Abstract
Background. Mannose-binding lectin (MBL) is a multifunctional protein involved in innate immunity. We tested whether MBL and elevated viral and bacterial antibodies were risk factors for acute coronary events.
Design. Controlled cohort study.
Methods. A total of 354 patients with unstable angina pectoris (UA) or acute myocardial infarction (AMI) were compared with 334 paired controls.
Results. Enterovirus titres were associated with increased risk of UA (odds ratio 10.04, P<0.001) and AMI (odds ratio 3.18, P=0.003), but titres did not correlate with either MBL concentration or genotype. Chlamydiapneumoniae heat shock protein 60 IgG concentrations were also associated with increased risk of UA (odds ratio 1.63, P=0.049). Compared to asymptomatic controls, patients had lower complement C3 serum concentrations (P<0.001), higher MBL serum concentration, and more frequently had MBL genotypes that determined high MBL levels (P<0.001). High MBL genotypes had odds ratios of 1.16 (P=0.010) for UA and 1.12 (P=0.007) for AMI. The elevation of MBL concentrations in the acute phase correlated with MBL concentrations after recovery (r=0.85, P<0.001).
Conclusions. Elevated microbial titres, indicating an on-going inflammation, were associated with cardiovascular events. MBL might have a dual role both decreasing susceptibility to infections and increasing the risk of acute coronary syndromes.
Introduction
Most cases of myocardial infarction result from an interaction of genetic and environmental factors that give rise to disease susceptibility but might not be the cause of the disease itself. There are conflicting data about the degree of risk that can be explained by classical risk factors in coronary heart disease (CHD); these factors are reported to explain 50%–90% of the risk Citation[1], Citation[2]. The heritable component of cardiovascular disease has been estimated to be 40%–60% in most populations Citation[3].
Chronic inflammation is an accepted risk factor for CHD. Abundant reports have demonstrated the role that infection plays as a risk factor for CHD. Accordingly, elevated antibody titres against several agents, including Chlamydia pneumoniae, Helicobacter pylori, herpes viridae (herpes simplex virus and cytomegalovirus) Citation[4], and enterovirus Citation[5], are reported to be associated with an increased incidence of CHD. Further, the number of pathogens involved in a simultaneous exposure (pathogen burden) has been reported to be an independent additive risk factor for CHD Citation[6], Citation[7]. The number of pathogens identified in CHD patients undergoing angiography was positively correlated to the degree of impairment in the coronary artery response to acetylcholine, an endothelium-dependent vasodilator Citation[8].
Key messages
There is an inflammatory stage in the body before acute coronary syndromes.
Elevated mannose-binding lectin (MBL) concentration is a risk factor for acute coronary syndromes.
MBL might have a dual role both decreasing susceptibility to infections but increasing the risk of acute coronary syndromes.
Mannose-binding lectin (MBL), surfactant protein (SP) A, and SP-D are multifunctional proteins that belong to the collectin family of proteins involved in innate immunity Citation[9], Citation[10]. Experimental research has shown that mice deficient in surfactant protein A (encoded by the SP-A1 and SP-A2 genes in human) and SP-D (encoded by the SFTPD gene) have a high susceptibility to a variety of bacterial and viral infections and exhibit elevated levels of proinflammatory cytokines Citation[11]. In addition, studies have shown that SP-D may be proatherogenic in mice Citation[12]. Apart from belonging to the collectin protein family, MBL is an acute-phase protein. Its concentrations increase slowly; for example, MBL rises during the first 10 days after an abdominal operation, while c- reactive protein (CRP) approaches its maximal concentration within 24 hours Citation[13]. Clinical data point out that the development and progression of coronary artery disease is associated with a modifying effect of MBL2, the gene encoding MBL Citation[14].
In this study, we analysed serum MBL concentrations and MBL2 and SFTPD genotypes. Our focus of interest was whether factors associated with infection resistance were also associated with acute myocardial infarction (AMI) or unstable angina pectoris (UA).
Patients and methods
Patients and sample collection
Patients with AMI or UA were admitted to the coronary care unit, Lund University Hospital, between March 1999 and April 2002. There were 354 patients included in the series: 277 men and 77 women. The inclusion criteria were as follows: age <80 years, no signs of cognitive intellectual disability, and sufficient competence to complete the research protocol. Of the originally invited patients 21 died before they could be interviewed, 48 chose not to participate, and after admission 12 patients were excluded from the study with the following diagnoses: unspecified precordial pain (n = 7), atrial fibrillation, pericarditis, myocarditis, pulmonary embolism, and aortic aneurysm. AMI was diagnosed in 241 patients and UA in 113 patients. It took a mean of 5 hours (median 3.3 hours, range 1–25 hours) from the first symptoms of the patient to get to the intensive care unit. Blood samples were taken soon after the patient's admission. Control serum samples taken 3–6 months after the acute coronary event were available from 168 patients.
Abbreviations
Control individuals were selected using the population register. There were 334 asymptomatic controls. They were chosen from the same area, usually from the same blocks, and were matched with age ±2 years, sex, and parish. Inclusion criteria were: no history of definite or suspected coronary heart disease or stroke, no operations or chemotherapy within the previous 4 weeks. Demographic data and levels of potential risk factors for coronary events of the study population are presented in .
Table I. Demographic and laboratory data for 354 patients with acute coronary syndromes and 334 controls. Patients that experienced a new event during the 6-year follow-up were significantly older (P < 0.001), but similar in other aspects to those without a new event.
The research ethics committee of Lund University approved the study. Written, informed consent was obtained from each patient for participation in the study.
AMI was diagnosed when two of the following criteria were fulfilled: the ST-segment elevation was followed by a T-wave inversion or new Q-waves in the electrocardiogram, chest pain that lasted >20 min, or an increase of creatine kinase MB (CK-MB) to >5 µg/L (more than twice the upper limit of the normal value). UA was diagnosed in patients with the following: 1) continuous ischemic chest pain and transient or persistent ST-segment depression (>1 mm), or 2) T-wave inversion (>1 mm), and/or 3) elevation of CK-MB (10 µg/L < CK-MB > 5 µg/L) or troponin T (0.10 µg/L < TNT > 0.05 µg/L). These samples were collected within 24 h of diagnosis and stored at -70°C and used for analyses of MBL, complement C3 and C4, CRP, and viral and bacterial antibodies.
Complement analysis
Concentrations of MBL in serum were determined by a sandwich enzyme-linked immunosorbent assay (ELISA) as described previously Citation[15]. Complement C3 and C4 were measured in patients and controls using turbidometry with polyclonal antibodies. The methods were calibrated against Human Serum Protein Calibrator, DAKO X908 (Dako A/S, Glostrup, Denmark).
Serology
Bacterial serology included measurement of IgG and IgA antibodies against C. pneumoniae and C. pneumoniae heat shock protein 60 (Cpn HSP60) and H. pylori. Viral serology included determination of antibody titres against cytomegalovirus, enterovirus, and herpes simplex virus as previously described by us Citation[16].
DNA extraction
Blood samples containing EDTA were collected and DNA extracted by a salting-out method Citation[17]. In brief, nucleated cells were lysed and treated with protease K solution. The proteins were precipitated with saturated NaCl, and then the DNA was precipitated with absolute ethanol.
Analysis of MBL gene polymorphism
MBL2 variants, including single nucleotide polymorphism (SNPs) at codon 52 (D), 54 (B), and 57 (C) in exon 1 of the MBL2 gene (rs5030737, rs1800450, and rs1800451, respectively) and promoter variants at position −550 (H/L) and −221 (X/Y) (rs11003125 and rs7096206) were determined by allele-specific polymerase chain reaction (PCR) amplification, essentially as described Citation[18]. Based on previously described associations between MBL genotype and serum concentrations, the genotypes were ranked in the order of their probable potential to induce high serum MBL concentrations in classes from 1 to 10, according to earlier findings Citation[18], Citation[19] ().
Table II. Mannose-binding lectin genotypes were assigned to classes that ranked their potential for inducing high serum MBL concentrations.
Analysis of SP-D gene (SFTPD) polymorphism
A synonymous exonic T/C (Met/Thr) SNP (rs721917) at codon 31 of the SFTPD gene was determined using restiction fragment length polymorphism PCR (PCR-RFLP). This polymorphism is presumed to be functional and has been shown to associate with serum SP-D concentration and oligomerization Citation[20]. DNA samples were amplified using the primers 5′-TCACCTCTAGAAGCTGAGCCAAGCC-3′ (sense) and 5′-ACAAAGTACCCAGAGTTGCTG-3′ (antisense), followed by a nested amplification with the primers 5′-GCATAAGCATGTCTTTTCTCTTTC-3′ (sense) and 5′-ACCAGGGTGCAAGCACTGCG-3′ (antisense). The latter antisense nested primer had a single nucleotide mismatch in the penultimate position to create an arbitrary FspI restriction enzyme recognition site to distinguish the FspI-cut allele T from the uncut allele C.
Statistical methods
Actual values and logarithmic transformations of the MBL concentrations and the genetic variants were used in the statistical analyses. Spearman's correlation coefficients were used to evaluate correlations between categorized and continuous variables. In case of non-normal distribution the Mann-Whitney test was used. The group means of continuous variables were compared using one-way ANOVA. Chi-square test was used for categorical variables. The MBL concentrations and the genetic variants were tested for correlations to the risk of UA or AMI with multinomial logistic regression. The Cox proportional hazards model was used to calculate hazard ratios (odds ratios) for hospitalization or death during the follow-up. The data were analysed using SPSS for Windows software version 15.0 (SPSS Inc., Chicago, IL, USA).
Results
Complement components C3, C4, and MBL
The serum concentration of C3 was lower in patients than in controls (1.64 versus 1.87, P = 0.009), while C4 concentrations did not differ between the two groups. Serum concentrations of MBL generally varied between 5 and 10,000 µg/L, but 19 samples had levels higher than 10,000 µg/L. MBL concentrations correlated significantly with CRP (P = 0.005) but did not correlate with serum troponin or complement C3 or C4. MBL serum concentrations were grouped into genotype classes ranked from 1 to 10, based on their potential to induce high serum concentration (). The MBL concentrations within each genotype class were normally distributed (). The MBL genotype did not correlate significantly with any of the bacterial or viral antibody titres investigated.
Figure 1. The logarithms of Mannose-binding lectin (MBL) concentrations (Ln[MBL]) were normally distributed in each MBL genotype class (MBL Score defined in ).
![Figure 1. The logarithms of Mannose-binding lectin (MBL) concentrations (Ln[MBL]) were normally distributed in each MBL genotype class (MBL Score defined in Table II).](/cms/asset/58f7e5b0-70a0-430f-85da-26e9c7bfa011/iann_a_411272_f0001_b.gif)
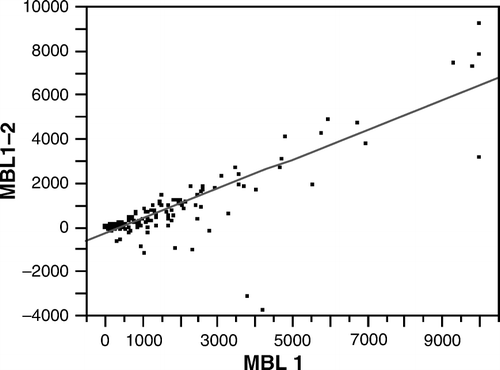
The MBL concentrations were higher in samples taken within 24 hours of the acute event than those taken at the follow-up visit (168 patients returned 3–6 months after the event; r=0.60, P < 0.001). The MBL concentrations were significantly correlated with genotypes known to effectively produce MBL (r=0.695, P < 0.001). The difference between the MBL concentrations observed in the acute and recovery phases were significantly correlated with the MBL concentration in the acute phase (r=0.85, P < 0.001) ().
Figure 2. The concentration of MBL in the acute phase (MBL 1) correlated significantly with the difference in MBL concentrations between the acute and recovery phases (MBL1–2) (r=0.85, P < 0.001).
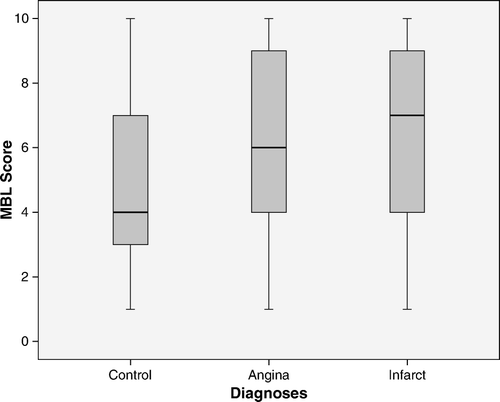
Patients with AMI had higher MBL concentrations than controls; the median for MBL was 855 µg/L in patients and 441 µg/L in controls (P < 0.0001) (A). The median genotype class was also higher in patients than in controls (A). The median genotype class was 6.11 (95% confidence interval (CI) 5.70–6.41) in patients and 5.2 (95% CI 4.87–5.61) in the controls. The odds ratio for UA was 1.25 (P = 0.006) based on the logarithm of MBL serum concentrations, and 1.16 (P = 0.010) based on the genotype. The odds ratios were almost the same for AMI, 1.28 (P < 0.001) and 1.12 (P < 0.007), respectively.
Figure 3. MBL genotype class (MBL Score) in controls and patients with unstable angina pectoris (Angina) and with acute myocardial infarction (Infarct). The box plots show medians, 75%, and 25% points and the ranges. Both patient groups fell into higher MBL genotype classes than controls P<0.001.
During the mean follow-up of 6 years, 156 of 354 patients experienced a new cardiovascular event ( and B). There was no significant difference between the genotypes of patients that had or did not have a new event.
Surfactant protein D
The SP-D allele and genotype distributions were similar across all the diagnostic groups (). Neither bacterial nor viral antibody titres were correlated with the genotypes.
Table III. Surfactant D genotype distribution in study groups.
Serology
The titres of antibodies against enterovirus were associated with an increased incidence of UA (odds ratio 10.04, P < 0.001) and AMI (odds ratio 3.18, P = 0.003). The concentration of IgG antibodies against C. pneumoniae heat shock protein 60 (HSP60 IgG) was also associated with increased risk for UA (odds ratio 1.63, P = 0.049).
Discussion
The high levels of enterovirus and C. pneumoniae HSP60 antibodies found in patients with acute coronary syndromes supported the role of infection. In addition, the elevated MBL concentrations suggested an on-going inflammatory activity, presumably due to a recent infection or reactivation of an infection. An increase in C3 typically occurs during the late acute-phase period, i.e. after 2–3 days. Since the samples were taken already during the early period after symptoms, it was possible to monitor probable disease-associated complement activation and consequent C3 consumption. Complement activation during myocardial infarction has been demonstrated in many studies. It appears that all complement activation pathways become activated, and since C3 is a component common to all pathways and main part of the amplification cascade, it is natural that its level would become most consumed during the acute activation stage. The reduction in C3 suggested that complement activation was associated with acute coronary syndromes (). This result contrasts with some other studies Citation[21], Citation[22] and might be explained by the time point of sample collection soon after admission to hospital. As complement consumption can be prominent at the time of the acute event, we would expect to observe a reduced C3 concentration at that time, even though a high C3 at base-line is a risk factor. During an acute coronary event, MBL concentrations were more elevated in patients with high base-line MBL than in those with low base-line MBL concentrations. This suggested that the MBL concentration before an acute event determined the degree of MBL elevation.
Compared to controls, patients more frequently had MBL2 genetic variants that determined high MBL concentrations and had higher actual concentrations of MBL. Moreover, there was a trend of MBL genotypes producing higher MBL concentrations in those that experienced a new cardiovascular event during the 6-year follow-up; this contradicted the hypothesis that low serum MBL concentrations might reduce infection resistance and would thus constitute an aetiological factor that predisposes to acute coronary syndromes. However, MBL might have a dual role both modulating susceptibility to infections and as the risk factor for acute coronary syndromes.
Studies linking infection and inflammation with accelerated atherosclerosis have accumulated during the past years. We observed high MBL concentrations shortly after the acute attack and a later decrease in concentrations. As basal MBL levels are genetically determined and rather stable throughout an individual's life, this result suggested that on-going inflammatory activity had commenced before the acute coronary event. Postoperatively, a significant increase in MBL could not be detectable until after 3 days Citation[13]. That the MBL concentrations were elevated from basal levels speaks for an inflammatory process being active for days before the final symptoms of coronary disease.
The collectins, MBL and SP-D, are important in the immune defence system Citation[23]. Collectins possess lectin domains that mediate interaction with a wide variety of pathogens. They aggregate a number of viruses and influence the chemotaxis and phagocytosis of specific bacteria. MBL may bind to microbial surfaces and activate the complement system via the lectin pathway. Other collectins, including SP-D, do not activate the complement system Citation[24]. When the innate immune response is weak, one would expect high bacterial and viral titres due to an increased susceptibility to infections. This expectation was borne out in a study with children that showed that low levels of MBL were associated with a syndrome of recurrent infections and a serum opsonin deficiency; this syndrome could be corrected by the administration of MBL Citation[25]. Furthermore, serum MBL levels were reported to be lower in children with sepsis compared to those without Citation[26]. MBL deficiency was associated with increased likelihood of death among patients with severe bacterial infection Citation[27].
There is a paradox of the role of MBL in cardiovascular disease. Some studies show it to be a protecting factor and some a risk factor. In more detail, MBL-deficient patients were found to have earlier onset or a more progressive atherosclerotic disease course than analogous MBL-competent patients Citation[28]. High MBL levels were associated with a low odds ratio for myocardial infarction in patients with type 2 diabetes or raised cholesterol, but not all patients, in a cohort of 19,381 in a Reykjavik study Citation[29]. In another study of patients with heart failure, MBL levels measured 1 month after a myocardial infarct were inversely associated with a high incidence of reinfarction Citation[30]. In three American Indian communities with a high prevalence of type 2 diabetes and experiencing an increased mortality and morbidity from coronary artery disease (CAD), variant MBL genotypes coding for markedly diminished levels of MBL were predictive of CAD Citation[31]. Infection susceptibility alleles (non-A) of MBL were interpreted to associate with increased carotid plaque area Citation[32]. In subjects carrying MBL variant alleles and being anti-C pneumoniae-positive, the odds ratio of development of new myocardial infarction and/or by-pass operation or cardiovascular death was over 3 as compared with the control group (14).
On the other hand, in some patient groups high MBL levels are reported to predict cardiovascular disease. Patients with type 1 diabetes and a history of cardiovascular disease had significantly elevated MBL levels Citation[33]. The rate of restenosis was high after carotid eversion endarterectomy in homozygous carriers of normal mannose-binding lectin genotype Citation[34]. High MBL concentrations were associated with increased risk of CAD in healthy men Citation[35]. In systemic lupus erythematosus patients, genotypes conferring MBL deficiency were not significantly associated with cerebrovascular, cardiovascular, or peripheral arterial damage Citation[36]. In patients with rheumatoid arthritis, high serum levels of MBL confered a risk for myocardial infarction but only in patients with high serum levels of agalactosyl IgG Citation[37].
According to the findings in the present study, both UA and AMI were associated with high MBL concentrations. Probably multiple, still unknown factors co-operate with MBL determining its effect on the vasculature.
Our results may seem to contradict previous reports that individuals with low MBL are more prone to develop atherosclerosis and acute coronary events. However, there are differences in the study settings between various studies. For example, in the before-mentioned Reykjavik heart study Citation[29] the design of the study differed in several respects from that of ours: MBL was not analysed in connection to the acute coronary event, and MBL genotypes were not studied. In the study of Ueland et al. Citation[30] MBL levels measured 1 month after a myocardial infarct were inversely associated with a high incidence of reinfarction, but they only studied patients with AMI complicated by heart failure, which in itself may alter the inflammatory response. However, there is a paradox that some studies show MBL being protective and some, like ours, a risk factor for vascular disease.
MBL might amplify the inflammatory reaction in vessels through complement interacting with damaged endothelial cells. The original damaging factor might be inflammation which is often induced by chronic and acute infections. The elevated MBL concentrations can only partially be explained by inflammation, because the gene variants that produce high MBL serum concentrations were more frequently observed in patients compared to controls. Thus, we found that a high MBL serum concentration was a risk factor for UA and AMI.
We hypothesize that the unique properties of MBL in host defence, pattern recognition, and complement activation could position this molecule as a factor that maintains the balance of other factors that influence the risk of coronary disease.
Conclusions
Enterovirus antibody titres, C. pneumoniae HSP60 titres, and MBL concentrations were elevated at the time of an acute coronary event, suggesting a history of infection and presence of inflammation days before the event. The decreased C3 concentrations observed in patients may be explained by complement activation causing C3 consumption; in turn, activation of the complement system might occur through the lectin pathway, when MBL binds to microbial surfaces.
High MBL serum concentration was a risk factor for UA and AMI. These results do not support the hypothesis that decreased serum concentrations of collectins would increase infection susceptibility, and by this mechanism increase the risk of CHD. Instead, MBL might have a dual role both by decreasing susceptibility to infections but also promoting persistent infection, inflammation, and tissue damage through, e.g., complement activation and thereby increasing the risk of acute coronary syndromes. The factors behind the paradox that some studies show MBL being protective and some a risk factor for vascular disease remain elusive.
Acknowledgements
We would like to express our gratitude to our dear friend Anders G. Sjöholm who initiated this work before he sadly passed away in June 2006. We wish to thank research nurses Laura Darcy and Annica Maxedius for interviewing the control subjects, collecting blood samples, collecting data, and registering data. The study was supported by grants from the Swedish Research Council (grant no. 15092), the Medical Faculty at Lund University, the Swedish National Association against Rheumatism, Alfred Österlund's Foundation, The Crafoord Foundation, Greta and Johan Kock's Foundation, Sigrid Juselius Foundation (M.H.), The King Gustaf V's 80th Birthday Fund, Academy of Finland (Grants 202491, 110340 and 119004 for MP), and Lund University Hospital.
Related Research Data
References
- Lusis AJ, Fogelman AM, Fonarow GC. Genetic basis of atherosclerosis: part I: new genes and pathways. Circulation. 2004; 110: 1868–73
- Futterman LG, Lemberg L. Fifty percent of patients with coronary artery disease do not have any of the conventional risk factors. Am J Crit Care. 1998; 7: 240–4
- Khot UN, Khot MB, Bajzer CT, Sapp SK, Ohman EM, Brener SJ, et al. Prevalence of conventional risk factors in patients with coronary heart disease. JAMA. 2003; 290: 898–904
- Epstein SE, Zhou YF, Zhu J. Infection and atherosclerosis: emerging mechanistic paradigms. Circulation. 1999; 100: 20–8
- Reunanen A, Roivainen M, Kleemola M, Saikku P, Leinonen M, Hovi T, et al. Enterovirus, mycoplasma and other infections as predictors for myocardial infarction. J Intern Med. 2002; 252: 421–9
- Rupprecht H, Blankenberg S, Bickel C. Impact of viral and bacterial infectious burden on long-term prognosis in patients with coronary artery disease. Circulation. 2001; 104: 25–31
- Espinola-Klein C, Rupprecht HJ, Blankenberg S, Bickel C, Kopp H, Rippin G, et al. Impact of infectious burden on extent and long-term prognosis of atherosclerosis. Circulation. 2002; 105: 15–21
- Prasad A, Zhu J, Halcox JP, Waclawiw MA, Epstein SE, Quyyumi AA. Predisposition to atherosclerosis by infections: role of endothelial dysfunction. Circulation. 2002; 106: 184–90
- Haagsman HP, Hogenkamp A, van Eijk M, Veldhuizen EJ. Surfactant collectins and innate immunity. Neonatology. 2008; 93: 288–94
- Thiel S. Complement activating soluble pattern recognition molecules with collagen-like regions, mannan-binding lectin, ficolins and associated proteins. Mol Immunol. 2007; 44: 3875–88
- Wright JR. Immunoregulatory functions of surfactant proteins. Nat Rev Immunol. 2005; 5: 58–68
- Sorensen GL, Madsen J, Kejling K, Tornoe I, Nielsen O, Townsend P, et al. Surfactant protein D is proatherogenic in mice. Am J Physiol Heart Circ Physiol. 2006; 290: 2286–94
- Van Till JW, Boermeester MA, Modderman PW, Van Sandick JW, Hart MH, Gisbertz SS, et al. Variable mannose-binding lectin expression during postoperative acute-phase response. Surg Infect (Larchmt) 2006; 7: 443–52
- Rugonfalvi-Kiss S, Endrész V, Madsen HO, Burián K, Duba J, Prohászka Z, et al. Association of Chlamydia pneumoniae with coronary artery disease and its progression is dependent on the modifying effect of mannose-binding lectin. Circulation. 2002; 106: 1071–6
- Sjöholm AG, Truedsson L, Jensenius JC. Complement pathways and meningococcal disease—diagnostic aspects. Methods in molecular medicine, AJ Pollard, MCJ Maiden. Humana Press Inc, Totowa 2001; 529–47
- Pesonen E, Andsberg E, Ohlin H, Puolakkainen M, Rautelin H, Sarna S, et al. Dual role of infections as risk factors for coronary heart disease. Atherosclerosis. 2007; 192: 370–5
- Miller SA, Dykes DD, Polesky HF. A simple salting out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res. 1988; 16: 1215
- Madsen HO, Garred P, Thiel S, Kurtzhals JA, Lamm LU, Ryder LP, et al. Interplay between promoter and structural gene variants control basal serum level of mannan-binding protein. J Immunol. 1995; 155: 3013–20
- Carlsson M, Sjöholm AG, Eriksson L, Thiel S, Jensenius JC, Segelmark M, et al. Deficiency of the MBL pathway of complement and poor outcome in cystic fibrosis: bacterial colonization may be decisive for a relationship. Clin Exp Immunol. 2005; 139: 306–13
- Leth-Larsen R, Garred P, Jensenius H, Meschi J, Hartshorn K, Madsen J, et al. A common polymorphism in the SFTPD gene influences assembly, function, and concentration of surfactant protein D. J Immunol. 2005; 174: 1532–8
- Engström G, Hedblad B, Janzon L, Lindgärde F. Complement C3 and C4 in plasma and incidence of myocardial infarction and stroke: a population-based cohort study. Eur J Cardiovasc Prev Rehabil. 2007; 14: 392–7
- Palikhe A, Sinisalo J, Seppänen M, Haario H, Meri S, Valtonen V, et al. Serum complement C3/C4 ratio, a novel marker for recurrent cardiovascular events. Am J Cardiol. 2007; 99: 890–5
- Gupta G, Surolia A. Collectins: sentinels of immunity. Bioessays. 2007; 29: 452–64
- Holmskov U, Thiel S, Jensenius JC. Collections and ficolins: humoral lectins of the innate immune defense. Annu Rev Immunol. 2003; 21: 547–78
- Super M, Thiel S, Lu J, Levinsky RJ, Turner MW. Association of low levels of mannan-binding protein with a common defect of opsonisation. Lancet. 1989; 2: 1236–9
- de Benedetti F, Auriti C, D'Urbano LE, Ronchetti MP, Ravà L, Tozzi A, et al. Low serum levels of mannose binding lectin are a risk factor for neonatal sepsis. Pediatr Res. 2007; 6: 325–8
- Eisen DP, Dean MM, Boermeester MA, Fidler KJ, Gordon AC, Kronborg G, et al. Low serum mannose-binding lectin level increases the risk of death due to pneumococcal infection. Clin Infect Dis. 2008; 47: 510–6
- Madsen HO, Videm V, Svejgaard A, Svennevig JL, Garred P. Asscociation of mannose-binding-lectin deficiency with severe atherosclerosis. Lancet. 1998; 352: 959–60
- Saevarsdottir S, Oskarsson OO, Aspelund T, Eiriksdottir G, Vikingsdottir T, Gudnason V, et al. Mannan binding lectin as an adjunct to risk assessment for myocardial infarction in individuals with enhanced risk. J Exp Med. 2005; 201: 117–25
- Ueland T, Espevik T, Kjekshus J, Gullestad L, Omland T, Squire IB, et al. Mannose binding lectin and soluble Toll-like receptor 2 in heart failure following acute myocardial infarction. J Card Fail. 2006; 12: 659–63
- Best LG, Davidson M, North KE, MacCluer JW, Zhang Y, Lee ET, et al. Prospective analysis of mannose-binding lectin genotypes and coronary artery disease in American Indians: the Strong Heart Study. Circulation. 2004; 109: 471–5
- Hegele RA, Ban MR, Anderson CM, Spence JD. Infection-susceptibility alleles of mannose-binding lectin are associated with increased carotid plaque area. J Investig Med. 2000; 48: 198–202
- Hansen TK, Tarnow L, Thiel S, Steffensen R, Stehouwer CD, Schalkwijk CG, et al. Association between mannose-binding lectin and vascular complications in type 1 diabetes. Diabetes. 2004; 53: 1570–6
- Rugonfalvi-Kiss S, Dósa E, Madsen HO, Endrész V, Prohászka Z, Laki J, et al. High rate of early restenosis after carotid eversion endarterectomy in homozygous carriers of the normal mannose-binding lectin genotype. Stroke. 2005; 36: 944–8
- Keller T, van Leuven S, Meuwese M, Wareham N, Luben R, Stroes E, et al. Levels of mannose-binding lectin and the risk of future coronary artery disease in apparently healthy men and women. Arterioscler Thromb Vasc Biol. 2006; 26: 2345–50
- Jönsen A, Gullstrand B, Güner N, Bengtsson AA, