-deficient mice Citation[28]. Finally, in cerebellar Purkinje cells from mdx mice, the frequency and amplitude of spontaneous inhibitory postsynaptic currents were reduced compared with littermate controls Citation[29]. This observation is consistent with the reported reduction in the number and size of GABAA receptor clusters described above. Although these molecules are not essential for GABAergic synaptogenesis, these findings suggest that dystrophin and the DGC are involved in modulating synapse function at a subset of GABAergic synapses in the hippocampus and cerebellum Citation[28], Citation[29].
Dystrophin has also been reported to be a component of the postsynaptic density (PSD) Citation[30]. The PSD is composed of a detergent-resistant matrix containing excitatory neurotransmitter receptors and their subsynaptic scaffolds. Whilst this finding appears to contradict the association of dystrophin with inhibitory synapses, differences in the methodologies for PSD preparation may result in the apparent association of dystrophin with the PSD. GABAB receptors have been detected in PSD preparations although GABAA receptors were not similarly identified Citation[19], Citation[31]. Furthermore, β-dystroglycan (see below) is not PSD-enriched and is effectively removed from synaptosomes with the non-ionic detergent Triton X-100 Citation[32]. Finally, in cultured hippocampal neurons, core DGC components are selectively associated with inhibitory synapses but were not found at excitatory glutamatergic synapses Citation[28]. Taken together, these data suggest that dystrophin and the DGC are found at GABAergic rather than glutamatergic synapses.
Behaviour and synaptic plasticity in dystrophin-deficient animal models
The vast majority of studies addressing the function of dystrophin and DGC in brain have utilized the dystrophin-deficient mdx mouse. The mdx mouse is a commonly used model of muscular dystrophy that also has biochemical, physiological, and behavioural abnormalities attributable to the absence of dystrophin in the brain. Mdx mice have impaired retention in a T-maze but no deficit in locomotor activity Citation[33]. The T-maze tests spatial working memory and is often used to assess hippocampal and forebrain function. Retention impairments at long delays in mdx mice suggest a role for dystrophin in long-term consolidation processes. By contrast, mdx3cv mice lacking dystrophin and its C-terminal isoforms (see below) showed no retention deficit in the T-maze but displayed increased anxiety and reduced locomotion compared to wild-type mice Citation[34].
Electrophysiological studies of synaptic function in the mdx mouse have generated important but conflicting insights into the role of dystrophin and its isoforms in synaptic plasticity. In the first experiments designed to examine synaptic plasticity in mdx mice, Sesay and colleagues found no deficit in hippocampal long-term potentiation (LTP) in mdx mice that lack only full-length dystrophin Citation[35]. This study also found no difference in hippocampal-dependent spatial learning tasks between mdx mice and wild-type controls Citation[35]. Using different training paradigms, Vaillend et al. found abnormal enhancement of hippocampal LTP in mdx mice and deficits in long-term spatial memory and object recognition Citation[36]. The same study found that short-term potentiation (STP) and depression (STD) are also enhanced in the mdx mouse Citation[37]. Furthermore, the GABAA antagonist bicuculline increased basal neurotransmission in wild-type mice but had no effect upon the mdx mouse. Interestingly, bicuculline was able to prevent enhanced STP and STD in the mdx mouse Citation[37]. Defects in long-term depression have been found in the cerebellum of mdx mice Citation[38]. Bicuculline did not alter the LTD deficit, suggesting that another class of receptor may be affected by the absence of dystrophin Citation[38].
Dystroglycan
Dystroglycan is a matrix receptor that spans the plasma membrane linking dystrophin and the cytoplasmic components of the DGC to the ECM. α-Dystroglycan and β-dystroglycan are produced by proteolytic cleavage and glycosylation of a single precursor propeptide. Dystroglycan proteolysis appears to be functionally important and occurs via the autocatalytic SEA (sea urchin, enterokinase, agrin)-module that contains the processing site for the precursor Citation[39], Citation[40]. α-Dystroglycan binds to a number of laminin G-domain-containing ECM proteins, including some laminins, perlecan, agrin, and the neurexins. Moreover, laminin-binding to α-dystroglycan is mediated by O-linked glycans, specifically O-linked mannose structures, in a Ca2 +-dependent manner (see below) Citation[41]. β-Dystroglycan associates with α-dystroglycan at the plasma membrane following proteolytic processing and binds to dystrophin and its isoforms on the cytoplasmic face of the membrane. Whilst it is beyond the scope of this review to describe in detail the function of dystroglycan, the reader is referred to the following reviews that cover this subject Citation[41], Citation[42].
No mutations have been described in the DAG1 gene that encodes dystroglycan in humans. It has been suggested that DAG1 is an essential gene in mammals. Indeed, mice lacking Dag1 (the murine dystroglycan gene) die in utero due to a defect in the formation of Reichert's membrane during early embryogenesis Citation[43]. Conditional knockout of dystroglycan specifically in transgenic mice have provided important insights into the function of dystroglycan during brain development (see below) Citation[44]. In addition to the neuronal migration abnormality in these mice, loss of dystroglycan in the hippocampus results in a severe deficit in LTP but has no effect upon basal neurotransmission and paired pulse facilitation, a measure of presynaptic neurotransmitter release Citation[44]. Whether this defect in LTP is caused by cortical disorganization or reflects a direct role of dystroglycan in synaptic function is at present unclear. Nevertheless, these studies explain how defects in dystroglycan can cause severe CNS abnormalities in congenital muscular dystrophy (CMD) patients (see below).
α-Dystroglycan glycosylation defects, CMD, and brain development
While DMD is the most common inherited muscle disease in humans, in the last few years the genetic defects underlying several rare CMDs have been identified. Perhaps the most surprising finding to emerge from these studies is that that many of the CMDs are associated with defects in the posttranslational processing of α-dystroglycan. Mutations in the genes encoding several confirmed or putative glycosyltransferases have been discovered in patients with different forms of CMD (). These mutations are associated with incomplete glycosylation of α-dystroglycan that abrogates its function as a matrix receptor. Many of the CMDs have a broad phenotypic overlap with mutations in several functionally related genes producing similar clinical diagnoses (). For example, FKTN mutations in the gene that encodes fukutin were originally found in Japanese families with Fukuyama congenital muscular dystrophy (FCMD) Citation[45]. Subsequently, FKTN mutations were found to cause a disease phenotypically resembling Walker-Warburg syndrome (WWS), steroid-responsive limb-girdle muscular dystrophy, and more recently dilated cardiomyopathy with mild muscle weakness Citation[46–48].
Table II. Muscular dystrophies associated with hypoglycosylation of α-dystroglycan. The Table lists the salient features of the diseases associated with a defect in α-dystroglycan glycosylation. Underlining indicates the original disease in which mutations were found. Severe structural changes include neuronal migration defects (causing lissencephaly), cerebellar hypoplasia, and cerebellar dysplasia Citation[48].
Mutations that cause FCMD, muscle-eye-brain disease (MEB), and WWS are commonly associated with defects in cortical migration, cerebellar cysts, and mental retardation Citation[49], Citation[50]. A subset of mutations in the gene encoding fukutin-related protein (FKRP) is also associated with CMD and similar brain pathologies Citation[51]. Mutations in fukutin and its relative FKRP can cause a broad range of phenotypes; however, the function of each of these proteins has still to be determined. Each protein is predicted to be a glycosyltransferase having weak homology to bacterial phosphoryl ligand transferases, some of which are involved in synthesis of capsular polysaccharides Citation[52], Citation[53].
The enzymes, POMT1 (protein O-mannosyltransferase-1), POMT2 (protein O-mannosyltransferase-2), and POMGnT1 (protein O-mannose β-1,2-N-acetyl glucosaminyltransferase), catalyse the initial steps in protein O-linked mannosylation within the endoplasmic reticulum (ER). O-linked mannose is further modified by as yet unknown glycosyltransferases to produce mucin-like modifications of the α-dystroglycan peptide. One of the enzymes that are thought to generate terminal modifications on α-dystroglycan is LARGE. LARGE has two distinct glycosyltransferase domains. The N-terminal domain is similar to the glycosyltransferase 8 and 24, (uridine diphosphate, UDP-glucose glycoprotein glucosyltransferase) families whereas the C-terminal domain has similarities to the glycosyltransferase 31 (β-1,3-N-acetylglucosaminyltransferase 6) family Citation[54], Citation[55]. Interestingly, ectopic expression of LARGE can restore α-dystroglycan functionality in cellular models of CMD caused by defects in other CMD-associated genes Citation[56].
Significant progress has been made in deciphering the role played by α-dystroglycan during brain development and neurogenesis. In addition to muscle, dystroglycan is also a laminin receptor in the brain. Laminin-α2 chain deficiency also causes CMD (merosin-negative CMD), and there is some evidence that this form of CMD is associated with mental retardation, structural abnormalities of the cortex and cerebellum, and white matter changes Citation[57]. By contrast, dy/dy (dystrophia muscularis) mice harbouring mutations in the Lama2 gene that encodes the α2 chain of laminin have muscle disease, white matter changes, but no evidence of a neuronal migration defect Citation[58]. Thus, laminin-2 may not be directly involved in the neuronal migration defects seen in the CMDs associated with hypoglycosylation of α-dystroglycan.
Myd (myodystrophy) mice homozygous for a deletion in the LARGE gene also have a neuronal migration defect that affects cortical lamination, neuronal placement in the hippocampal formation, and the organization of the laminated folia in the cerebellum Citation[44], Citation[59]. Conditional deletion of dystroglycan in the brain causes a similar neuronal migration defect suggesting that the deficiency of glycosyltransferases results in loss of dystroglycan function as a matrix receptor Citation[44]. Clearly the identity of the brain-expressed ECM ligands for α-dystroglycan would contribute to our understanding of the neuropathology of CMD. These ligands may include other forms of laminin, perlecan, or agrin. Dystroglycan is also a receptor for the synaptic adhesion molecule neurexin Citation[60]. Neurexins function as transsynaptic adhesion molecules that play important roles in synapse specification and postsynaptic differentiation Citation[61]. Dystroglycan has also been found to influence the differentiation of neuroepithelial cells during CNS development Citation[62]. The mechanism for this effect is as yet unknown; however, these studies raise the possibility that a defect in neuroepithelial proliferation may contribute to the severe brain abnormalities in some forms of CMD.
Cytosolic components of DGC in the brain
In addition to dystrophin and dystroglycan, the dystrobrevins and syntrophins are also components of the core DGC that are expressed in the brain (B and 1C). The dystrobrevins are a family of dystrophin-related proteins encoded by two separate genes: DTNA encoding the α-dystrobrevins and DTNB encoding β-dystrobrevin. The dystrobrevins bind directly to dystrophin through reciprocal coiled-coil interactions. In addition, the dystrobrevins and dystrophin bind to the syntrophin protein family Citation[63]. Whilst the α-dystrobrevins are part of the DGC in striated muscle, the related protein β-dystrobrevin is found in many non-muscle DGC-like complexes, including those found in the brain Citation[64]. Mice lacking α-dystrobrevin have a mild form of muscular dystrophy, which does not impair the assembly of the core DGC Citation[65]. By contrast mice lacking β-dystrobrevin appear to be phenotypically normal Citation[66]. However, mice lacking both α- and β-dystrobrevin have impaired motor function, performing poorly in tests assessing sensorimotor function such as the ability to balance on a rotating rotorod Citation[67]. Double knockout (dko) mice also have a reduction in cerebellar GABAA receptors that is similar in magnitude to the deficit in the mdx mouse described above Citation[67]. Dystrophin was also absent from GABAA clusters in dko mice, demonstrating the interdependence of each protein for physiological synaptic targeting Citation[67].
In addition to the DGC, several dystrobrevin-binding proteins have been described. These proteins include syncoilin, dysbindin (dystrobrevin-binding protein-1 (DTNBP1)), and neuronal kinesin heavy chain 5A Citation[68]. Interestingly, dysbindin has been shown to be an important schizophrenia susceptibility gene Citation[69]. Variants in the DTNBP1 gene that are associated with an increased risk of schizophrenia are also associated with poor educational performance and the so-called negative symptoms of cognitive decline in patients with schizophrenia Citation[70]. Although the function of dysbindin in the context of the DGC has not been determined, dysbindin is part of another protein complex, the biogenesis of lysosome-related organelles complex-1 (BLOC-1) that is involved in protein trafficking between the endosomal and lysosomal compartments Citation[71]. Thus BLOC-1 may play a role in the trafficking and assembly of DGC-like complexes in the brain.
The syntrophin family of adaptor proteins is composed of five members, α-, β1-, β2-, γ1-, and γ2-syntrophin Citation[72–74]. Each syntrophin contains a PDZ (PSD-95 (postsynaptic density protein 95) discs–large and ZO-1 (zonula occludens-1)) domain and two PH (plecstrin homology) domains (the first PH domain of each syntrophin is split by the insertion of the PDZ domain). PDZ domains bind to the C-terminal tails of many different classes of transmembrane protein but can also participate in homotypic interactions with other PDZ domain-containing proteins including neuronal nitric oxide synthase (nNOS) Citation[75]. The different syntrophins have been found to interact with a wide variety of proteins in the brain (see for details). Many of these proteins have been identified in yeast two-hybrid screens for proteins interacting with the tails of transmembrane receptors. In most cases, the PDZ domain of the syntrophins is responsible for binding to specific conserved sequences at the tails of these proteins (). Many of the syntrophin-associated proteins, such as nNOS, aquaporin-4 (AQP4), and the neuroligins, have important functions in the brain (). However, in the majority of studies reporting these syntrophin-dependent interactions, the effect on the DGC has not been explored.
