Abstract
Mast cells (MCs) are perivascularly located resident cells of hematopoietic origin, recognized as effectors in inflammation and immunity. Their subendothelial location at the boundary between the intravascular and extravascular milieus, and their ability to rapidly respond to blood- and tissue-borne stimuli via release of potent vasodilatatory, proteolytic, fibrinolytic, and proinflammatory mediators, render MCs with a unique status to act in the first-line defense in various pathologies. We review experimental evidence suggesting a role for MCs in the pathophysiology of brain ischemia and hemorrhage. In new-born rats, MCs contributed to brain damage in hypoxic-ischemic insults. In experimental cerebral ischemia/reperfusion, MCs regulated permeability of the blood-brain barrier, brain edema formation, and the intensity of local neutrophil infiltration. MCs were reported to play a role in the tissue plasminogen activator-mediated cerebral hemorrhages after experimental ischemic stroke, and to be involved in the expansion of hematoma and edema following intracerebral hemorrhage. Importantly, the MC-stabilizing drug cromoglycate inhibited MC-mediated adverse effects on brain pathology and improved survival of experimental animals. This brings us to a position to consider MC stabilization as a novel initial adjuvant therapy in the prevention of brain injuries in hypoxia-ischemia in new-borns, as well as in ischemic stroke and intracerebral hemorrhage in adults.
Introduction
Ischemic and hemorrhagic strokes are among the foremost causes of death and disability worldwide. Massive brain edema is the leading cause of death in large hemispheric strokes Citation[1] and is only modestly alleviated by the limited conservative treatments available today. The two main detrimental effects of brain edema are: 1) mass effect with compression of normal brain structures (herniation), and 2) decrease in cerebral blood flow. Both effects damage the penumbra's (tissue-at-risk) metabolism and facilitate its transformation into an infarction.
Aimed at fast restoration of circulation, thrombolysis with tissue plasminogen activator (tPA) is the only approved therapy in acute ischemic stroke within 4.5 hours after symptom onset Citation[2]. Fear of tPA-mediated hemorrhage is often a reason for withholding this otherwise beneficial treatment. In addition, recanalization of the occluded artery (spontaneously or with thrombolysis) may cause reperfusion injury (depending on severity and duration of the preceding ischemia), a dominant cause of which seems to be disruption of the blood-brain barrier (BBB). This leads to extravasation of plasma and erythrocytes, i.e. to formation of edema and of hemorrhage, respectively, and to inflammatory cell infiltration, as well. Moreover, tPA-treated patients with acute myocardial infarction have had a 1.1% Citation[3] and with pulmonary embolism a 3% Citation[4] risk for cerebral hemorrhage, so adding to the overall burden of this often fatal condition.
In contrast to ischemic stroke, no widely approved clinical therapy Citation[5], Citation[6] exists for intracerebral hemorrhage (ICH), which is associated with poor outcome with only 40%–50% of the patients surviving through the first year Citation[7], Citation[8], and many of them with chronic disability. The dismal outcome derives from early brain damage involving direct tissue damage, evolving mass effect of hematoma and edema, and also from delayed brain damage. The latter is mediated by toxins associated with blood break-down products, thrombin, inflammation Citation[9–11], excitotoxicity Citation[12], and disruption of the BBB Citation[13].
Key messages
Mast cell mediators are capable of disrupting the blood-brain barrier.
Mast cells aggravate brain edema and inflammatory cell infiltration in ischemic stroke.
Mast cells increase brain damage in hypoxic-ischemic brain damage.
Mast cells play a role in the pathophysiology of hemorrhage formation following thrombolysis of acute ischemic stroke, as well as of spontaneous intracerebral hemorrhage.
Hypoxic-ischemic brain damage is associated with 20%–50% mortality of asphyxiated new-borns during the perinatal period, and up to 25% of the survivors suffer from permanent disability Citation[14].
Here, we review the recent experimental evidence of mast cell involvement in the above-described pathologies of cerebral ischemia and hemorrhage.
Mast cells—basic facts
Mast cells (MCs) are effector cells of inflammation and immunity Citation[15], Citation[16]. They contain granules that store preformed mediators, which are released upon activation Citation[17]. Derived from pluripotential stem cells of the bone-marrow Citation[18], Citation[19], MCs enter the circulation as committed immature precursor cells Citation[20]. Thereafter, they home in to specific tissues, where they undergo differentiation under the specific microenvironmental conditions of the homing tissue Citation[18], Citation[21], MC differentiation being principally stem-cell-factor-dependent Citation[22]. In the rodent brain, however, other factors are likely to be necessary for MC differentiation and survival including interleukin (IL)-3 and nerve growth factor Citation[23–25]. The latter was found to be synthesized and stored by MCs themselves, suggesting an effective autoregulatory mechanism Citation[26].
Abbreviations
Since MC differentiation occurs within the homing tissue under local microenvironmental conditions, MCs have a noteworthy heterogeneity allowing them to express different functions in health and disease Citation[18], Citation[19]. Based on the preformed mediator content, two types of mature MCs are recognized in rodents: serosal and connective tissue-type MCs in the pleura, the peritoneum, and the skin, and mucosal MCs in the nasal and gastrointestinal mucosa Citation[27]. It is not yet known whether the brain MCs represent a third phenotype Citation[27–30]. However, the various subpopulations of brain MCs—dural, perivascular, and parenchymal—may represent different phenotypes which express, to a variable degree, some features of either main phenotype Citation[31–34]. Some differences exist in the mediator palette between rodents and humans ().
Table I. Main differences in the preformed mediators between rodents and humans.
Mast cells—activation and mediators
Activation of MCs is a response to a wide range of various biological, chemical, and physical stimuli Citation[16], Citation[35], Citation[36]. Such activation leads to the release of a vast array of mediators. Observation of pH-dependent activation of MCs is relevant for conditions of local lactate acidosis after cerebral ischemia Citation[16]. Mediator release happens in two phases with the first fast-response phase involving release of preformed mediators stored in the granules of MCs and typically including histamine, serotonin, heparin, chymase, tryptase, and cathepsin G. Other types of mediators such as prostaglandins, leukotrienes, thromboxanes, and cytokines are synthesized de novo and secreted by the activated MCs. Excellent reviews on this topic have been published recently Citation[16], Citation[22], Citation[37]. Here, we only discuss possible roles of individual mediators in the context of the pathophysiology of brain hemorrhage and ischemia.
It is important to note that MCs react to activation not only stereotypically with the explosive (anaphylactic) degranulation leading to acute release of mediators into the extracellular space, but also in a more controlled fashion resulting in differential release of mediators, i.e. the so-called piecemeal degranulation Citation[37–40]. This allows stimulus-specific release of individual mediators, which may also vary among the different populations of MCs Citation[35], Citation[36]. However, whether or not piecemeal degranulation of mast cell products exists is still under debate. Nevertheless, different mast cell triggers, e.g. IL-1, IL-33, CD30, lipopolysaccharide, can induce a specific set of mediators, independent of exocytosis Citation[37].
Mast cell location and abundance in the brain
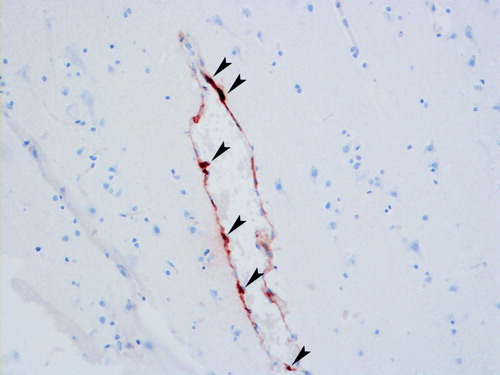
The presence of MCs within the nervous system was noticed more than 100 years ago Citation[41], and since then the presence of MCs in the vertebrate brain has been studied in various experimental settings Citation[18], Citation[27], Citation[28], Citation[36], Citation[42]. MCs are found in the rat pia mater already in late fetal life, and they also enter the central nervous system (CNS) postnatally during days 7–8 Citation[43]. MC progenitors enter the CNS during development via penetrating blood vessels, with which they remain associated () Citation[31], Citation[40], Citation[43]. The known association of MCs with the vascular bed (preferentially at branching points) during development is dependent on contact of the blood vessel with astroglial processes Citation[44]. In the mammalian brain, MCs are typically found in the dura mater, leptomeninges, choroid plexus, thalamus, hypothalamus, olfactory bulb, and the mesencephalon Citation[27], Citation[28], Citation[32], Citation[33], Citation[42], Citation[45]. They are also present in the cerebral cortex Citation[27], Citation[28], Citation[46], often at the branching points of cortical penetrating arterioles, localized deep in the basal lamina nesting between glial processes Citation[47].
Figure 1. Tryptase-stained section of human brain showing mast cells (arrow-heads) in typical perivascular position.
Role of mast cells in brain ischemia and hemorrhage—cell cultures, animal models, and potential clinical applications
The first evidence suggesting MC involvement in the pathophysiology of ischemic stroke came from our laboratory Citation[48], Citation[49], and later also from other laboratories Citation[50]. To pharmacologically activate or stabilize MCs in the rats, we systematically used compound 48/80 and sodium cromoglycate, respectively.
Cell cultures with mast cells
In an in vitro model of ischemia involving oxygen-glucose deprivation, activation of MCs could be induced Citation[51]. Acute ischemic stroke in selected patients can currently be managed with tPA thrombolysis, this mode of pharmaceutical treatment being the only currently approved mode of therapy. Thus, it is of importance to note that tPA can activate MCs in vitro in a dose-dependent manner Citation[52]. When co-cultured with hippocampal neurons, activated MCs potentiated glutamate excitotoxicity, presumably by histamine release, suggesting a role for MCs under conditions of cerebral ischemia with enhanced glutamatergic neurotransmission Citation[53].
Cerebral palsy and cerebral ischemia in neonates
In a retrospective study of children with cerebral palsy, the investigators detected higher levels of several perinatal circulating cytokines, including IL-9 Citation[54]. In addition, in a new-born mouse model of cerebral palsy, IL-9 pretreatment of the mice exacerbated brain damage Citation[55], an effect which was later shown to be MC-dependent Citation[56]. IL-9, in turn, has been shown to be a growth factor of human mast cells Citation[57]. Histological changes in cerebral palsy include ischemic-like and hypoxic-ischemic cortical damage Citation[58], Citation[59]. In the immature rat, hypoxic-ischemic brain damage led to a rapid increase of the cerebral population of MCs and their activation, the activated MCs being present in the pia, parenchyma, and, importantly, in the regions showing neuronal loss Citation[60]. Importantly, MC stabilization reduced MC count in the latter region and limited brain damage more than 50% compared with controls. In this study, it was noted that only hypoxia-ischemia—but not hypoxia alone—led to the brain damage, confirming stimulus-specific reactions of MCs. A similar study by a different group Citation[61] showed contribution of MC-derived histamine to ischemia-induced neuronal death in the postnatal day 7 new-born rats. Observation of ipsilaterally upregulated expression of MC-associated genes after hypoxic-ischemic brain damage Citation[62] provides another body of evidence supporting the involvement of MCs in this particular condition.
Focal transient cerebral ischemia—brain edema, hemorrhage formation, and inflammation
Vasogenic brain edema, hemorrhage formation, and inflammatory response are all associated with disruption of the BBB. Before we address the possible interactions between MCs and the BBB at the molecular level, we provide the available experimental data showing MC involvement in these various brain ischemia-dependent processes.
Brain edema is a major determinant of acute stroke mortality in man Citation[1]. In the rat, we showed MC involvement in ischemic brain edema early after focal cerebral ischemia onset Citation[63]. Specifically, we found almost 90% increase after treatment with the MC-degranulating agent compound 48/80, and almost 40% reduction of ischemic brain edema after treatment with the MC-stabilizing agent sodium cromoglycate. The role of MCs as a major determinant of brain edema in this animal model was confirmed in genetically MC-deficient rats, which had 60% less brain edema compared with their wild-type MC-competent litter-mates Citation[63]. Moreover, a role for histamine in the development of brain edema has been observed in a rat model of bilateral common carotid artery occlusion, which represents global rather than focal cerebral ischemia Citation[64], Citation[65]. Since histamine is the major MC mediator during acute stimulation of MCs, the above finding implies MC involvement also in this scenario.
Thrombolysis-mediated hemorrhage formation can devastate clinical prognosis following successful stroke thrombolysis and may occur even in patients with acute myocardial infarction or with pulmonary embolism, i.e. in patients treated with thrombolytic agents without any on-going cerebral ischemia Citation[3], Citation[4]. We reported a robust tPA-mediated activation of MCs in vitroCitation[52], a finding not reported for any other fibrinolytic drug so far. Importantly, in in vivo experiments in rats, which underwent focal cerebral ischemia-reperfusion and postischemic tPA administration, we found a 70- to 100-fold increase in hemorrhage formation. Pharmacological MC stabilization with cromoglycate led to significant reduction in the tPA-mediated hemorrhage formation at 3 (97%), 6 (76%), and 24 hours (96%) compared with controls. Moreover, MC-deficient rats showed robust reduction of tPA-mediated hemorrhage compared with their wild-type MC-competent litter-mates. Importantly, these reductions in hemorrhage formation translated into better neurological outcomes and reduced the mortality. Specifically, in the control group 30% and in the tPA-alone-treated group 65% of animals died, while no deaths occurred in the tPA-treated group if the animals had been treated with the MC-stabilizing drug sodium cromoglycate. Again, a similar beneficial effect was observed in the MC-deficient rats.
MCs contribute to CNS inflammation Citation[36], Citation[66], Citation[67], particularly by recruiting leukocytes to sites of vascular inflammation Citation[68–70]. Recently, we reported MCs to play a significant role in neutrophil infiltration following experimental cerebral ischemia-reperfusion injury in the rat Citation[63]. Thus, MC stabilization reduced neutrophil infiltration by 40%, and the reduction was even stronger (almost 50%) in MC-deficient rats, while MC degranulation enhanced neutrophil infiltration. In a different study Citation[52], an intravenous administration of tPA was accompanied by a 2- to 3-fold increase in neutrophil infiltration in ischemic hemispheres and by an up to 6-fold increase in the intact hemisphere, suggesting an independent proinflammatory effect of tPA. Moreover, the numbers of brain tissue neutrophils in MC-deficient rats subjected to focal cerebral ischemia and subsequent tPA treatment were less than 40% of those in their wild-type litter-mates.
Blood-brain barrier as the main target of mast cells
Disruption of the BBB in the above-mentioned scenarios is a consequence of ischemia-reperfusion injury. Outside the CNS, MCs have been shown to be involved in the pathophysiology of elements of ischemia-reperfusion injury in the myocardium Citation[71–75], kidney Citation[76], intestine Citation[77–81], skeletal muscle Citation[82–84], and the lungs Citation[83], Citation[85], Citation[86].
Differentiation or migration of intraparenchymal MCs in the developing brain occurs during the formation of the BBB Citation[87], and the time-course of MC migration from the choroid fissure to the thalamus coincides with the formation of the vascular network Citation[31]. Since MCs not only show characteristic perivascular location in the brain, but many of their mediators are profoundly vasoactive Citation[28], Citation[31], Citation[32], Citation[34], it is reasonable to hypothesize that MCs play a role in the regulation of the BBB. MCs were shown to be involved in the regulation of the BBB in healthy doves Citation[88], and they also regulated the BBB under conditions of acute stress challenge in the rat Citation[89]. In that latter study, the authors noticed that a relatively limited population of MCs may be sufficient to mount an effective alteration of the BBB permeability. Furthermore, MC-mediated BBB disruption has been documented to precede the onset of clinical symptoms in multiple sclerosis Citation[90], Citation[91].
Our group addressed the role of MCs in the BBB disruption following focal cerebral ischemia Citation[63]. In the rat model of focal cerebral ischemia, we observed a 50% increase in the BBB disruption after triggering of MC degranulation, while pharmacological stabilization of MCs reduced it by 50%. The role of MCs in the regulation of the BBB disruption was confirmed by demonstrating 50% less BBB disruption in genetically MC-deficient rats compared with their wild-type MC-competent litter-mates. Similar experiments with additional administration of tPA supported the involvement of MCs in mediating the BBB disruption Citation[52]. Most probably, MCs with their armamentarium of bioactive mediators influence all the described consequences of ischemia-reperfusion injury by targeting the BBB and the basal lamina (), both of which normally secure the integrity of the cerebral microvasculature and the extravascular environment of the CNS.
Figure 2. Schematic illustration of the neurovascular unit (middle) and the components of the blood-brain barrier (bottom left) and the basal lamina (bottom right) (ECM = extracellular matrix; ZO = zona occludens; AF6 = afadin; 7H6 = tight junction associated phosphoprotein).
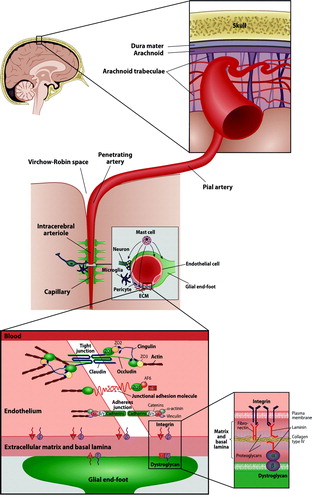
The BBB is composed of tight interendothelial junctions in capillaries and in postcapillary venules (). Furthermore, several integral transmembrane proteins contribute to the BBB integrity (e.g. claudin, occludin, and junctional adhesion molecules). The second barrier—the basal lamina—is a specialized part of the extracellular matrix that connects the endothelial cells to the adjoining cell layers and further to the smooth muscle layer of the media (). The basal lamina loses its integrity very early after the onset of ischemia, as demonstrated by rapid disappearances of the three main constituents of the basal lamina, i.e. laminin, fibronectin, and collagen type IV Citation[92], Citation[93].
Some evidence shows involvement of proteases, e.g. matrix metalloproteinases (MMPs), plasminogen activators, and cathepsins, in the disruption of the matrix of the basal lamina. The MMPs comprise a large family of proteolytic proenzymes, which require activation. Once activated, they can degrade most of the protein constituents of the neurovascular matrix, i.e. collagen, elastin, fibronectin, vitronectin, and gelatine Citation[94]. Involvement of the gelatinases MMP-2 and MMP-9 in the degradation of the components of the basal lamina, with ensuing BBB disruption and development of edema and hemorrhage, has been demonstrated experimentally in several pieces of work Citation[92], Citation[95–100]. Furthermore, MMP-mediated disruption of the tight junction proteins occludin and claudin () was reported in the rat Citation[100], Citation[101]. Finally, clinical data have revealed a positive correlation between the degree of brain edema and the level of MMP-9 in the cerebrospinal fluid Citation[102], suggesting the involvement of this MMP also in human CNS pathology involving BBB damage.
In view of the processes described above, it is of relevance that peripheral MCs are able to migrate across tissues. Thus MCs in the bovine gall-bladder migrated from the adjacent connective tissue across the basal membrane to enter the epithelium and so to reach the luminal surface of the gall-bladder Citation[103]. MCs are also able to enhance the antibody-mediated injury of the skin basement membrane in mice Citation[104]. Moreover, in vitro data show that both resting and activated MCs can attach to and move across a variety of extracellular matrix proteins, including fibronectin and laminin, for which MCs have receptors Citation[105–107]. The involvement of MCs in the proteolytic degradation of endothelial basement membrane with ensuing detachment of the endothelial cell, i.e. endothelial erosions, has been extensively studied in the context of atherosclerosis and its thrombotic complications Citation[108], Citation[109].
How can MCs disrupt the BBB and degrade the components of the basal lamina ()? There are several possible mechanisms involved, in which the vasoactive and the matrix-degrading components of MCs act in concert. MCs produce a potent vasodilatatory mediator, histamine, which contributes up to 90% of the histamine content in the thalamus and up to 50% of that in the whole brain Citation[110]. It is of fundamental importance for the pathogenic mechanisms of acute ischemic brain conditions that, of the resident cells in the brain, only MCs are able to acutely respond by releasing massive amounts of preformed vasodilatatory mediators, such as histamine. Under experimental conditions in vitro, histamine binding to its specific receptors on the endothelial surface leads to increased phosphorylation of adherens junction and loosening of vascular-endothelial-cadherin-mediated adhesion of adjacent endothelial cells Citation[111], Citation[112]. The same effect can be induced by MC-derived protease tryptase Citation[113], a process which can be aggravated by other MC-neutral proteases—chymase, and cathepsin G Citation[114]. Again, all these three MC proteases are acutely released from activated MCs along with histamine. Moreover, cerebral MC-derived chymase Citation[31] is a potent protease which cleaves fibronectin and occludin and activates procollagenases Citation[115–117] even in the presence of the MMP-inhibiting protein TIMP (tissue inhibitor of metalloproteinase)-1 Citation[118]. Indeed, MC-derived chymase can degrade TIMP-1 Citation[119], and a key role for MC chymase in the activation of pro-MMP-2 and pro-MMP-9 was demonstrated recently by Pejler and co-workers Citation[120]. Similarly, MC tryptase is also able to activate MMPs Citation[121] and to degrade TIMP-1 Citation[119]. Moreover, MCs themselves can release gelatinase A (MMP-2) and gelatinase B (MMP-9) Citation[122]. Regarding cathepsin G, this chymotrypsin-like neutral protease present in some MCs is able to cleave many components of the extracellular and pericellular matrix, including fibronectin and vitronectin Citation[109], Citation[123]. The role of cathepsins in microvascular matrix degradation following focal cerebral ischemia has been noticed Citation[124]. Finally, apart from proteases, MCs can release tumor necrosis factor (TNF)-α and IL-1, two cytokines shown to be involved in the modulation of BBB permeability and consequently in ischemic brain edema formation Citation[125–127]. Indeed, MCs do possess a potent armamentarium to target the components of the BBB and basal lamina shortly after their activation, whereas de novo production of mediators reactivates and maintains the process.
The mechanism of postthrombolytic hemorrhage formation still awaits detailed understanding. It is of interest to note that it occurs with any of the fibrinolytic substances in clinical use, i.e. tPA, streptokinase, prourokinase (can be activated by human MC tryptase Citation[128]), and reteplase Citation[129], Citation[130]. They all are potent serine proteinases, which, in addition to their local plasminogen-activating effect to dissolve fibrin locally, also induce systemic plasminemia Citation[131]. Plasmin, again, has an independent proinflammatory activity that causes direct brain tissue damage Citation[132], Citation[133], and in addition it degrades a range of extracellular matrix proteins by activating MMPs Citation[134]. Interestingly, inhibition of MMPs reduced tPA-mediated mortality in experimental ischemia-reperfusion Citation[135]. Perhaps even more important in the setting of thrombolysis of an acute ischemic stroke is the observation that the development of hemorrhage may be further promoted by heparin, also acutely released by activated MCs, which are the only source of this strong anticoagulant Citation[136], Citation[137]. Heparin released locally from perivascularly positioned MCs may prevent the formation of hemostatic plugs, which otherwise would patch any BBB breaches. Thus, the endogenously released heparin may aggravate erythrocyte extravasation and contribute to hemorrhage formation. In dogs bearing MC tumors, intense activation of the fibrinolytic system has been observed after injection of minute amounts the MC-degranulating agent 48/80 Citation[138]. Superimposed on the fact that (in addition to heparin) tPA itself belongs to the palette of MC-derived mediators Citation[139], this confirms the antithrombotic and fibrinolytic potential of MCs.
Infiltration of neutrophilic granulocytes is held to compromise microcirculation and BBB integrity and to contribute to the release of free radicals. One of the most potent mediators possibly involved in the recruitment of granulocytes is TNF-α, which represents a distinct type of MC mediators, since after MC stimulation it is both acutely released from preformed pools stored in the cytoplasmic secretory granules, and then continuously synthesized de novo and secreted Citation[22]. Being probably the only cell type containing preformed stores of TNF-α Citation[140], MCs are likely to represent a critical initial cellular source of TNF-α during inflammation Citation[16]. Later during the inflammatory response, TNF-α is also produced by neutrophils, eosinophils, T cells and B cells, and macrophages Citation[141]. The role of MC-derived TNF-α in inflammatory cell recruitment was reported in various non-cerebral tissues Citation[141–143]. However, as much as 50% of the recruitment of neutrophils during MC-dependent cutaneous late phase reactions in the mouse was independent of MC-derived TNF-α, suggesting that other MC-associated mediators also contributed to this response Citation[142]. Indeed, MCs are a source of other mediators that could also promote leukocytic infiltration, including IL-1, members of the macrophage inflammatory protein-1 family Citation[141], Citation[144], Citation[145], and other potent chemotactic substances, such as platelet-activating factor, IL-4, IL-5, IL-8, and neutrophil, eosinophil, and macrophage chemotactic factors. Together, these chemoattractants are likely to contribute to the early steep chemotactic gradient, after which neutrophils start to transmigrate. Since MC stabilization reduced both the number of transmigrated neutrophils and of those still adherent to the endothelial wall Citation[63], MC blocking not only reduced passive neutrophil trafficking through BBB breaches, but probably also attenuated the perivascular chemotactic gradient up which the neutrophils migrate. The proposed proinflammatory effect of tPA might be mediated not only by the described MC activation and the released of MC-derived mediators, but also directly by plasmin, which is able to induce the synthesis of platelet-activating factor, activate the terminal complement cascade Citation[146], and to recruit neutrophils when injected into the brain Citation[133].
The fact that clinical trials based on inhibition of neutrophils have failed to have therapeutic success Citation[147], Citation[148] may be explained by the neutrophils arriving at the scene too late to be able to influence the early damage Citation[149]. In contrast, the resident MCs are already present in the brain tissue at the onset of ischemia, and may thus offer an alternative target for early antichemotactic intervention before the involvement of any circulating inflammatory cells.
Brain hemorrhage
MC involvement in the pathophysiology of ICH was shown in a rat model in our laboratory Citation[150]. In that study, MC inhibition led to significant reductions in the growth of ICH volume during a 24-hour follow-up. Furthermore, depending on the route of administration of the MC-stabilizing agent sodium cromoglycate, brain edema was reduced by 50%–75%. MC degranulation, again, led to opposite effects. The specific role for MCs in this setting was supported by similar findings in the genetically MC-deficient rats. Importantly, the degree of brain edema corresponded with mortality of the animals. Thus, all rats treated with the MC-stabilizing agent survived and showed good outcome, whereas those treated with saline or with the MC-degranulating agent had mortality rates of 45% and 55%, respectively. This striking result could be observed again, when we repeated the experiment using the genetically MC-deficient rats: all MC-deficient rats stayed alive, while 25% of their wild-type litter-mates died.
The pathophysiological basis of MC-dependent brain edema during ICH is uncertain. We propose that the release of MC-derived vasoactive substances (histamine) and proteolytic enzymes (tryptase, chymase, and cathepsin G) increases vascular permeability and disrupts the basal lamina of the vasculature along with the surrounding extracellular tissue matrix, leading to secondary aggravation of extravasated blood cells and further extravascular spreading of serum proteins. Furthermore, mast cell heparin markedly accelerates the generation of another potent vasoactive substance, bradykinin Citation[151]. Moreover, several MC-mediated cytokines (e.g. TNF-α and various ILs) are likely to be involved in this scenario. Indeed, some human studies show a correlation between plasma concentrations of TNF-α and IL-6 and magnitude of perihematomal brain edema and hematoma growth Citation[152], Citation[153]. Although the cellular sources of these cytokines was not known in the human studies, hematoma growth has been attributed not only to continued bleeding from the primary source Citation[154] but also to a mechanical disruption of the surrounding vessels in the peripheral zone around the ICH Citation[154], Citation[155], which is a possible stimulus for enhanced local MC activation. As discussed above, upon activation MCs acutely release heparin, an endogenous anticoagulant, which may promote impaired hemostasis and prolonged extravasation of erythrocytes and the occurrence of secondary hemorrhage. This hypothesis is in line with our recent observations in a rat intracerebral hemorrhage model (injection of autologous blood into the basal ganglia), in which the significant hematoma growth observed in saline- and compound 48/80-treated groups could be significantly inhibited by pharmacological stabilization of MCs or by using MC-deficient rats in the experiment Citation[150].
Conclusions
To sum up the current evidence about the emerging role of MCs in cerebral ischemia and hemorrhage, it has become evident only in recent experimental studies that MCs, which represent a stationary, resident cell population in the mammalian brain and meninges, participate in several clinically important sequelae of the progression of ischemic and hemorrhagic stroke, namely in BBB damage, brain edema, hemorrhage formation, and in the inflammatory response to such events. The observed effects are well in line with the biphasic phlogistic responses of MCs described in type-I hypersensitivity reactions and the ensuing late phase reactions. MCs reside in the pivotal perivascular position and contain an abundance of vasoactive, proinflammatory, anticoagulant, and proteolytic mediators released immediately after activation, and many of these mediators are released also during the late phase response. This provides MCs with a multitude of strategic opportunities to interact at the level of the neurovascular unit. Hence, MC stabilization may represent an efficient strategy by blocking simultaneously several molecular cascades that participate in the progressive tissue damage associated with ischemic and hemorrhagic stroke. This contrasts with the prevailing approach of blocking just one single pathway, which has repeatedly failed in clinical trials employing neuroprotective agents in ischemic stroke.
In addition to a role in ischemic stroke, MCs seem to participate also in hemorrhagic cerebral events due to enhancement of vascular injury and aggravated bleeding. Indeed, all described antihemostatic effects of MCs superimposed on their vasculopathic proteolytic potential afford to MCs a fibrinolytic capacity in the microvascular milieu. Then, the activated MCs clearly have the potential to exert unwanted side-effects as an acute response to therapeutic administration of thrombolytics after ischemic stroke. Stabilization of MCs, again, is a potential novel adjuvant therapy to prevent these occasionally devastating complications of thrombolysis. This bold proposal is mainly based on the experimental data in rats, which can differ from the human situation. This issue clearly requires further studies to establish its potential role as an adjunct mode of the acute therapy of cerebral ischemia and hemorrhage in man. Fortunately, MC-stabilizing pharmacological agents are safe in clinical use, and so we are ready to explore novel indications for this old drug, such as acute ischemic stroke and intracerebral hemorrhage.
Acknowledgements
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Ayata C, Ropper A. Ischaemic brain oedema. J Clin Neurosci. 2002; 9: 113–24
- Hacke W, Kaste M, Bluhmki E, Brozman M, Davalos A, Guidetti D, et al. Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N Engl J Med. 2008; 359: 1317–29
- Grines CL, Serruys P, O'Neill WW. Fibrinolytic therapy: is it a treatment of the past?. Circulation. 2003; 107: 2538–42
- Han S, Chaya C, Hoo GW. Thrombolytic therapy for massive pulmonary embolism in a patient with a known intracranial tumor. J Intensive Care Med. 2006; 21: 240–5
- Mayer SA, Brun NC, Begtrup K, Broderick J, Davis S, Diringer MN, et al. Efficacy and safety of recombinant activated factor VII for acute intracerebral hemorrhage. N Engl J Med. 2008; 358: 2127–37
- Mendelow AD, Gregson BA, Fernandes HM, Murray GD, Teasdale GM, Hope DT, et al. Early surgery versus initial conservative treatment in patients with spontaneous supratentorial intracerebral haematomas in the International Surgical Trial in Intracerebral Haemorrhage (STICH): a randomised trial. Lancet. 2005; 365: 387–97
- Nilsson OG, Lindgren A, Brandt L, Saveland H. Prediction of death in patients with primary intracerebral hemorrhage: a prospective study of a defined population. J Neurosurg. 2002; 97: 531–6
- Vermeer SE, Algra A, Franke CL, Koudstaal PJ, Rinkel GJ. Long-term prognosis after recovery from primary intracerebral hemorrhage. Neurology. 2002; 59: 205–9
- Lee KR, Colon GP, Betz AL, Keep RF, Kim S, Hoff JT. Edema from intracerebral hemorrhage: the role of thrombin. J Neurosurg. 1996; 84: 91–6
- Lee KR, Betz AL, Kim S, Keep RF, Hoff JT. The role of the coagulation cascade in brain edema formation after intracerebral hemorrhage. Acta Neurochir. 1996; 138: 396–401
- Xi GH, Hua Y, Bhasin RR, Ennis SR, Keep RF, Hoff JT. Mechanisms of edema formation after intracerebral hemorrhage: Effects of extravasated red blood cells on blood flow and blood-brain barrier integrity. Stroke. 2001; 32: 2932–8
- Qureshi AI, Ali Z, Suri MF, Shuaib A, Baker G, Todd K, et al. Extracellular glutamate and other amino acids in experimental intracerebral hemorrhage: an in vivo microdialysis study. Crit Care Med. 2003; 31: 1482–9
- Power C, Henry S, del Bigio MR, Larsen PH, Corbett D, Imai Y, et al. Intracerebral hemorrhage induces macrophage activation and matrix metalloproteinases. Ann Neurol. 2003; 53: 731–42
- Vannucci RC. Hypoxic-ischemic encephalopathy. Am J Perinatol. 2000; 17: 113–20
- Galli SJ. Mast cells and basophils. Curr Opin Hematol. 2000; 7: 32–9
- Costa JJ, Weller PF, Galli SJ. The cells of the allergic response: mast cells, basophils, and eosinophils. JAMA. 1997; 278: 1815–22
- Dvorak AM. Biochemical contents of granules and lipid bodies – two distinctive organelles found in basophils and mast cells. Basophil and mast cell degranulation and recovery, JR Harris. Plenum Press, New York 1991; 4: 27–65
- Galli SJ. New insights into ‘the riddle of the mast cells’: microenvironmental regulation of mast cell development and phenotypic heterogeneity. Lab Invest. 1990; 62: 5–33
- Kitamura Y. Heterogeneity of mast cells and phenotypic change between subpopulations. Annu Rev Immunol. 1989; 7: 59–76
- Rodewald HR, Dessing M, Dvorak AM, Galli SJ. Identification of a committed precursor for the mast cell lineage. Science. 1996; 271: 818–22
- Kitamura Y, Go S, Hatanaka K. Decrease of mast-cells in W/Wv mice and their increase by bone-marrow transplantation. Blood. 1978; 52: 447–52
- Galli SJ. New concepts about the mast cell. N Engl J Med. 1993; 328: 257–65
- Mekori YA, Oh CK, Metcalfe DD. IL-3-dependent murine mast cells undergo apoptosis on removal of IL-3. Prevention of apoptosis by c-kit ligand. J Immunol. 1993; 151: 3775–84
- Horigome K, Bullock ED, Johnson EM, Jr. Effects of nerve growth factor on rat peritoneal mast cells. Survival promotion and immediate-early gene induction. J Biol Chem. 1994; 269: 2695–702
- Shanas U, Bhasin R, Sutherland AK, Silverman AJ, Silver R. Brain mast cells lack the c-kit receptor: immunocytochemical evidence. J Neuroimmunol. 1998; 90: 207–11
- Leon A, Buriani A, Dal Toso R, Fabris M, Romanello S, Aloe L, et al. Mast cells synthesize, store, and release nerve growth factor. Proc Natl Acad Sci U S A. 1994; 91: 3739–43
- Theoharides TC. Mast cells: the immune gate to the brain. Life Sci. 1990; 46: 607–17
- Silver R, Silverman AJ, Vitkovic L, Lederhendler II. Mast cells in the brain: evidence and functional significance. Trends Neurosci. 1996; 19: 25–31
- Pang X, Letourneau R, Rozniecki JJ, Wang L, Theoharides TC. Definitive characterization of rat hypothalamic mast cells. Neuroscience. 1996; 73: 889–902
- Cocchiara R, Bongiovanni A, Albeggiani G, Azzolina A, Geraci D. Evidence that brain mast cells can modulate neuroinflammatory responses by tumour necrosis factor-alpha production. Neuroreport. 1998; 9: 95–8
- Dimitriadou V, Lambracht-Hall M, Reichler J, Theoharides TC. Histochemical and ultrastructural characteristics of rat brain perivascular mast cells stimulated with compound 48/80 and carbachol. Neuroscience. 1990; 39: 209–24
- Goldschmidt RC, Hough LB, Glick SD, Padawer J. Mast cells in rat thalamus: nuclear localization, sex difference and left-right asymmetry. Brain Res. 1984; 323: 209–17
- Dropp, JJ. Mast cells in mammalian brain. Acta Anat. 1976:1–21.
- Ibrahim MZ. The mast cells of the mammalian central nervous system. Part I. Morphology, distribution and histochemistry. J Neurol Sci. 1974; 21: 431–78
- Holgate ST, Robinson C, Church MK. Mediators of immediate hypersensitivity. Allergy: Principles and practice, E Middleton, Jr, CE Reed, EF Ellis, NF Adkinson, Jr, JW Yuninger. Mosby, St Louis 1988; 135–63
- Johnson D, Krenger W. Interactions of mast cells with the nervous system—recent advances. Neurochem Res. 1992; 9: 939–51
- Theoharides TC, Kempuraj D, Tagen M, Conti P, Kalogeromitros D. Differential release of mast cell mediators and the pathogenesis of inflammation. Immunol Rev. 2007; 217: 65–78
- Theoharides TC, Bondy PK, Tsakalos ND, Askenase PW. Differential release of serotonin and histamine from mast cells. Nature. 1982; 297: 229–31
- Dvorak AM, McLeod RS, Onderdonk A, Monahan-Earley RA, Cullen JB, Antonioli DA, et al. Ultrastructural evidence for piecemeal and anaphylactic degranulation of human gut mucosal mast cells in vivo. Int Arch Allergy Immunol. 1992; 99: 74–83
- Letourneau R, Rozniecki JJ, Dimitriadou V, Theoharides TC. Ultrastructural evidence of brain mast cell activation without degranulation in monkey experimental allergic encephalomyelitis. J Neuroimmunol. 2003; 145: 18–26
- Neuman J. Ueber das Vorkommen der sogenannten ‘mastzellen’ bei pathologischen Veraenderungen des Gehirns. Arch Pathol Anat Physiol Virchows. 1890; 122: 378
- Manning KA, Pienkowski TP, Uhlrich DJ. Histaminergic and non-histamine-immunoreactive mast cells within the cat lateral geniculate complex examined with light and electron microscopy. Neuroscience. 1994; 63: 191–206
- Lambracht-Hall M, Dimitriadou V, Theoharides TC. Migration of mast cells in the developing rat brain. Brain Res Dev Brain Res. 1990; 56: 151–9
- Khalil M, Ronda J, Weintraub M, Jain K, Silver R, Silverman AJ. Brain mast cell relationship to neurovasculature during development. Brain Res. 2007; 1171: 18–29
- Florenzano F, Bentivoglio M. Degranulation, density, and distribution of mast cells in the rat thalamus: a light and electron microscopic study in basal conditions and after intracerebroventricular administration of nerve growth factor. J Comp Neurol. 2000; 424: 651–69
- Hough L. Cellular localization and possible functions for brain histamine: recent progress. Prog Neurobiol. 1988; 30: 469–505
- Silverman AJ, Sutherland AK, Wilhelm M, Silver R. Mast cells migrate from blood to brain. J Neurosci. 2000; 20: 401–8
- Karjalainen-Lindsberg, M-L, Tatlisumak, T, Lindsberg, PJ. Mast cells in ischemic rat brain. Society for Neuroscience, Poster presentation, San Diego. 2001:Abstract No:330.11.
- Strbian D, Tatlisumak T, Karjalainen-Lindsberg M-L, Lindsberg PJ. Mast cells regulate ischemic brain edema (abstract). J Cereb Blood Flow Metab. 2003; 23(Suppl 1)166
- Hu W, Xu L, Pan J, Zheng X, Chen Z. Effect of cerebral ischemia on brain mast cells in rats. Brain Res. 2004; 1019: 275–80
- Hu W, Shen Y, Fu Q, Dai H, Tu H, Wei E, et al. Effect of oxygen-glucose deprivation on degranulation and histamine release of mast cells. Cell Tissue Res. 2005; 322: 437–41
- Strbian D, Karjalainen-Lindsberg ML, Kovanen PT, Tatlisumak T, Lindsberg PJ. Mast cell stabilization reduces hemorrhage formation and mortality after administration of thrombolytics in experimental ischemic stroke. Circulation. 2007; 116: 411–8
- Skaper SD, Facci L, Kee WJ, Strijbos PJ. Potentiation by histamine of synaptically mediated excitotoxicity in cultured hippocampal neurones: a possible role for mast cells. J Neurochem. 2001; 76: 47–55
- Nelson KB, Dambrosia JM, Grether JK, Phillips TM. Neonatal cytokines and coagulation factors in children with cerebral palsy. Ann Neurol. 1998; 44: 665–75
- Dommergues MA, Patkai J, Renauld JC, Evrard P, Gressens P. Proinflammatory cytokines and interleukin-9 exacerbate excitotoxic lesions of the newborn murine neopallium. Ann Neurol. 2000; 47: 54–63
- Patkai J, Mesples B, Dommergues MA, Fromont G, Thornton EM, Renauld JC, et al. Deleterious effects of IL-9-activated mast cells and neuroprotection by antihistamine drugs in the developing mouse brain. Pediatr Res. 2001; 50: 222–30
- Matsuzawa S, Sakashita K, Kinoshita T, Ito S, Yamashita T, Koike K. IL-9 enhances the growth of human mast cell progenitors under stimulation with stem cell factor. J Immunol. 2003; 170: 3461–7
- Gressens P, Marret S, Hill JM, Brenneman DE, Gozes I, Fridkin M, et al. Vasoactive intestinal peptide prevents excitotoxic cell death in the murine developing brain. J Clin Invest. 1997; 100: 390–7
- Marret S, Mukendi R, Gadisseux JF, Gressens P, Evrard P. Effect of ibotenate on brain development: an excitotoxic mouse model of microgyria and posthypoxic-like lesions. J Neuropathol Exp Neurol. 1995; 54: 358–70
- Jin Y, Silverman AJ, Vannucci SJ. Mast cell stabilization limits hypoxic-ischemic brain damage in the immature rat. Dev Neurosci. 2007; 29: 373–84
- Biran V, Cochois V, Karroubi A, Arrang JM, Charriaut-Marlangue C, Heron A. Stroke induces histamine accumulation and mast cell degranulation in the neonatal rat brain. Brain Pathol. 2008; 18: 1–9
- Hedtjarn M, Mallard C, Hagberg H. Inflammatory gene profiling in the developing mouse brain after hypoxia-ischemia. J Cereb Blood Flow Metab. 2004; 24: 1333–51
- Strbian D, Karjalainen-Lindsberg ML, Tatlisumak T, Lindsberg PJ. Cerebral mast cells regulate early ischemic brain swelling and neutrophil accumulation. J Cereb Blood Flow Metab. 2006; 26: 605–12
- Joo F, Tosaki A, Olah Z, Koltai M. Inhibition by H-7 of the protein kinase C prevents formation of brain edema in Sprague-Dawley CFY rats. Brain Res. 1989; 490: 141–3
- Tosaki A, Szerdahelyi P, Joo F. Treatment with ranitidine of ischemic brain edema. Eur J Pharmacol. 1994; 264: 455–8
- Hickey WF. Migration of hematogenous cells through the blood-brain barrier and the initiation of CNS inflammation. Brain Pathol. 1991; 1: 97–105
- Theoharides TC, Dimitriadou V, Letourneau R, Rozniecki JJ, Vliagoftis H, Boucher W. Synergistic action of estradiol and myelin basic protein on mast cell secretion and brain myelin changes resembling early stages of demyelination. Neuroscience. 1993; 57: 861–71
- Gaboury JP, Johnston B, Niu XF, Kubes P. Mechanisms underlying acute mast cell-induced leukocyte rolling and adhesion in vivo. J Immunol. 1995; 154: 804–13
- Kubes P, Gaboury JP. Rapid mast cell activation causes leukocyte-dependent and -independent permeability alterations. Am J Physiol. 1996; 271: H2438–46
- Kubes P, Granger DN. Leukocyte-endothelial cell interactions evoked by mast cells. Cardiovasc Res. 1996; 32: 699–708
- Gilles S, Zahler S, Welsch U, Sommerhoff CP, Becker BF. Release of TNF-alpha during myocardial reperfusion depends on oxidative stress and is prevented by mast cell stabilizers. Cardiovasc Res. 2003; 60: 608–16
- Singh M, Saini HK. Resident cardiac mast cells and ischemia-reperfusion injury. J Cardiovasc Pharmacol Ther. 2003; 8: 135–48
- Frangogiannis NG, Lindsey ML, Michael LH, Youker KA, Bressler RB, Mendoza LH, et al. Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation. 1998; 98: 699–710
- Horwitz LD, Kaufman D, Kong Y. An antibody to leukocyte integrins attenuates coronary vascular injury due to ischemia and reperfusion in dogs. Am J Physiol. 1997; 272: H618–24
- Lefer DJ, Flynn DM, Buda AJ. Effects of a monoclonal antibody directed against P-selectin after myocardial ischemia and reperfusion. Am J Physiol. 1996; 270: H88–98
- Morikawa T, Imanishi M, Suzuki H, Okada N, Okumura M, Konishi Y, et al. Mast cell chymase in the ischemic kidney of severe unilateral renovascular hypertension. Am J Kidney Dis 2005; 45: e45–50
- Kalia N, Brown NJ, Wood RF, Pockley AG. Ketotifen abrogates local and systemic consequences of rat intestinal ischemia-reperfusion injury. J Gastroenterol Hepatol. 2005; 20: 1032–8
- Szalay L, Kaszaki J, Nagy S, Boros M. Endothelin-1 induces mucosal mast cell degranulation in the rat small intestine. Life Sci. 2000; 67: 1947–58
- Boros M, Kaszaki J, Ordogh B, Nagy S. Mast cell degranulation prior to ischemia decreases ischemia-reperfusion injury in the canine small intestine. Inflamm Res. 1999; 48: 193–8
- Kanwar S, Kubes P. Ischemia/reperfusion-induced granulocyte influx is a multistep process mediated by mast cells. Microcirculation. 1994; 1: 175–82
- Kurose I, Anderson DC, Miyasaka M, Tamatani T, Paulson JC, Todd RF, et al. Molecular determinants of reperfusion-induced leukocyte adhesion and vascular protein leakage. Circ Res. 1994; 74: 336–43
- Abonia JP, Friend DS, Austen WG, Jr, Moore FD, Jr, Carroll MC, Chan R, et al. Mast cell protease 5 mediates ischemia-reperfusion injury of mouse skeletal muscle. J Immunol. 2005; 174: 7285–91
- Mukundan C, Gurish MF, Austen KF, Hechtman HB, Friend DS. Mast cell mediation of muscle and pulmonary injury following hindlimb ischemia-reperfusion. J Histochem Cytochem. 2001; 49: 1055–6
- Korthuis RJ, Carden DL, Kvietys PR, Shepro D, Fuseler J. Phalloidin attenuates postischemic neutrophil infiltration and increased microvascular permeability. J Appl Physiol. 1991; 71: 1261–9
- Vural KM, Liao H, Oz MC, Pinsky DJ. Effects of mast cell membrane stabilizing agents in a rat lung ischemia-reperfusion model. Ann Thorac Surg. 2000; 69: 228–32
- Goldman G, Welbourn R, Klausner JM, Kobzik L, Valeri CR, Shepro D, et al. Mast cells and leukotrienes mediate neutrophil sequestration and lung edema after remote ischemia in rodents. Surgery. 1992; 112: 578–86
