Abstract
Therapeutic monoclonal antibodies (mAbs) are the fastest growing class of new therapeutic molecules. They hold great promises for the treatment of a variety of diseases, including chronic inflammatory diseases and cancer. However, the current manufacturing and purification processes cause limitations in the production capacity of therapeutic antibodies, leading to an increase in cost. Genetic delivery of therapeutic monoclonal antibodies by in vivo production offers a new potential solution to these problems. Firstly, therapeutic efficacy can be improved by maintaining stable therapeutic, non-toxic levels within the blood circulation over a long period of time. Repeated high-dose bolus injections could be avoided, thereby reducing the possibility of side-effects. Secondly, the high cost of manufacturing and purification of the therapeutic antibodies could be reduced, making an in vivo/ex vivo mAb gene transfer an economically viable and attractive option.
In general, three approaches can be used for the stable long-term expression and secretion of therapeutic antibodies in vivo: 1) direct in vivo administration of integrating vectors carrying a mAb gene, 2) grafting of ex vivo genetically modified autologous cells, and 3) implantation of an encapsulated antibody producing heterologous or autologous cells.
This paper describes the key factors and problems associated with the current antibody-based immunotherapies and reviews prospects for genetic in vivo delivery of therapeutic antibodies.
Introduction
Behring and his colleagues probably conducted the first antibody-based therapy in 1890. They immunized rats, guinea-pigs, and rabbits with attenuated forms of infectious agents causing diphtheria and tetanus. Serum produced in these animals was injected into non-immunized animals that were previously infected with the fully virulent bacteria. By doing so, the ill animals could be cured through the administration of the serum, and it was already in the following year the first successful therapeutic serum treatment of a child suffering from diphtheria occurred. Since then γ-globulin and related therapies have become clinical practice.
But it was not until 1975 that it became possible to produce identical antibodies specific to a given antigen using the mouse hybridoma technology described by Köhler and Milstein Citation[1]. It paved the way for the emergence of therapeutic monoclonal antibodies (mAbs). However, the utility of these murine antibodies as human therapeutics turned out to be limited. Two major problems plagued the use of murine mAbs: First, although they had great specificity for their targets, they did not always trigger human effector functions (e.g. complement system or professional phagocytes). Secondly, murine antibodies were recognized by the immune system, and they stimulated the production of human anti-mouse antibodies (HAMA response). Also, the therapeutic use of murine antibodies was limited by their short serum half-life Citation[2]. An exception to these is the mouse monoclonal antibody orthoclone OKT3 used in the prevention of organ graft rejection. It also became obvious that the incidence of immunogenicity is variable and depends on the nature of the target antigen, disease process being treated, and the schedule of administration. It was shown that about 50%–70% of patients with solid tumours develop human anti-mouse antibodies after exposure to mouse antibodies, whereas only about 30% of patients with relapsed B-cell malignancies treated with mouse antibodies developed HAMA.
Key messages
Therapeutic monoclonal antibodies (mAbs) are the fastest growing class of new therapeutic molecules.
Current manufacturing and purification processes cause limitations in the production capacity of therapeutic antibodies.
Genetic delivery of therapeutic monoclonal antibodies by in vivo production offers a new potential solution in antibody therapy.
Current therapeutic antibodies
Monoclonal antibodies now comprise the majority of recombinant proteins in clinical testing, with more than 150 trials worldwide Citation[2]. About a quarter of new biotech drugs in development are monoclonal antibodies Citation[3]. These entities represent also the fastest growing class of new therapeutic molecules Citation[4]. Seven of the approved monoclonal antibody products each had global sales of over US$1 billion in 2006. Three of the products, infliximab and adalimumab, both indicated for the treatment of autoimmune disorders, and rituximab indicated for non-Hodgkin's lymphoma, B-cell leukaemia, and autoimmune disorders, had sales over US$2 billion each. Global sales expectation for monoclonal antibodies is around US$16 billion by 2010 Citation[5] ().
Table I. Monoclonal antibodies approved by the US Food and Drug Administration (FDA).
The first mAb approved for human therapy was the murine monoclonal antibody, muromonab-CD3 (Orthoclone OKT3) in June 1986, for the treatment of transplant rejection. The platelet aggregation inhibitor, abciximab (ReoPro) was the first chimeric and the first mAb fragment to be approved by the Food and Drug Administration (FDA), in December 1994. The first humanized mAb approved was daclizumab (Zenapax) in December 1997 for the prevention of transplant rejection. The first human mAb approved by FDA was adalimumab (Humira) in December 2002, for the treatment of rheumatoid arthritis and other inflammatory conditions. Of the 21 FDA-approved mAbs, 3 are murine, 5 are chimeric, 11 are humanized, and 2 are human mAbs (). Gemtuzumab ozogamicin conjugated with immunotoxin calicheamicin was approved in 2000, and ibritumomab tiuxetan (Zevalin) was the first approved mAb conjugated with the radionuclide yttrium-90 in 2002, for the treatment of acute myelogenous leukaemia and non-Hodgkin's lymphoma, respectively. In 1997, rituximab (Rituxan), the first mAb for the treatment of cancer, was approved by the FDA ().
By March 2008, 21 therapeutic mAbs had been approved by the FDA for human therapy in both haematological and solid tumours, and autoimmune, inflammatory, infectious, and cardiovascular diseases Citation[6–8] (). Non-Hodgkin's lymphoma, metastatic breast cancer, acute myeloid leukaemia, chronic lymphocytic leukaemia, and metastatic colorectal cancer are some of the cancers where mAbs are indicated for therapy Citation[2], Citation[8], Citation[9]. Acute transplant rejection is the major autoimmune indication, with Crohn's disease and rheumatoid arthritis being inflammatory indications for therapy. In addition to these, mAbs are used for the treatment of respiratory syncytial viral infection, prevention of blood clotting, in refractory unstable angina, and as a snake venom antidote Citation[8], Citation[9].
Genetic engineering of mAbs
In an attempt to reduce immunogenicity of mouse antibodies, genetic engineering was used to generate chimeric antibodies, that is, antibodies with human constant regions and mouse variable regions. This technique has been central to the clinical use of antibodies. Chimeric antibodies were less immunogenic, with the ability to trigger human effector functions and increased circulation half-life. Even though chimeric antibodies were perceived as less foreign, and therefore less immunogenic, human anti-chimeric antibody responses (HACAs) have been observed Citation[10]. Further minimization of the mouse component of antibodies was achieved through complementarity-determining region (CDR) grafting. In such ‘humanized’ antibodies, only the CDR loops that are responsible for antigen binding are inserted into the human variable-domain framework. The ability to manipulate antibodies into more human-like variants finally made antibodies in the main stream of clinical use. With the isolation of genes encoding for human variable regions, their successful expression in Escherichia coli, and the introduction of phage-display technology along with the developments in hybridoma technology, the task of selecting fully human variable domains has been greatly simplified Citation[11]. Recent advancements in transgenic animal techniques too enabled the production of fully human mAbs in animals such as humanized mouse Citation[12].
Technical advances in antibody development further enabled the generation of modified antibodies with enhanced properties and less immunogenicity Citation[4]. Different types of variable domain architectures, such as diabodies, triabodies, and bispecific diabodies, are engineered to better serve specific therapeutic applications. Bivalent antibody-binding fragments (F(ab)2), single chain variable fragments (scFv), minibodies with two scFv regions fused to heavy-chain constant region (CH3), and bispecific diabodies with two different scFv regions are some examples of modified mAbs.
Limitations of current antibody therapies
Production
Different methods using hybridoma technology in bacterial, yeast, and mammalian cell lines and transgenic animals are used in the production of different types of mAbs. In all these methods the biological materials used in the production process have to be free of microbial contamination by mycoplasma, fungi, bacteria, viruses, and prions. Animals have to be specific pathogen-free (SPF) quality. Currently, recombinant expression technology in mammalian cell culture is the principal means for the commercial production of antibodies. Human mAbs produced in cell lines immortalized by introduction of Epstein-Barr virus (EBV) could carry a potential risk of cancer. Risk of contamination of different cell lines, production of endogenous immunoglobulin chains, genetic instability of the cell line, and level and consistency of the expression of both heavy and light chains are other aspects that need further attention. Another problem encountered during mAb production is to find ways of generating stoichiometric amounts of both heavy and light chains in the production cell lines, because such imbalances in the chain production can be toxic to cells or can result in non-functional immunoglobulin chains in the supernatant that can complicate the purification process Citation[13].
Tedious purifications, characterizations, and validations have to be done during the production process for the establishment of consistency with regard to identity, purity, and potency between different batches. Changes or adaptations in the production process such as transition of in vivo production to in vitro production, changes in culture procedure or culture conditions, changes in purification procedure, different types of fermentation technology in different plants, hollow fibre versus suspension culture, or additional modifications of the antibody molecule can lead to an altered form of the antibody with compromised specificity Citation[5], Citation[14].