Table III. Syntrophin-binding proteins. The table collates the repertoire of proteins that bind to the syntrophin protein family in different tissues. The majority of these interactions occur between the PDZ domain in syntrophin and the C-terminal tails of membrane associated receptors. For clarity, components of the core DGC and DGC-related complexes, such as Dp71 and the dystrophin paralogue utrophin, have been omitted.
Mice lacking α-syntrophin have muscle hypertrophy and abnormal neuromuscular junctions but do not appear to have any gross brain pathology or behavioural abnormalities Citation[76], Citation[77]. In common with muscle, the assembly of syntrophin-based complexes in the brain is likely to be dependent upon dystrophin, dystrophin isoforms, and the DGC. Mdx-βgeo mice that are devoid of dystrophin and all of its isoforms have a compound deficiency of syntrophins in the brain (Esapa and Blake, unpublished results) Citation[78]. It is therefore possible that some of CNS deficits in DMD are related to the mistargeting of syntrophin-interacting proteins in the brain (see below). The lack of overt CNS phenotypes in mice lacking individual syntrophins may be due to genetic redundancy such that other syntrophin family members may have a compensatory role.
Involvement of the DGC in H2O channel clustering
As mentioned above, the syntrophins can interact with the C-terminal tails of several different membrane-associated channels (). Components of the DGC, notably Dp71, the syntrophins, and dystroglycan are highly expressed in perivascular astrocytes Citation[19], Citation[79], Citation[80]. In perivascular astrocytes the DGC has been shown to be part of a protein complex that regulates intracellular water transport (C). Aquaporin-4 (AQP4) is the major water-channel in the central nervous system and is ostensibly found at the end-feet of perivascular astrocytes and in ependymal cells that line the brain ventricles. Mice lacking AQP4 are resistant to brain oedema induced following acute water intoxication or ischaemic stroke Citation[81]. The C-terminal tail of AQP4 contains the consensus sequence for PDZ binding (). Although a direct interaction between the PDZ domain of α-syntrophin and the tail of AQP4 has not been demonstrated, AQP4 co-purifies with syntrophin and Dp71 from mouse in a cross-linked protein complex. Furthermore, a pool of AQP4 is mislocalized in α-syntrophin-deficient mice suggesting that α-syntrophin is required to anchor AQP4 to the membrane in perivascular astrocytes but not the endothelium Citation[82]. AQP4 localization is also dependent upon the major brain isoform of dystrophin, Dp71. Dp71 null mice fail to concentrate AQP4 at glial end-feet, possibly due to impaired assembly of a DGC-like complex containing α-syntrophin. Furthermore, mdx-βgeo mice have an attenuated response to brain oedema compared to controls following water intoxication and fail to target AQP4 to astroglial end-feet surrounding capillaries Citation[83]. Taken together these results suggest that DGC-like complexes in perivascular astrocytes are required for membrane targeting of AQP4. It should be noted that there is also a DGC-independent pool of AQP4 in the brain found in the granule cell layer of the cerebellum, in subpial glial cells and in ependymal cells of the ventricles Citation[84].
The sarcoglycan complex in the brain
The sarcoglycans are a family of plasma membrane proteins that are expressed predominantly in striated and smooth muscle and peripheral nerve. The sarcoglycan family is composed of six members (α-, β-, γ-, δ-, ε-, and ζ-) that form a heterotetrameric complex that associates with dystroglycan at the plasma membrane. Although the precise function of the sarcoglycan complex is unknown, mutations in α-, β-, γ-, and δ-sarcoglycan cause different forms of autosomal recessive limb-girdle muscular dystrophies (LGMD), demonstrating its importance for normal muscle function Citation[85]. Initial studies on the sarcoglycans suggested that their expression was ostensibly limited to striated and smooth muscle and to a lesser extent peripheral nerve. However, recent research has shown that ε-sarcoglycan has an important role in the brain and is mutated in myoclonus-dystonia syndrome (MDS, DYT11) Citation[86]. In addition, the most recently described member of the sarcoglycan family, ζ-sarcoglycan, is also highly expressed in the brain Citation[87].
ε-Sarcoglycan mutations and MDS
In 2001, Zimprich and colleagues identified heterozygous mutations in the SGCE gene, encoding ε-sarcoglycan, in five families with MDS Citation[86]. MDS is a dystonia-plus syndrome characterized by myoclonic jerks, which predominantly affect the neck and upper limbs, in combination with focal or segmental dystonia. Symptoms often develop during childhood or early adolescence with myoclonus as the predominant feature, frequently accompanied with mild dystonia. Alcohol ingestion is frequently found to suppress myoclonic symptoms. In addition to the motor features, psychiatric abnormalities such as obsessive-compulsive disorder (OCD), alcohol misuse, depression, anxiety, and panic attacks have also been reported in some individuals Citation[88], Citation[89].
SGCE is a maternally imprinted gene thus, in most tissues, only one allele is expressed Citation[86]. The majority of MDS cases are caused by nonsense mutations in the SGCE gene, with a lower incidence of missense mutations Citation[90], Citation[91]. Thus, although MDS is inherited in an autosomal dominant manner (with reduced penetrance on maternal transmission), most patients with heterozygous mutations in SGCE fail to produce any functional ε-sarcoglycan. Functional analysis of a small set of SGCE missense mutations has shown that mutations in the extracellular domain of ε-sarcoglycan impair membrane trafficking of the mutant proteins in cultured cells Citation[92]. These missense mutations are less stable than the wild-type protein and are degraded by the ubiquitin proteasome system Citation[92].
Whilst the function of ε-sarcoglycan has not been determined, defects in brain development and synaptic function have been implicated in the pathophysiology of the dystonias Citation[93]. ε-Sarcoglycan is found in many brain regions and is associated with dopaminergic neurons in the substantia nigra and ventral tegmental area Citation[94], Citation[95]. Altered dopaminergic neurotransmission has been implicated in other forms of dystonia such as DOPA (l-3,4-dihydroxyphenylalanine)-responsive dystonia (DRD, GCH1) caused by mutations in the gene for guanosine triphosphate (GTP) cyclohydrolase I Citation[96]. Preliminary neurochemical analysis of ε-sarcoglycan-deficient mice showed increased levels of dopamine and its metabolites within the striatum Citation[97]. Thus, ε-sarcoglycan may be involved in dopaminergic neurotransmission.
A sarcoglycan complex in the brain?
A plethora of in vivo and in vitro studies have analysed the trafficking and assembly of the sarcoglycan complex. Despite evidence for assembly and trafficking of incomplete sarcoglycan complexes Citation[98], a tetrameric assembly seems to be functionally important for endogenous sarcoglycans in all tissues and cell lines studied to date Citation[99]. All currently described complexes consist of a βδ-sarcoglycan core complex, which then associates with α-/ε-sarcoglycan and γ-/ζ-sarcoglycan to form a tetrameric complex Citation[98], Citation[100]. Although α- and γ-sarcoglycan are predominantly expressed in striated muscle, the other sarcoglycans are more widely expressed. It is therefore possible that the tetrameric εβδζ-complex could be present in the brain. This complex has already been described in Schwann cells of peripheral nerve Citation[101]. However, although theoretically possible, the existence of a prototypical tetrameric sarcoglycan complex in the brain is unlikely. No motor phenotypes have been described in animal models of LGMD that lack β- and δ-sarcoglycan Citation[102], Citation[103]. Conversely, Hjermind and colleagues have recently found that MDS patients with mutations in the SGCE gene do not appear to have any muscle involvement Citation[104]. These data suggest that ε-sarcoglycan may function independently of the other sarcoglycans in the brain. Interestingly, ε-sarcoglycan does not require the presence of other sarcoglycans in order to reach the plasma membrane in heterologous cells Citation[92], Citation[105]. The existence of brain-specific ε-sarcoglycan isoforms generated by alternative splicing may suggest that ε-sarcoglycan has acquired additional functions independent of the conventional sarcoglycan complex. Further studies are required to determine sarcoglycan function and synaptic physiology in animal models of MDS.
Conclusion
D/BMD are complex diseases that in many cases can have both cognitive and neuropsychiatric components. Although little is known about the molecular mechanisms that cause cognitive impairment in D/BMD patients, considerable progress has been made in identifying the proteins and biochemical pathways that may contribute to this abnormality. Recent studies implicate dystrophin and the DGC in GABAA receptor clustering. It is therefore tempting to speculate that GABAergic dysfunction may contribute to some of the behavioural and cognitive abnormalities in patients with D/BMD. Beyond dystrophin, there is now a considerable body of evidence showing that DGC-like complexes have an important role in the brain. For example, dystroglycan plays a role in neuronal migration during brain development, whereas other DGC-like complexes are involved in channel anchoring in the adult brain. Finally, SGCE mutations in MDS define an important and unexpected role for ε-sarcoglycan and the putative neuronal sarcoglycan complex in the brain. Whilst some of these interactions may contribute to the neuropathology of DMD and allied disorders, it remains to be established whether proteins such as the syntrophins and ε-sarcoglycan can function independently of the DGC. A detailed understanding of the functional role of dystrophin and the DGC in the brain will augment our understanding of the complex pathogenesis of muscular dystrophy.
Acknowledgements
Due to space constraints, it has sadly not been possible to cite all of the primary literature in this review. The work was generously supported by grants from the Wellcome Trust (#061225, Senior Research Fellowship to DJB) and the Medical Research Council. AW and ML are funded by an MRC Studentship and an Usher Cunningham Scholarship from Exeter College, Oxford respectively. Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Davies KE, Nowak KJ. Molecular mechanisms of muscular dystrophies: old and new players. Nat Rev Mol Cell Biol 2006; 7: 762–73
- Yoshida M, Suzuki A, Yamamoto H, Noguchi S, Mizuno Y, Ozawa E. Dissociation of the complex of dystrophin and its associated proteins into several unique groups by n-octyl beta-D-glucoside. Eur J Biochem 1994; 222: 1055–61
- Blake DJ, Weir A, Newey SE, Davies KE. Function and genetics of dystrophin and dystrophin-related proteins in muscle. Physiol Rev 2002; 82: 291–329
- Hopf FW, Turner PR, Steinhardt RA. Calcium misregulation and the pathogenesis of muscular dystrophy. Subcell Biochem 2007; 45: 429–64
- Batchelor CL, Winder SJ. Sparks, signals and shock absorbers: how dystrophin loss causes muscular dystrophy. Trends Cell Biol 2006; 16: 198–205
- Ervasti JM. Dystrophin, its interactions with other proteins, and implications for muscular dystrophy. Biochim Biophys Acta 2007; 1772: 108–17
- Nowak KJ, Davies KE. Duchenne muscular dystrophy and dystrophin: pathogenesis and opportunities for treatment. EMBO Rep 2004; 5: 872–6
- Mehler MF. Brain dystrophin, neurogenetics and mental retardation. Brain Res Brain Res Rev 2000; 32: 277–307
- Cotton SM, Voudouris NJ, Greenwood KM. Association between intellectual functioning and age in children and young adults with Duchenne muscular dystrophy: further results from a meta-analysis. Dev Med Child Neurol 2005; 47: 257–65
- Billard C, Gillet P, Barthez M, Hommet C, Bertrand P. Reading ability and processing in Duchenne muscular dystrophy and spinal muscular atrophy. Dev Med Child Neurol 1998; 40: 12–20
- Moizard MP, Toutain A, Fournier D, Berret F, Raynaud M, Billard C, et al. Severe cognitive impairment in DMD: obvious clinical indication for Dp71 isoform point mutation screening. Eur J Hum Genet 2000; 8: 552–6
- Lenk U, Hanke R, Thiele H, Speer A. Point mutations at the carboxy terminus of the human dystrophin gene: implications for an association with mental retardation in DMD patients. Hum Mol Genet 1993; 2: 1877–81
- Wibawa T, Takeshima Y, Mitsuyoshi I, Wada H, Surono A, Nakamura H, et al. Complete skipping of exon 66 due to novel mutations of the dystrophin gene was identified in two Japanese families of Duchenne muscular dystrophy with severe mental retardation. Brain Dev 2000; 22: 107–12
- Hendriksen JG, Vles JS. Neuropsychiatric disorders in males with duchenne muscular dystrophy: frequency rate of attention-deficit hyperactivity disorder (ADHD), autism spectrum disorder, and obsessive-compulsive disorder. J Child Neurol 2008; 23: 477–81
- Hinton VJ, Nereo NE, Fee RJ, Cyrulnik SE. Social behavior problems in boys with Duchenne muscular dystrophy. J Dev Behav Pediatr 2006; 27: 470–6
- Poysky J. Behavior patterns in Duchenne muscular dystrophy: report on the Parent Project Muscular Dystrophy behavior workshop 8–9 of December 2006, Philadelphia, USA. Neuromuscul Disord 2007; 17: 986–94
- Lee JS, Pfund Z, Juhasz C, Behen ME, Muzik O, Chugani DC, et al. Altered regional brain glucose metabolism in Duchenne muscular dystrophy: a pet study. Muscle Nerve 2002; 26: 506–12
- Tracey I, Scott RB, Thompson CH, Dunn JF, Barnes PR, Styles P, et al. Brain abnormalities in Duchenne muscular dystrophy: phosphorus-31 magnetic resonance spectroscopy and neuropsychological study. Lancet 1995; 345: 1260–4
- Blake DJ, Hawkes R, Benson MA, Beesley PW. Different dystrophin-like complexes are expressed in neurons and glia. J Cell Biol 1999; 147: 645–58
- Moukhles H, Carbonetto S. Dystroglycan contributes to the formation of multiple dystrophin-like complexes in brain. J Neurochem 2001; 78: 824–34
- Gorecki DC, Monaco AP, Derry JM, Walker AP, Barnard EA, Barnard PJ. Expression of four alternative dystrophin transcripts in brain regions regulated by different promoters. Hum Mol Genet 1992; 1: 505–10
- Blake DJ, Love DR, Tinsley J, Morris GE, Turley H, Gatter K, et al. Characterization of a 4.8kb transcript from the Duchenne muscular dystrophy locus expressed in Schwannoma cells. Hum Mol Genet 1992; 1: 103–9
- Lederfein D, Yaffe D, Nudel U. A housekeeping type promoter, located in the 3' region of the Duchenne muscular dystrophy gene, controls the expression of Dp71, a major product of the gene. Hum Mol Genet 1993; 2: 1883–8
- Lidov HG, Selig S, Kunkel LM. Dp140: a novel 140 kDa CNS transcript from the dystrophin locus. Hum Mol Genet 1995; 4: 329–35
- Lidov HG, Byers TJ, Watkins SC, Kunkel LM. Localization of dystrophin to postsynaptic regions of central nervous system cortical neurons. Nature 1990; 348: 725–8
- Knuesel I, Mastrocola M, Zuellig RA, Bornhauser B, Schaub MC, Fritschy JM. Short communication: altered synaptic clustering of GABAA receptors in mice lacking dystrophin (mdx mice). Eur J Neurosci 1999; 11: 4457–62
- Brunig I, Suter A, Knuesel I, Luscher B, Fritschy JM. GABAergic terminals are required for postsynaptic clustering of dystrophin but not of GABA(A) receptors and gephyrin. J Neurosci 2002; 22: 4805–13
- Levi S, Grady RM, Henry MD, Campbell KP, Sanes JR, Craig AM. Dystroglycan is selectively associated with inhibitory GABAergic synapses but is dispensable for their differentiation. J Neurosci 2002; 22: 4274–85
- Kueh SL, Head SI, Morley JW. GABA(A) receptor expression and inhibitory post-synaptic currents in cerebellar Purkinje cells in dystrophin-deficient mdx mice. Clin Exp Pharmacol Physiol 2008; 35: 207–10
- Kim TW, Wu K, Xu JL, Black IB. Detection of dystrophin in the postsynaptic density of rat brain and deficiency in a mouse model of Duchenne muscular dystrophy. Proc Natl Acad Sci U S A 1992; 89: 11642–4
- Peng J, Kim MJ, Cheng D, Duong DM, Gygi SP, Sheng M. Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J Biol Chem 2004; 279: 21003–11
- Mummery R, Sessay A, Lai FA, Beesley PW. Beta-dystroglycan: subcellular localisation in rat brain and detection of a novel immunologically related, postsynaptic density-enriched protein. J Neurochem 1996; 66: 2455–9
- Vaillend C, Rendon A, Misslin R, Ungerer A. Influence of dystrophin-gene mutation on mdx mouse behavior. I. Retention deficits at long delays in spontaneous alternation and bar-pressing tasks. Behav Genet 1995; 25: 569–79
- Vaillend C, Ungerer A. Behavioral characterization of mdx3cv mice deficient in C-terminal dystrophins. Neuromuscul Disord 1999; 9: 296–304
- Sesay AK, Errington ML, Levita L, Bliss TV. Spatial learning and hippocampal long-term potentiation are not impaired in mdx mice. Neurosci Lett 1996; 211: 207–10
- Vaillend C, Billard JM, Laroche S. Impaired long-term spatial and recognition memory and enhanced CA1 hippocampal LTP in the dystrophin-deficient Dmd(mdx) mouse. Neurobiol Dis 2004; 17: 10–20
- Vaillend C, Billard JM. Facilitated CA1 hippocampal synaptic plasticity in dystrophin-deficient mice: role for GABAA receptors?. Hippocampus 2002; 12: 713–7
- Anderson JL, Head SI, Morley JW. Long-term depression is reduced in cerebellar Purkinje cells of dystrophin-deficient mdx mice. Brain Res 2004; 1019: 289–92
- Akhavan A, Crivelli SN, Singh M, Lingappa VR, Muschler JL. SEA domain proteolysis determines the functional composition of dystroglycan. FASEB J 2008; 22: 612–21
- Esapa CT, Bentham GR, Schroder JE, Kroger S, Blake DJ. The effects of post-translational processing on dystroglycan synthesis and trafficking. FEBS Lett 2003; 555: 209–16
- Michele DE, Campbell KP. Dystrophin-glycoprotein complex: post-translational processing and dystroglycan function. J Biol Chem 2003; 278: 15457–60
- Cohn RD. Dystroglycan: important player in skeletal muscle and beyond. Neuromuscul Disord 2005; 15: 207–17
- Williamson RA, Henry MD, Daniels KJ, Hrstka RF, Lee JC, Sunada Y, et al. Dystroglycan is essential for early embryonic development: disruption of Reichert's membrane in Dag1-null mice. Hum Mol Genet 1997; 6: 831–41
- Moore SA, Saito F, Chen J, Michele DE, Henry MD. Messing A, et al. Deletion of brain dystroglycan recapitulates aspects of congenital muscular dystrophy. Nature 2002; 418: 422–5
- Kobayashi K, Nakahori Y, Miyake M, Matsumura K, Kondo-Iida E, Nomura Y, et al. An ancient retrotransposal insertion causes Fukuyama-type congenital muscular dystrophy. Nature 1998; 394: 388–92
- de Bernabe DB, van Bokhoven H, van Beusekom E, Van den Akker W, Kant S, Dobyns WB, et al. A homozygous nonsense mutation in the fukutin gene causes a Walker-Warburg syndrome phenotype. J Med Genet 2003; 40: 845–8
- Murakami T, Hayashi YK, Noguchi S, Ogawa M, Nonaka I, Tanabe Y, et al. Fukutin gene mutations cause dilated cardiomyopathy with minimal muscle weakness. Ann Neurol 2006; 60: 597–602
- Godfrey C, Clement E, Mein R, Brockington M, Smith J, Talim B, et al. Refining genotype phenotype correlations in muscular dystrophies with defective glycosylation of dystroglycan. Brain 2007; 130: 2725–35
- Martin-Rendon E, Blake DJ. Protein glycosylation in disease: new insights into the congenital muscular dystrophies. Trends Pharmacol Sci 2003; 24: 178–83
- Martin PT. Congenital muscular dystrophies involving the O-mannose pathway. Curr Mol Med 2007; 7: 417–25
- Topaloglu H, Brockington M, Yuva Y, Talim B, Haliloglu G, Blake D, et al. FKRP gene mutations cause congenital muscular dystrophy, mental retardation, and cerebellar cysts. Neurology 2003; 60: 988–92
- Aravind L, Koonin EV. The fukutin protein family—predicted enzymes modifying cell-surface molecules. Curr Biol 1999; 9: R836–7
- Brockington M, Blake DJ, Prandini P, Brown SC, Torelli S, Benson MA, et al. Mutations in the fukutin-related protein gene (FKRP) cause a form of congenital muscular dystrophy with secondary laminin alpha2 deficiency and abnormal glycosylation of alpha-dystroglycan. Am J Hum Genet 2001; 69: 1198–209
- Patnaik SK, Stanley P. Mouse large can modify complex N- and mucin O-glycans on alpha-dystroglycan to induce laminin binding. J Biol Chem 2005; 280: 20851–9
- Peyrard M, Seroussi E, Sandberg-Nordqvist AC, Xie YG, Han FY, Fransson I, et al. The human LARGE gene from 22q12.3-q13.1 is a new, distinct member of the glycosyltransferase gene family. Proc Natl Acad Sci U S A 1999; 96: 598–603
- Barresi R, Michele DE, Kanagawa M, Harper HA, Dovico SA, Satz JS, et al. LARGE can functionally bypass alpha-dystroglycan glycosylation defects in distinct congenital muscular dystrophies. Nat Med 2004; 10: 696–703
- Philpot J, Cowan F, Pennock J, Sewry C, Dubowitz V, Bydder G, et al. Merosin-deficient congenital muscular dystrophy: the spectrum of brain involvement on magnetic resonance imaging. Neuromuscul Disord 1999; 9: 81–5
- Chun SJ, Rasband MN, Sidman RL, Habib AA, Vartanian T. Integrin-linked kinase is required for laminin-2-induced oligodendrocyte cell spreading and CNS myelination. J Cell Biol 2003; 163: 397–408
- Holzfeind PJ, Grewal PK, Reitsamer HA, Kechvar J, Lassmann H, Hoeger H, et al. Skeletal, cardiac and tongue muscle pathology, defective retinal transmission, and neuronal migration defects in the Large(myd) mouse defines a natural model for glycosylation-deficient muscle-eye-brain disorders. Hum Mol Genet 2002; 11: 2673–87
- Sugita S, Saito F, Tang J, Satz J, Campbell K, Sudhof TC. A stoichiometric complex of neurexins and dystroglycan in brain. J Cell Biol 2001; 154: 435–45
- Craig AM, Kang Y. Neurexin-neuroligin signaling in synapse development. Curr Opin Neurobiol 2007; 17: 43–52
- Schroder JE, Tegeler MR, Grosshans U, Porten E, Blank M, Lee J, et al. Dystroglycan regulates structure, proliferation and differentiation of neuroepithelial cells in the developing vertebrate CNS. Dev Biol 2007; 307: 62–78
- Blake DJ, Tinsley JM, Davies KE, Knight AE, Winder SJ, Kendrick-Jones J. Coiled-coil regions in the carboxy-terminal domains of dystrophin and related proteins: potentials for protein-protein interactions. Trends Biochem Sci 1995; 20: 133–5
- Blake DJ, Nawrotzki R, Loh NY, Gorecki DC, Davies KE. beta-dystrobrevin, a member of the dystrophin-related protein family. Proc Natl Acad Sci U S A 1998; 95: 241–6
- Grady RM, Grange RW, Lau KS, Maimone MM, Nichol MC, Stull JT, et al. Role for alpha-dystrobrevin in the pathogenesis of dystrophin-dependent muscular dystrophies. Nat Cell Biol 1999; 1: 215–20
- Loh NY, Nebenius-Oosthuizen D, Blake DJ, Smith AJ, Davies KE. Role of beta-dystrobrevin in nonmuscle dystrophin-associated protein complex-like complexes in kidney and liver. Mol Cell Biol 2001; 21: 7442–8
- Grady RM, Wozniak DF, Ohlemiller KK, Sanes JR. Cerebellar synaptic defects and abnormal motor behavior in mice lacking alpha- and beta-dystrobrevin. J Neurosci 2006; 26: 2841–51
- Rees ML, Lien CF, Gorecki DC. Dystrobrevins in muscle and non-muscle tissues. Neuromuscul Disord 2007; 17: 123–34
- Benson MA, Sillitoe RV, Blake DJ. Schizophrenia genetics: dysbindin under the microscope. Trends Neurosci 2004; 27: 516–9
- Burdick KE, Lencz T, Funke B, Finn CT, Szeszko PR, Kane JM, et al. Genetic variation in DTNBP1 influences general cognitive ability. Hum Mol Genet 2006; 15: 1563–8
- Newell-Litwa K, Seong E, Burmeister M, Faundez V. Neuronal and non-neuronal functions of the AP-3 sorting machinery. J Cell Sci 2007; 120: 531–41
- Ahn AH, Yoshida M, Anderson MS, Feener CA, Selig S, Hagiwara Y, et al. Cloning of human basic A1, a distinct 59-kDa dystrophin-associated protein encoded on chromosome 8q23-24. Proc Natl Acad Sci U S A 1994; 91: 4446–50
- Adams ME, Butler MH, Dwyer TM, Peters MF, Murnane AA, Froehner SC. Two forms of mouse syntrophin, a 58 kd dystrophin-associated protein, differ in primary structure and tissue distribution. Neuron 1993; 11: 531–40
- Piluso G, Mirabella M, Ricci E, Belsito A, Abbondanza C, Servidei S, et al. Gamma1- and gamma2-syntrophins, two novel dystrophin-binding proteins localized in neuronal cells. J Biol Chem 2000; 275: 15851–60
- Brenman JE, Chao DS, Gee SH, McGee AW, Craven SE, Santillano DR, et al. Interaction of nitric oxide synthase with the postsynaptic density protein PSD-95 and alpha1-syntrophin mediated by PDZ domains. Cell 1996; 84: 757–67
- Adams ME, Kramarcy N, Krall SP, Rossi SG, Rotundo RL, Sealock R, et al. Absence of alpha-syntrophin leads to structurally aberrant neuromuscular synapses deficient in utrophin. J Cell Biol 2000; 150: 1385–98
- Hosaka Y, Yokota T, Miyagoe-Suzuki Y, Yuasa K, Imamura M, Matsuda R, et al. Alpha1-syntrophin-deficient skeletal muscle exhibits hypertrophy and aberrant formation of neuromuscular junctions during regeneration. J Cell Biol 2002; 158: 1097–107
- Wertz K, Fuchtbauer EM. Dmd(mdx-beta geo): a new allele for the mouse dystrophin gene. Dev Dyn 1998; 212: 229–41
- Haenggi T, Soontornmalai A, Schaub MC, Fritschy JM. The role of utrophin and Dp71 for assembly of different dystrophin-associated protein complexes (DPCs) in the choroid plexus and microvasculature of the brain. Neuroscience 2004; 129: 403–13
- Tian M, Jacobson C, Gee SH, Campbell KP, Carbonetto S, Jucker M. Dystroglycan in the cerebellum is a laminin alpha 2-chain binding protein at the glial-vascular interface and is expressed in Purkinje cells. Eur J Neurosci 1996; 8: 2739–47
- Manley GT, Fujimura M, Ma T, Noshita N, Filiz F, Bollen AW, et al. Aquaporin-4 deletion in mice reduces brain edema after acute water intoxication and ischemic stroke. Nat Med 2000; 6: 159–63
- Amiry-Moghaddam M, Xue R, Haug FM, Neely JD, Bhardwaj A, Agre P, et al. Alpha-syntrophin deletion removes the perivascular but not endothelial pool of aquaporin-4 at the blood-brain barrier and delays the development of brain edema in an experimental model of acute hyponatremia. FASEB J 2004; 18: 542–4
- Vajda Z, Pedersen M, Fuchtbauer EM, Wertz K, Stodkilde-Jorgensen H, Sulyok E, et al. Delayed onset of brain edema and mislocalization of aquaporin-4 in dystrophin-null transgenic mice. Proc Natl Acad Sci U S A 2002; 99: 13131–6
- Nicchia GP, Rossi A, Nudel U, Svelto M, Frigeri A. Dystrophin-dependent and -independent AQP4 pools are expressed in the mouse brain. Glia 2008; 56: 869–76
- Ozawa E, Mizuno Y, Hagiwara Y, Sasaoka T, Yoshida M. Molecular and cell biology of the sarcoglycan complex. Muscle Nerve 2005; 32: 563–76
- Zimprich A, Grabowski M, Asmus F, Naumann M, Berg D, Bertram M, et al. Mutations in the gene encoding epsilon-sarcoglycan cause myoclonus-dystonia syndrome. Nat Genet 2001; 29: 66–9
- Shiga K, Yoshioka H, Matsumiya T, Kimura I, Takeda S, Imamura M. Zeta-sarcoglycan is a functional homologue of gamma-sarcoglycan in the formation of the sarcoglycan complex. Exp Cell Res 2006; 312: 2083–92
- Doheny DO, Brin MF, Morrison CE, Smith CJ, Walker RH, Abbasi S, et al. Phenotypic features of myoclonus-dystonia in three kindreds. Neurology 2002; 59: 1187–96
- Misbahuddin A, Placzek M, Lennox G, Taanman JW, Warner TT. Myoclonus-dystonia syndrome with severe depression is caused by an exon-skipping mutation in the epsilon-sarcoglycan gene. Mov Disord 2007; 22: 1173–5
- Grunewald A, Djarmati A, Lohmann-Hedrich K, Farrell K, Zeller JA, Allert N, et al. Myoclonus-dystonia: significance of large SGCE deletions. Hum Mutat 2008; 29: 331–2
- Tezenas du Montcel S, Clot F, Vidailhet M, Roze E, Damier P, Jedynak CP, et al. Epsilon sarcoglycan mutations and phenotype in French patients with myoclonic syndromes. J Med Genet 2006; 43: 394–400
- Esapa CT, Waite A, Locke M, Benson MA, Kraus M, McIlhinney RA, et al. SGCE missense mutations that cause myoclonus-dystonia syndrome impair epsilon-sarcoglycan trafficking to the plasma membrane: modulation by ubiquitination and torsinA. Hum Mol Genet 2007; 16: 327–42
- Breakefield XO, Blood AJ, Li Y, Hallett M, Hanson PI, Standaert DG. The pathophysiological basis of dystonias. Nat Rev Neurosci 2008; 9: 222–34
- Chan P, Gonzalez-Maeso J, Ruf F, Bishop DF, Hof PR, Sealfon SC. Epsilon-sarcoglycan immunoreactivity and mRNA expression in mouse brain. J Comp Neurol 2005; 482: 50–73
- Nishiyama A, Endo T, Takeda S, Imamura M. Identification and characterization of epsilon-sarcoglycans in the central nervous system. Brain Res Mol Brain Res 2004; 125: 1–12
- Ichinose H, Ohye T, Takahashi E, Seki N, Hori T, Segawa M, et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet 1994; 8: 236–42
- Yokoi F, Dang MT, Li J, Li Y. Myoclonus, motor deficits, alterations in emotional responses and monoamine metabolism in epsilon-sarcoglycan deficient mice. J Biochem 2006; 140: 141–6
- Draviam RA, Shand SH, Watkins SC. The beta-delta-core of sarcoglycan is essential for deposition at the plasma membrane. Muscle Nerve 2006; 34: 691–701
- Allikian MJ, McNally EM. Processing and assembly of the dystrophin glycoprotein complex. Traffic 2007; 8: 177–83
- Chan YM, Bonnemann CG, Lidov HG, Kunkel LM. Molecular organization of sarcoglycan complex in mouse myotubes in culture. J Cell Biol 1998; 143: 2033–44
- Cai H, Erdman RA, Zweier L, Chen J, Shaw JH 4th, Baylor KA, et al. The sarcoglycan complex in Schwann cells and its role in myelin stability. Exp Neurol 2007; 205: 257–69
- Durbeej M, Cohn RD, Hrstka RF, Moore SA, Allamand V, Davidson BL, et al. Disruption of the beta-sarcoglycan gene reveals pathogenetic complexity of limb-girdle muscular dystrophy type 2E. Mol Cell 2000; 5: 141–51
- Sakamoto A, Ono K, Abe M, Jasmin G, Eki T, Murakami Y, et al. Both hypertrophic and dilated cardiomyopathies are caused by mutation of the same gene, delta-sarcoglycan, in hamster: an animal model of disrupted dystrophin-associated glycoprotein complex. Proc Natl Acad Sci U S A 1997; 94: 13873–8
- Hjermind LE, Vissing J, Asmus F, Krag T, Lochmuller H, Walter MC, et al. No muscle involvement in myoclonus-dystonia caused by epsilon-sarcoglycan gene mutations. Eur J Neurol 2008; 15: 525–9
- Draviam RA, Wang B, Shand SH, Xiao X, Watkins SC. Alpha-sarcoglycan is recycled from the plasma membrane in the absence of sarcoglycan complex assembly. Traffic 2006; 7: 793–810
- Breitkreutz BJ, Stark C, Tyers M. Osprey: a network visualization system. Genome Biol 2003; 4: R22
- Bresolin N, Castelli E, Comi GP, Felisari G, Bardoni A, Perani D, et al. Cognitive impairment in Duchenne muscular dystrophy. Neuromuscul Disord 1994; 4: 359–69
- Hinton VJ, De Vivo DC, Fee R, Goldstein E, Stern Y. Investigation of poor academic achievement in children with Duchenne muscular dystrophy. Learn Disabil Res Pract 2004; 19: 146–54
- Young HK, Barton BA, Waisbren S, Portales Dale L, Ryan MM, Webster RI, et al. Cognitive and psychological profile of males with Becker muscular dystrophy. J Child Neurol 2008; 23: 155–62
- Wu JY, Kuban KC, Allred E, Shapiro F, Darras BT. Association of Duchenne muscular dystrophy with autism spectrum disorder. J Child Neurol 2005; 20: 790–5
- Okumura A, Nagai K, Okumura N. Interaction of alpha1-syntrophin with multiple isoforms of heterotrimeric G protein alpha subunits. FEBS J 2008; 275: 22–33
- Yamakawa H, Oyama S, Mitsuhashi H, Sasagawa N, Uchino S, Kohsaka S, et al. Neuroligins 3 and 4X interact with syntrophin-gamma2, and the interactions are affected by autism-related mutations. Biochem Biophys Res Commun 2007; 355: 41–6
- Garcia RA, Vasudevan K, Buonanno A. The neuregulin receptor ErbB-4 interacts with PDZ-containing proteins at neuronal synapses. Proc Natl Acad Sci U S A 2000; 97: 3596–601
- Vandebrouck A, Sabourin J, Rivet J, Balghi H, Sebille S, Kitzis A, et al. Regulation of capacitative calcium entries by alpha1-syntrophin: association of TRPC1 with dystrophin complex and the PDZ domain of alpha1-syntrophin. FASEB J 2007; 21: 608–17
- Chen Z, Hague C, Hall RA, Minneman KP. Syntrophins regulate alpha1D-adrenergic receptors through a PDZ domain-mediated interaction. J Biol Chem 2006; 281: 12414–20
- Buechler C, Boettcher A, Bared SM, Probst MC, Schmitz G. The carboxyterminus of the ATP-binding cassette transporter A1 interacts with a beta2-syntrophin/utrophin complex. Biochem Biophys Res Commun 2002; 293: 759–65
- Hogan A, Yakubchyk Y, Chabot J, Obagi C, Daher E, Maekawa K, et al. The phosphoinositol 3,4-bisphosphate-binding protein TAPP1 interacts with syntrophins and regulates actin cytoskeletal organization. J Biol Chem 2004; 279: 53717–24
- Hogan A, Shepherd L, Chabot J, Quenneville S, Prescott SM, Topham MK, et al. Interaction of gamma 1-syntrophin with diacylglycerol kinase-zeta. Regulation of nuclear localization by PDZ interactions. J Biol Chem 2001; 276: 26526–33
- Leonoudakis D, Conti LR, Anderson S, Radeke CM, McGuire LM, Adams ME, et al. Protein trafficking and anchoring complexes revealed by proteomic analysis of inward rectifier potassium channel (Kir2.x)-associated proteins. J Biol Chem 2004; 279: 22331–46
- Connors NC, Adams ME, Froehner SC, Kofuji P. The potassium channel Kir4.1 associates with the dystrophin-glycoprotein complex via alpha-syntrophin in glia. J Biol Chem 2004; 279: 28387–92
- Neely JD, Amiry-Moghaddam M, Ottersen OP, Froehner SC, Agre P, Adams ME. Syntrophin-dependent expression and localization of Aquaporin-4 water channel protein. Proc Natl Acad Sci U S A 2001; 98: 14108–13
- Lumeng C, Phelps S, Crawford GE, Walden PD, Barald K, Chamberlain JS. Interactions between beta 2-syntrophin and a family of microtubule-associated serine/threonine kinases. Nat Neurosci 1999; 2: 611–7
- Hasegawa M, Cuenda A, Spillantini MG, Thomas GM, Buee-Scherrer V, Cohen P, et al. Stress-activated protein kinase-3 interacts with the PDZ domain of alpha1-syntrophin. A mechanism for specific substrate recognition. J Biol Chem 1999; 274: 12626–31
- Gee SH, Madhavan R, Levinson SR, Caldwell JH, Sealock R, Froehner SC. Interaction of muscle and brain sodium channels with multiple members of the syntrophin family of dystrophin-associated proteins. J Neurosci 1998; 18: 128–37
- Williams JC, Armesilla AL, Mohamed TM, Hagarty CL, McIntyre FH, Schomburg S, et al. The sarcolemmal calcium pump, alpha-1 syntrophin, and neuronal nitric-oxide synthase are parts of a macromolecular protein complex. J Biol Chem 2006; 281: 23341–8
- Iwata Y, Pan Y, Yoshida T, Hanada H, Shigekawa M. Alpha1-syntrophin has distinct binding sites for actin and calmodulin. FEBS Lett 1998; 423: 173–7
- Newbell BJ, Anderson JT, Jarrett HW. Ca2 + -calmodulin binding to mouse alpha1 syntrophin: syntrophin is also a Ca2 + -binding protein. Biochemistry 1997; 36: 1295–305
- Olalla L, Aledo JC, Bannenberg G, Marquez J. The C-terminus of human glutaminase L mediates association with PDZ domain-containing proteins. FEBS Lett 2001; 488: 116–22