- Dimitriadou V, Rouleau A, Tuong MD, Ligneau X, Newlands GF, Miller HR, et al. Rat cerebral mast cells undergo phenotypic changes during development. Brain Res Dev Brain Res. 1996; 97: 29–41
- Zhuang X, Silverman AJ, Silver R. Brain mast cell degranulation regulates blood-brain barrier. J Neurobiol. 1996; 31: 393–403
- Esposito P, Gheorghe D, Kandere K, Pang X, Connoly R, Jacobson S, et al. Acute stress increases permeability of the blood-brain barrier through activation of brain mast cells. Brain Res. 2001; 888: 117–27
- Goodin DS, Ebers GC, Johnson KP, Rodriguez M, Sibley WA, Wolinsky JS. The relationship of MS to physical trauma and psychological stress: report of the Therapeutics and Technology Assessment Subcommittee of the American Academy of Neurology. Neurology. 1999; 52: 1737–45
- Kermode AG, Thompson AJ, Tofts P, MacManus DG, Kendall BE, Kingsley DP, et al. Breakdown of the blood-brain barrier precedes symptoms and other MRI signs of new lesions in multiple sclerosis. Pathogenetic and clinical implications. Brain. 1990; 113(Pt 5)1477–89
- Hamann GF, Okada Y, Fitridge R, del Zoppo GJ. Microvascular basal lamina antigens disappear during cerebral ischemia and reperfusion. Stroke. 1995; 26: 2120–6
- Tagaya M, Haring H-P, Stuiver I, Wagner S, Abumiya T, Lucero J, et al. Rapid loss of microvascular integrin expression during focal brain ischemia reflects neuron injury. J Cereb Blood Flow Metab. 2001; 21: 835–46
- Yong VW, Power C, Forsyth P, Edwards DR. Metalloproteinases in biology and pathology of the nervous system. Nat Rev Neurosci. 2001; 2: 502–11
- Asahi M, Wang X, Mori T, Sumii T, Jung JC, Moskowitz MA, et al. Effects of matrix metalloproteinase-9 gene knock-out on the proteolysis of blood-brain barrier and white matter components after cerebral ischemia. J Neurosci. 2001; 21: 7724–32
- Hamann GF, Okada Y, del Zoppo GJ. Hemorrhagic transformation and microvascular integrity during focal cerebral ischemia/reperfusion. J Cereb Blood Flow Metab. 1996; 16: 1373–8
- Heo JH, Lucero J, Abumiya T, Koziol JA, Copeland BR, del Zoppo GJ. Matrix metalloproteinases increase very early during experimental focal cerebral ischemia. J Cereb Blood Flow Metab. 1999; 19: 624–33
- Sumii T, Lo EH. Involvement of matrix metalloproteinase in thrombolysis-associated hemorrhagic transformation after embolic focal ischemia in rats. Stroke. 2002; 33: 831–6
- Lo EH, Wang X, Cuzner ML. Extracellular proteolysis in brain injury and inflammation: role for plasminogen activators and matrix metalloproteinases. J Neurosci Res. 2002; 69: 1–9
- Rosenberg GA, Estrada EY, Dencoff JE. Matrix metalloproteinases and TIMPs are associated with blood-brain barrier opening after reperfusion in rat brain. Stroke. 1998; 29: 2189–95
- Yang Y, Estrada EY, Thompson JF, Liu W, Rosenberg GA. Matrix metalloproteinase-mediated disruption of tight junction proteins in cerebral vessels is reversed by synthetic matrix metalloproteinase inhibitor in focal ischemia in rat. J Cereb Blood Flow Metab. 2007; 27: 697–709
- Miller A, Ben-Yosef Y, Braker C, Shapiro S. Matrix metalloproteinases and their inhibitors in hypoxia/reoxygenation and stroke. Inflammation and stroke, GZ Feuerstein. Birkhäuser Verlag, Basel 2001; 275–85
- Morales CR, Pereyra LA, Toledo OM, Montes GS. Histochemical and morphological characterization of migrating mast cells in the bovine gallbladder epithelium. Histochemistry. 1980; 68: 159–68
- Zhang Y, Ramos BF, Jakschik BA. Mast cells enhance the antibody-mediated injury of skin basement membrane in mice. J Immunol. 1992; 149: 2482–7
- Metcalfe DD. Interaction of mast cells with extracellular matrix proteins. Int Arch Allergy Immunol. 1995; 107: 60–2
- Thompson HL, Thomas L, Metcalfe DD. Murine mast cells attach to and migrate on laminin-, fibronectin-, and matrigel-coated surfaces in response to Fc epsilon RI-mediated signals. Clin Exp Allergy. 1993; 23: 270–5
- Thompson HL, Burbelo PD, Yamada Y, Kleinman HK, Metcalfe DD. Identification of an amino acid sequence in the laminin A chain mediating mast cell attachment and spreading. Immunology. 1991; 72: 144–9
- Lehtonen-Smeds EM, Mäyränpää M, Lindsberg PJ, Soinne L, Saimanen E, Järvinen AA, et al. Carotid plaque mast cells associate with atherogenic serum lipids, high grade carotid stenosis and symptomatic carotid artery disease. Results from the helsinki carotid endarterectomy study. Cerebrovasc Dis. 2005; 19: 291–301
- Mäyränpää MI, Heikkilä HM, Lindstedt KA, Walls AF, Kovanen PT. Desquamation of human coronary artery endothelium by human mast cell proteases: implications for plaque erosion. Coron Artery Dis. 2006; 17: 611–21
- Goldschmidt RC, Hough LB, Glick SD. Rat brain mast cells: contribution to brain histamine levels. J Neurochem. 1985; 44: 1943–7
- Andriopoulou P, Navarro P, Zanetti A, Lampugnani MG, Dejana E. Histamine induces tyrosine phosphorylation of endothelial cell-to-cell adherens junctions. Arterioscler Thromb Vasc Biol. 1999; 19: 2286–97
- Winter MC, Shasby SS, Ries DR, Shasby DM. Histamine selectively interrupts VE-cadherin adhesion independently of capacitive calcium entry. Am J Physiol Lung Cell Mol Physiol. 2004; 287: L816–23
- Itoh Y, Sendo T, Oishi R. Physiology and pathophysiology of proteinase-activated receptors (PARs): role of tryptase/PAR-2 in vascular endothelial barrier function. J Pharmacol Sci. 2005; 97: 14–9
- Schechter NM, Brass LF, Lavker RM, Jensen PJ. Reaction of mast cell proteases tryptase and chymase with protease activated receptors (PARs) on keratinocytes and fibroblasts. J Cell Physiol. 1998; 176: 365–73
- Ebihara N, Funaki T, Murakami A, Takai S, Miyazaki M. Mast cell chymase decreases the barrier function and inhibits the migration of corneal epithelial cells. Curr Eye Res. 2005; 30: 1061–9
- Saarinen J, Kalkkinen N, Welgus H, Kovanen P. Activation of human interstitial procollagenase through direct cleavage of the Leu83-Thr84 bond by mast cell chymase. J Biol Chem. 1994; 269: 18134–40
- Tetlow LC, Harper N, Dunningham T, Morris MA, Bertfield H, Woolley DE. Effects of induced mast cell activation on prostaglandin E and metalloproteinase production by rheumatoid synovial tissue in vitro. Ann Rheum Dis. 1998; 57: 25–32
- Frank BT, Rossall JC, Caughey GH, Fang KC. Mast cell tissue inhibitor of metalloproteinase-1 is cleaved and inactivated extracellularly by alpha-chymase. J Immunol. 2001; 166: 2783–92
- Di Girolamo N, Indoh I, Jackson N, Wakefield D, McNeil HP, Yan W, et al. Human mast cell-derived gelatinase B (matrix metalloproteinase-9) is regulated by inflammatory cytokines: role in cell migration. J Immunol. 2006; 177: 2638–50
- Tchougounova E, Lundequist A, Fajardo I, Winberg JO, Abrink M, Pejler G. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. J Biol Chem. 2005; 280: 9291–6
- Lohi J, Harvima I, Keski-Oja J. Pericellular substrates of human mast cell tryptase: 72,000 dalton gelatinase and fibronectin. J Cell Biochem. 1992; 50: 337–49
- Fang KC, Wolters PJ, Steinhoff M, Bidgol A, Blount JL, Caughey GH. Mast cell expression of gelatinases A and B is regulated by kit ligand and TGF-beta. J Immunol. 1999; 162: 5528–35
- Helske S, Syväranta S, Kupari M, Lappalainen J, Laine M, Lommi J, et al. Possible role for mast cell-derived cathepsin G in the adverse remodelling of stenotic aortic valves. Eur Heart J. 2006; 27: 1495–504
- Fukuda S, Fini CA, Mabuchi T, Koziol JA, Eggleston LL, Jr, del Zoppo GJ. Focal cerebral ischemia induces active proteases that degrade microvascular matrix. Stroke. 2004; 35: 998–1004
- Rosenberg GA, Estrada EY, Dencoff JE, Stetler-Stevenson WG. Tumor necrosis factor-alpha-induced gelatinase B causes delayed opening of the blood-brain barrier: an expanded therapeutic window. Brain Res. 1995; 703: 151–5
- Kim KS, Wass CA, Cross AS, Opal SM. Modulation of blood-brain barrier permeability by tumor necrosis factor and antibody to tumor necrosis factor in the rat. Lymphokine Cytokine Res. 1992; 11: 293–8
- Yamasaki Y, Suzuki T, Yamaya H, Matsuura N, Onodera H, Kogure K. Possible involvement of interleukin-1 in ischemic brain edema formation. Neurosci Lett. 1992; 142: 45–7
- Stack MS, Johnson DA. Human mast cell tryptase activates single-chain urinary-type plasminogen activator (pro-urokinase). J Biol Chem. 1994; 269: 9416–9
- Furlan AJ, Higashida R, Wechsler L, Gent M, Rowley H, Kase C, et al. Intra-arterial prourokinase for acute ischemic stroke: The PROACT II study—A randomized controlled trial: Prolyse in Acute Cerebral Thromboembolism. JAMA. 1999; 282: 2003–11
- Qureshi AI, Ali Z, Suri MF, Kim SH, Shatla AA, Ringer AJ, et al. Intra-arterial third-generation recombinant tissue plasminogen activator (reteplase) for acute ischemic stroke. Neurosurgery. 2001; 49: 41–50
- Sobel BE. Intracranial bleeding fibrinolysis and anticoagulation: Causal connections and clinical implications. Circulation. 1994; 90: 2147–52
- Asahi M, Asahi K, Jung JC, del Zoppo GJ, Fini ME, Lo EH. Role for matrix metalloproteinase 9 after focal cerebral ischemia: effects of gene knockout and enzyme inhibition with BB-94. J Cereb Blood Flow Metab. 2000; 20: 1681–9
- Xue M, Del Bigio MR. Acute tissue damage after injections of thrombin and plasmin into rat striatum. Stroke. 2001; 32: 2164–9
- Castellino FJ. Plasmin. Handbook of proteolytic enzymes, AJ Barrett, ND Rawlings, JF Woessner. California Academic Press, San Diego 1998; 190–9
- Pfefferkorn T, Rosenberg GA. Closure of the blood-brain barrier by matrix metalloproteinase inhibition reduces rtPA-mediated mortality in cerebral ischemia with delayed reperfusion. Stroke. 2003; 34: 2025–30
- Lane DA, Björk I. Heparin and related polysaccharides. Advances in Experimental Medicine and Biology, DA Lane, I Björk, U Lindahl. Springer, UppsalaSweden 1992; 313: 138–142
- Rosenberg RD, Bauer KA. The heparin-antithrombin system: a natural anticoagulant mechanism. Hemostasis and thrombosis: basic principles and clinical practice, RW Colman, J Hirsh, VJ Marder, EW Salzman. Lippincott, Philadelphia 1994; 837–60
- Ende N, Auditore JV. Activation of a fibrinolytic system in a dog with mast cell tumor. Am J Physiol. 1964; 206: 567–72
- Valent P. Role of mast cells in endogeneous fibrinolysis and related (patho) physiological processes. Mast cells and basophils. London: Academic Press, G Marone, LM Lichtenstein, SJ Galli, 2000; 497–505
- Gordon JR, Galli SJ. Release of both preformed and newly synthesized tumor necrosis factor alpha (TNF-alpha)/cachectin by mouse mast cells stimulated via the Fc epsilon RI. A mechanism for the sustained action of mast cell-derived TNF-alpha during IgE-dependent biological responses. J Exp Med. 1991; 174: 103–7
- Galli SJ, Gordon JR, Wershil BK. Cytokine production by mast cells and basophils. Curr Opin Immunol. 1991; 3: 865–73
- Wershil BK, Wang ZS, Gordon JR, Galli SJ. Recruitment of neutrophils during IgE-dependent cutaneous late phase reactions in the mouse is mast cell-dependent. Partial inhibition of the reaction with antiserum against tumor necrosis factor-alpha. J Clin Invest. 1991; 87: 446–53
- Zhang Y, Ramos BF, Jakschik BA. Neutrophil recruitment by tumor necrosis factor from mast cells in immune complex peritonitis. Science. 1992; 258: 1957–9
- Gordon JR, Burd PR, Galli SJ. Mast cells as a source of multifunctional cytokines. Immunol Today. 1990; 11: 458–64
- Ehrlich, P. Beitrage zur Theorie und Praxis der histologischer Farbung. In: Thesis. Vol. Ph.D. Leipzig: University of Leipzig; 1878.
- Lupia E, Del Sorbo L, Bergerone S, Emanuelli G, Camussi G, Montrucchio G. The membrane attack complex of complement contributes to plasmin-induced synthesis of platelet-activating factor by endothelial cells and neutrophils. Immunology. 2003; 109: 557–63
- Enlimomab Acute Stroke Trial Investigators. Use of anti-ICAM-1 therapy in ischemic stroke: Results of the Enlimomab Acute Stroke Trial. Neurology. 2001;57:1428–34.
- Krams M, Lees K, Hacke W, Grieve A, Orgogozo J, Ford G. Acute Stroke Therapy by Inhibition of Neutrophils (ASTIN): an adaptive dose-response study of UK-279,276 in acute ischemic stroke. Stroke. 2003; 34: 2543–8
- Emerich DF, Dean RL, Bartus RT. The role of leukocytes following cerebral ischemia: pathogenic variable or bystander reaction to emerging infarct?. Exp Neurol. 2002; 173: 168–81
- Strbian D, Tatlisumak T, Ramadan UA, Lindsberg PJ. Mast cell blocking reduces brain edema and hematoma volume and improves outcome after experimental intracerebral hemorrhage. J Cereb Blood Flow Metab. 2007; 27: 795–802
- Reddigari S, Silverberg M, Kaplan AP. Assembly of the human plasma kinin-forming cascade along the surface of vascular endothelial cells. Int Arch Allergy Immunol. 1995; 107: 93–4
- Dziedzic T, Bartus S, Klimkowicz A, Motyl M, Slowik A, Szczudlik A. Intracerebral hemorrhage triggers interleukin-6 and interleukin-10 release in blood. Stroke. 2002; 33: 2334–5
- Silva Y, Leira R, Tejada J, Lainez JM, Castillo J, Davalos A. Molecular signatures of vascular injury are associated with early growth of intracerebral hemorrhage. Stroke. 2005; 36: 86–91
- Qureshi AI, Tuhrim S, Broderick JP, Batjer HH, Hondo H, Hanley DF. Spontaneuos intracerebral hemorrhage. N Engl J Med. 2001; 344: 1450–60
- Rosand J, Eskey C, Chang Y, Gonzalez RG, Greenberg SM, Koroshetz WJ. Dynamic single-section CT demonstrates reduced cerebral blood flow in acute intracerebral hemorrhage. Cerebrovasc Dis. 2002; 14: 214–20