Apart from the high material cost and long cycle times, the mammalian cell culture-based production systems provide a limited control over the glycosylation of the mAbs Citation[15]. The glycosylation profile is a major determinant of the stability, biological activity including receptor binding and effector function, pharmacokinetics, biodistribution, and solubility of mAbs. The glycosylation pattern is dependent on the protein's three-dimensional structure, host cell enzymatic system in the endoplasmic reticulum and Golgi apparatus, efficiency of protein expression, physiological status of the host cells, availability of the sugar-nucleotide donors, and the fermentation conditions Citation[16–18].
Due to the need for high doses over a long period of time, production capacity of mAbs in mammalian cell cultures may become a limiting factor Citation[5], Citation[19–22] with forecasts of a shortage of the capacity for the production in near future Citation[5], Citation[8]. Transgenic animals provide an alternative and potentially a more economical source for the production as compared to traditional cell culture and fermentation systems especially when needed in high doses for a longer period of time Citation[23].
Cost
The start-up cost for six 15,000-litre bioreactors will be in the range of 100 million euros. In addition, investments in the range of 300–500 million euros along with the need to hire and train 400–500 employees will be required every 5–10 years Citation[5]. Large-scale production of monoclonal antibodies needs economical purification processes with high throughput that can deliver a high yield and process reliability while producing an extremely pure product Citation[24]. The success of production systems depend on intrinsic characteristics of hybridomas such as cell growth and antibody production ability in large-scale production Citation[3].
The cost of therapy is a major limiting factor associated with mAbs, with monthly costs of certain drugs reaching nearly US$10,000 and annual costs of about US$100,000 per patient leading to serious concerns Citation[25]. Both the high doses and the longer period of time have aggravated the issue within the industry justifying the high cost as the inherent value of life-sustaining therapies Citation[6], Citation[8], Citation[25]. Monoclonal antibodies are also differentially priced according to the therapeutic category. Monoclonal antibodies for oncological conditions with a smaller market are priced much more highly than those for immunological diseases with a larger market. High treatment costs have raised the necessity for biosimilars, although these have not been successful yet due to complexity of production, lack of regulatory pathways for approval, and patent protection covering all the recently approved products Citation[2].
Pharmacokinetics
Many factors, such as antigen distribution and antigen concentration, antibody structure, size and engineering, host factors, concurrent medications and immunogenicity, are involved in the pharmacokinetics of monoclonal antibodies Citation[7], Citation[26], Citation[27]. The half-lives of murine antibodies are much shorter than chimeric or humanized antibodies in man: murine (2–3 days) < chimeric (8–10 days) < humanized (20–23 days) Citation[7]. Smaller modified antibodies are rapidly cleared from plasma (half-life 0.5–30 hours) due to the lack of Fc domain, though they have a better penetration ability especially in the treatment of cancer Citation[9], Citation[28], Citation[29]. In fact, only 0.001%–0.01% of the injected antibody dose accumulates per gram of solid tumour in patients Citation[30].
Elimination of mAbs might be altered by interactions with the target antigen which manifests as a dose-dependent clearance rate and half-life, with a shorter half-life at low antibody concentrations that do not saturate the antigens, whereas an increased half-life and decreased clearance rate is observed with a higher concentration that can saturate the antigens Citation[7]. Monoclonal antibodies show non-linear pharmacokinetic and pharmacodynamic properties which are not properly understood with individual variability Citation[31]. Effects of demographic variables, such as age, gender, and renal and hepatic function, on the pharmacokinetics of monoclonal antibodies are somewhat controversial Citation[7]. Large proteins in general have slower kinetics than small molecules with limited tissue penetration properties. Some concomitant medications, such as methotrexate, azothioprine, and mycophenolate mofetil, have been reported to reduce the clearance of some antibodies, and paclitaxel has been shown to increase serum concentrations of some antibodies Citation[3].
Safety
Most of the current mAbs have to be administered in large doses, ranging from 5 to 20 milligrams per kilogram body weight at a time, to achieve a high and sustained therapeutic plasma concentration of around several hundred µg per millilitre. This is not only leading to problems such as high cost, but also to problems related to administration, bioavailability, and increased immunogenicity Citation[3], Citation[5], Citation[8], Citation[22], Citation[25], Citation[32–38]. Most mAbs have to be given as hour-long intra-venous (i.v.) infusions needing hospital environments with skilled staff and equipment, and repeated over a long period of time Citation[8]. Due to infusion-based treatment the concentration of the molecule varies between subtoxic to subtherapeutic levels resulting in variations in bioavailability with a higher probability of neutralizing anti-idiotypic immune response Citation[8].
HAMA response results in rapid elimination of mouse antibodies from the host with a reduced efficiency, possible immune complex damage to the kidneys, anaphylaxis, and allergic reactions Citation[2], Citation[3], Citation[7], Citation[9], Citation[13], Citation[39], Citation[40]. These were reduced with the development of chimeric antibodies, humanized antibodies Citation[41], and the development of fully human antibodies through transgenic mice Citation[42], Citation[43] or through generation of human hybridomas Citation[9], Citation[39], Citation[41]. Two types of HAMA responses have been reported: anti-isotypic and anti-idiotypic Citation[3]. All mAbs in clinical use have exhibited some level of immunogenicity Citation[7]. Chimeric, humanized, and human mAbs have demonstrated immunogenicity rates of 5%–10% Citation[2], Citation[43]. The protein structure, immune modulatory effect of protein, formulation, contaminants and impurities, route, dose, frequency and duration of administration, major histocompatibility complex (MHC) genotype of the patient, associated diseases, and concomitant therapy are factors that directly affect immunogenicity, whereas timing and frequency of sampling, assay method, and expression of titres are factors that affect the measured immune response Citation[38]. Identification of anti-monoclonal antibodies is highly dependent on the sensitivity and specificity of the assay, making comparisons difficult Citation[2]. Lack of precise methods to determine immunogenicity and lack of specific guide-lines for immunogenicity assay validation are major limitations in the antibody therapy Citation[44].
Like all other classes of therapeutic agents, monoclonal antibodies can cause side-effects. These effects are related to one of three major mechanisms: xenogenic nature of the antibodies, suppression of physiological functions in line with the specificity of the antibody, and activation of inflammatory cells and mediators after binding to the target Citation[3]. Monoclonal antibodies are generally considered less toxic than cytotoxic chemotherapeutic agents used in cancer treatment. Mechanism-independent toxicities are due to hypersensitivity reactions and can rarely be severe enough to cause anaphylactic reactions. Mechanism-dependent toxicities include cardiac toxicity with trastuzumab Citation[40], Citation[45], first-dose toxicity related to rapid lysis of malignant cells by rituximab Citation[46], skin eruptions with cetuximab Citation[47], Citation[48], and hypertension, bleeding, thrombosis, and proteinuria with bevacizumab Citation[49]. Hypersensitivity reactions leading to serum sickness are generally rare, but fatal infusion reactions have been reported with some mAbs Citation[3].
Potential improvements of future antibody therapies
Because of the limiting factors described above, as well as the inconvenience of weekly infusions over a long period of time, new technological solutions have been explored. One solution to the problems mentioned above could be the use of gene transfer technology for the production of therapeutic antibodies in vivo.
The aim of genetic delivery of therapeutic mAb genes is to avoid the costly and laborious procedure of repeated intravenous mAb infusions. The genetic delivery of mAb genes can be achieved with viral and non-viral gene transfer methods. In an optimal situation, a single vector injection would result in a continuous and stable release of low but therapeutically relevant quantities of mAbs within the patient. Monoclonal antibodies produced by the patient have the advantage of being potentially less immunogenic than recombinantly produced mAbs with an improved bioavailability of the therapeutic proteins. Possible side-effects and immune responses against therapeutic proteins could be minimized by avoiding repeated administration of high doses of these therapeutic antibodies.
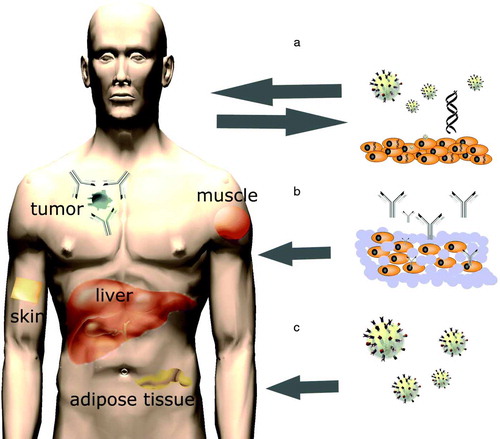
Stable long-term expression and secretion of therapeutic antibodies in vivo can be achieved in vivo by three approaches: 1) direct in vivo administration of mAb gene carrying vectors, 2) grafting of ex vivo genetically modified autologous cells, and 3) implantation of ex vivo transduced cells, encapsulated within artificial semi-permeable membranes ().
Figure 1. There are three approaches through which stable long-term expression and secretion of therapeutic antibodies in vivo could be achieved: A) grafting of ex vivo genetically modified autologous cells, B) implantation of ex vivo transduced cells encapsulated within artificial semi-permeable membranes, and C) direct in vivo administration of mAb gene-carrying vectors.
The ability to regulate the antibody production very tightly and consistently is essential if the therapy is no longer required or in the case of development of undesired side-effects. Tetracycline-dependent transcriptional switches, lac operator, rapamycin and progesterone analogue-inducible systems have been successfully employed in several preclinical gene therapy studies using different vector systems Citation[50], though the authors could not find specific examples of regulation of in vivo antibody production. However, the leakiness of the regulation systems, lack of penetration of the inducer drug to the target tissue/organ, immune responses against the activator proteins, and modulation of endogenous gene expression are some of the drawbacks of these systems Citation[50]. Another possibility is to incorporate a suicide gene such as herpes simplex virus thymidine kinase in the transgene cassette along with the antibody gene. Administration of the pro-drug such as ganciclovir will specifically kill the cells expressing the transgene.
Ex vivo gene transfer
Ex vivo gene transfer involves the collection of cells which will be genetically modified, their maintenance in cell culture, and in vitro gene transfer. Successfully transduced cells will be selected, expanded and introduced into the recipient (a). Once transplanted, the gene-modified cells produce therapeutic proteins. Benefits of this approach include the ability to target gene transfer to a specific cell type and the possibility to select for correctly modified cells before transplantation. Possible vector inactivation by the host immune system is also avoided. Ex vivo gene transfer to the patient-derived cells is, however, only applicable to those cells and tissues that can be safely removed, are susceptible to gene transfer, and can be reimplanted.
In addition to the patient's own cells, transplantation of ex vivo modified non-self cells is possible if cells are microencapsulated prior to transplantation. In microencapsulation, the modified cells are enclosed within artificial semi-permeable membranes. The membrane is designed to allow bidirectional diffusion of nutrients, oxygen, and waste products, while preventing the immune cells from destroying the enclosed cells (b). Many types of polymers can be used for microencapsulation to allow long-term function of the graft. Citation[51], Citation[52].
In vivo gene transfer
Several studies have shown that production and secretion of antibodies can also be achieved through direct in vivo gene transfer using different viral vectors and plasmid DNA (pDNA) Citation[8], Citation[22], Citation[53], Citation[54] (c; ). In animal studies, vectors have mostly been injected into skeletal muscles (i.m.) and blood circulation. After i.v. vector injection, the main site of transduction is the liver, thus making hepatocytes key players in antibody expression. Transgene proteins expressed by hepatocytes may actually be less immunogenic than proteins expressed by other cells, which is an advantage of i.v. injections Citation[55]. Muscle cells are also known to be capable of expressing correctly folded and functional antibody molecules Citation[56–58] (). The data available from animal studies imply that after i.m. vector injection antibody blood concentrations have generally been lower and more variable than after i.v. administration Citation[54], Citation[57], Citation[59]. Adenovirus (Ad) vectors do not integrate, which leads to declining antibody expression after the initial production peaks at around one week Citation[59–61]. The majority of adeno-associated virus (AAV) vector genomes persist in infected cells as extrachromosomal episomes Citation[62–64] which promote more sustained antibody expression than Ads Citation[22], Citation[58]. Lentivirus vectors efficiently catalyse transduction and transgene integration in various cell types, and intravenously injected they have promoted high and rather stable blood mAb concentrations, the major site of transduction being the liver Citation[54]. Intravenously administered lentiviral vectors or rAAVs thus seem to be the most suitable vectors for long-term in vivo mAb production. For short-term production, non-integrating Ads or plasmid DNA would be more suitable. The use of integrating vectors for in vivo mAb gene transfer raises safety issues and ethical concerns, because the unpredictable integration pattern of the currently available vectors can adversely result in insertional mutagenesis and disruption of cellular genes Citation[65], Citation[66]. This risk can be overcome if the transduced cells are selected in vitro before implantation into the recipient. The development of site-specifically integrating vectors would also enable safer in vivo mAb gene transfer. Also, by using non-integrating vectors the risks of insertional mutagenesis can be minimized, and rather long-term mAb secretion can be achieved if non-dividing cells and tissues, such as the muscle, are targeted for gene transfer Citation[67].
Table II. In vivo antibody production by different vectors in animal models.
Conclusion
Therapeutic mAbs are the fastest growing class of new therapeutic molecules. They hold great promise for the treatment of a variety of diseases. However, with the rising demand for these therapeutics, there is a need for new technological innovations regarding mAb manufacturing and the therapeutical approach itself. The idea of the genetic delivery of therapeutic mAbs either by ex vivo gene transfer or in vivo gene transfer represents an exciting technique to further improve monoclonal antibody-based therapies. Genetic delivery of therapeutic proteins have clear advantages: First the therapeutic efficacy of the therapeutic antibody could most likely be improved by maintaining stable therapeutic and non-toxic levels of the therapeutic antibody within the blood circulation over a long period of time. Repeated high-dose bolus injections are avoided, thereby reducing the possibility of side-effects. Secondly, highly expensive production and manufacturing costs of the therapeutic proteins can be reduced, making a one-time gene transfer procedure available to the patients.
Acknowledgements
This work was supported by grants from Finnish Academy and Ark Therapeutics group Plc. Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper. Haritha Samaranayake and Thomas Wirth contributed equally to the writing.
References
- Kohler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975; 256: 495–7
- Reichert JM, Rosensweig CJ, Faden LB, Dewitz MC. Monoclonal antibody successes in the clinic. Nat Biotechnol. 2005; 23: 1073–8
- Breedveld FC. Therapeutic monoclonal antibodies. Lancet. 2000; 355: 735–40
- Stocks M. Intrabodies as drug discovery tools and therapeutics. Curr Opin Chem Biol. 2005; 9: 359–65
- Werner RG. Economic aspects of commercial manufacture of biopharmaceuticals. J Biotechnol. 2004; 113: 171–82
- Marasco WA. Therapeutic antibody gene transfer. Nat Biotechnol. 2005; 23: 551–2
- Tabrizi MA, Tseng CM, Roskos LK. Elimination mechanisms of therapeutic monoclonal antibodies. Drug Discov Today. 2006; 11: 81–8
- Pelegrin M, Gros L, Dreja H, Piechaczyk M. Monoclonal antibody-based genetic immunotherapy. Curr Gene Ther. 2004; 4: 347–56
- Schrama D, Reisfeld RA, Becker JC. Antibody targeted drugs as cancer therapeutics. Nat Rev Drug Discov. 2006; 5: 147–59
- Baert F, Noman M, Vermeire S, Van Assche G, D'Haens G, Carbonez A, et al. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn's disease. N Engl J Med. 2003; 348: 601–8
- Knappik A, Ge L, Honegger A, Pack P, Fischer M, Wellnhofer G, et al. Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J Mol Biol. 2000; 296: 57–86
- Lonberg, N. Human monoclonal antibodies from transgenic mice. Handb Exp Pharmacol. 2008:69–97.
- Iannello A, Ahmad A. Role of antibody-dependent cell-mediated cytotoxicity in the efficacy of therapeutic anti-cancer monoclonal antibodies. Cancer Metastasis Rev. 2005; 24: 487–99
- Directive 75/318/EEC. Production and quality control of monoclonal antibodies. 2007. Ref Type: Generic., http://www.emea.europa.eu/pdfs/human/bwp/3ab4aen.pdf
- Sethuraman N, Stadheim TA. Challenges in therapeutic glycoprotein production. Curr Opin Biotechnol. 2006; 17: 341–6
- Butler M. Optimisation of the cellular metabolism of glycosylation for recombinant proteins produced by Mammalian cell systems. Cytotechnology. 2006; 50: 57–76
- Werner RG, Noe W, Kopp K, Schluter M. Appropriate mammalian expression systems for biopharmaceuticals. Arzneimittelforschung. 1998; 48: 870–80
- Werner RG, Kopp K, Schlueter M. Glycosylation of therapeutic proteins in different production systems. Acta Paediatr Suppl. 2007; 96: 17–22
- Costello JC, Halverson PB. A new era in rheumatoid arthritis treatment. WMJ. 2003; 102: 29–33
- Finesilver AG. Newer approaches to the treatment of rheumatoid arthritis. WMJ. 2003; 102: 34–7
- Toi M, Takada M, Bando H, Toyama K, Yamashiro H, Horiguchi S, et al. Current status of antibody therapy for breast cancer. Breast Cancer. 2004; 11: 10–4
- Fang J, Qian JJ, Yi S, Harding TC, Tu GH, VanRoey M, et al. Stable antibody expression at therapeutic levels using the 2A peptide. Nat Biotechnol. 2005; 23: 584–90
- Das, RC. Proteins and antibodies make advances as therapeutic products. Am Clin Lab. 2001;20:8, 10–8, 14.
- Zhou JX, Tressel T. Basic concepts in Q membrane chromatography for large-scale antibody production. Biotechnol Prog. 2006; 22: 341–9
- Berenson, A. A cancer drug shows promise, at a price that many can't pay. New York Times. 2006, Jun 15.
- Lobo ED, Hansen RJ, Balthasar JP. Antibody pharmacokinetics and pharmacodynamics. J Pharm Sci. 2004; 93: 2645–68
- Tang L, Persky AM, Hochhaus G, Meibohm B. Pharmacokinetic aspects of biotechnology products. J Pharm Sci. 2004; 93: 2184–204
- Metzler B, Li C, Hu Y, Sturm G, Ghaffari-Tabrizi N, Xu Q. LDL stimulates mitogen-activated protein kinase phosphatase-1 expression, independent of LDL receptors, in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 1999; 19: 1862–71
- Yokota T, Milenic DE, Whitlow M, Schlom J. Rapid tumor penetration of a single-chain Fv and comparison with other immunoglobulin forms. Cancer Res. 1992; 52: 3402–8
- Sanz L, Blanco B, Alvarez-Vallina L. Antibodies and gene therapy: teaching old ‘magic bullets’ new tricks. Trends Immunol. 2004; 25: 85–91
- Ternant D, Paintaud G. Pharmacokinetics and concentration-effect relationships of therapeutic monoclonal antibodies and fusion proteins. Expert Opin Biol Ther. 2005; 5((Suppl 1))S37–47
- Gruber R, van Haarlem LJ, Warnaar SO, Holz E, Riethmuller G. The human antimouse immunoglobulin response and the anti-idiotypic network have no influence on clinical outcome in patients with minimal residual colorectal cancer treated with monoclonal antibody CO17-1A. Cancer Res. 2000; 60: 1921–6
- Isaacs JD. The antiglobulin response to therapeutic antibodies. Semin Immunol. 1990; 2: 449–56
- Kuus-Reichel K, Grauer LS, Karavodin LM, Knott C, Krusemeier M, Kay NE. Will immunogenicity limit the use, efficacy, and future development of therapeutic monoclonal antibodies?. Clin Diagn Lab Immunol. 1994; 1: 365–72
- Davis TA, Grillo-Lopez AJ, White CA, McLaughlin P, Czuczman MS, Link BK, et al. Rituximab anti-CD20 monoclonal antibody therapy in non-Hodgkin's lymphoma: safety and efficacy of re-treatment. J Clin Oncol. 2000; 18: 3135–43
- Lin YS, Nguyen C, Mendoza JL, Escandon E, Fei D, Meng YG, et al. Preclinical pharmacokinetics, interspecies scaling, and tissue distribution of a humanized monoclonal antibody against vascular endothelial growth factor. J Pharmacol Exp Ther. 1999; 288: 371–8
- Armbruster C, Stiegler GM, Vcelar BA, Jager W, Michael NL, Vetter N, et al. A phase I trial with two human monoclonal antibodies (hMAb 2F5, 2G12) against HIV-1. AIDS. 2002; 16: 227–33
- Hermeling S, Crommelin DJ, Schellekens H, Jiskoot W. Structure-immunogenicity relationships of therapeutic proteins. Pharm Res. 2004; 21: 897–903
- Waldmann TA. Immunotherapy: past, present and future. Nat Med. 2003; 9: 269–77
- Adams GP, Weiner LM. Monoclonal antibody therapy of cancer. Nat Biotechnol. 2005; 23: 1147–57
- Wawrzynczak, EJ. Antibody Therapy. Oxford: BIOS Scientific Publishers Ltd; 1995.
- Bruggemann M, Taussig MJ. Production of human antibody repertoires in transgenic mice. Curr Opin Biotechnol. 1997; 8: 455–8
- Mendez MJ, Green LL, Corvalan JR, Jia XC, Maynard-Currie CE, Yang XD, et al. Functional transplant of megabase human immunoglobulin loci recapitulates human antibody response in mice. Nat Genet. 1997; 15: 146–56
- Geng D, Shankar G, Schantz A, Rajadhyaksha M, Davis H, Wagner C. Validation of immunoassays used to assess immunogenicity to therapeutic monoclonal antibodies. J Pharm Biomed Anal. 2005; 39: 364–75
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001; 344: 783–92
- Lowndes S, Darby A, Mead G, Lister A. Stevens-Johnson syndrome after treatment with rituximab. Ann Oncol. 2002; 13: 1948–50
- Robert F, Ezekiel MP, Spencer SA, Meredith RF, Bonner JA, Khazaeli MB, et al. Phase I study of anti—epidermal growth factor receptor antibody cetuximab in combination with radiation therapy in patients with advanced head and neck cancer. J Clin Oncol. 2001; 19: 3234–43
- Herbst RS, Langer CJ. Epidermal growth factor receptors as a target for cancer treatment: the emerging role of IMC-C225 in the treatment of lung and head and neck cancers. Semin Oncol. 2002; 29((Suppl 4))27–36
- Hurwitz H, Fehrenbacher L, Novotny W, Cartwright T, Hainsworth J, Heim W, et al. Bevacizumab plus irinotecan, fluorouracil, and leucovorin for metastatic colorectal cancer. N Engl J Med. 2004; 350: 2335–42
- Goverdhana S, Puntel M, Xiong W, Zirger JM, Barcia C, Curtin JF, et al. Regulatable gene expression systems for gene therapy applications: progress and future challenges. Mol Ther. 2005; 12: 189–211
- Orive G, Gascon AR, Hernandez RM, Igartua M, Luis PJ. Cell microencapsulation technology for biomedical purposes: novel insights and challenges. Trends Pharmacol Sci. 2003; 24: 207–10
- Orive G, Hernandez RM, Gascon AR, Igartua M, Pedraz JL. Encapsulated cell technology: from research to market. Trends Biotechnol. 2002; 20: 382–7
- Pelegrin M, Marin M, Oates A, Noel D, Saller R, Salmons B, et al. Immunotherapy of a viral disease by in vivo production of therapeutic monoclonal antibodies. Hum Gene Ther. 2000; 11: 1407–15
- Lamikanra A, Myers KA, Ferris N, Mitrophanous KA, Carroll MW. In vivo evaluation of an EIAV vector for the systemic genetic delivery of therapeutic antibodies. Gene Ther. 2005; 12: 988–98
- Herzog RW. Recent advances in hepatic gene transfer: more efficacy and less immunogenicity. Curr Opin Drug Discov Devel. 2005; 8: 199–206
- Noel D, Pelegrin M, Marin M, Biard-Piechaczyk M, Ourlin JC, Mani JC, et al. In vitro and in vivo secretion of cloned antibodies by genetically modified myogenic cells. Hum Gene Ther. 1997; 8: 1219–29
- Noel D, Pelegrin M, Kramer S, Jacquet C, Skander N, Piechaczyk M. High in vivo production of a model monoclonal antibody on adenoviral gene transfer. Hum Gene Ther. 2002; 13: 1483–93
- Lewis AD, Chen R, Montefiori DC, Johnson PR, Clark KR. Generation of neutralizing activity against human immunodeficiency virus type 1 in serum by antibody gene transfer. J Virol. 2002; 76: 8769–75
- Arafat WO, Gomez-Navarro J, Buchsbaum DJ, Xiang J, Wang M, Casado E, et al. Effective single chain antibody (scFv) concentrations in vivo via adenoviral vector mediated expression of secretory scFv. Gene Ther. 2002; 9: 256–62
- Whittington HA, Ashworth LJ, Hawkins RE. Recombinant adenoviral delivery for in vivo expression of scFv antibody fusion proteins. Gene Ther. 1998; 5: 770–7
- Jiang M, Shi W, Zhang Q, Wang X, Guo M, Cui Z, et al. Gene therapy using adenovirus-mediated full-length anti-HER-2 antibody for HER-2 overexpression cancers. Clin Cancer Res. 2006; 12((20 Pt 1))6179–85
- Schnepp BC, Jensen RL, Chen CL, Johnson PR, Clark KR. Characterization of adeno-associated virus genomes isolated from human tissues. J Virol. 2005; 79: 14793–803
- Schnepp BC, Clark KR, Klemanski DL, Pacak CA, Johnson PR. Genetic fate of recombinant adeno-associated virus vector genomes in muscle. J Virol. 2003; 77: 3495–504
- Duan L, Laughlin MA, Oakes JW, Pomerantz RJ. Potent inhibition of human immunodeficiency virus type 1 replication by an intracellular anti-Rev single-chain antibody. Proc Natl Acad Sci U S A. 1998; 95: 10344
- Hacein-Bey-Abina S, Von Kalle C, Schmidt M, McCormack MP, Wulffraat N, Leboulch P, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science. 2003; 302: 415–9
- Schmidt M, Hacein-Bey-Abina S, Wissler M, Carlier F, Lim A, Prinz C, et al. Clonal evidence for the transduction of CD34+ cells with lymphomyeloid differentiation potential and self-renewal capacity in the SCID-X1 gene therapy trial. Blood. 2005; 105: 2699–706
- Tjelle TE, Corthay A, Lunde E, Sandlie I, Michaelsen TE, Mathiesen I, et al. Monoclonal antibodies produced by muscle after plasmid injection and electroporation. Mol Ther. 2004; 9: 328–36
- Whittington HA, Ashworth LJ, Hawkins RE. Recombinant adenoviral delivery for in vivo expression of scFv antibody fusion proteins. Gene Ther. 1998; 5: 770–7
- Perez N, Bigey P, Scherman D, Danos O, Piechaczyk M, Pelegrin M. Regulatable systemic production of monoclonal antibodies by in vivo muscle electroporation. Genet Vaccines Ther. 2004; 2: 2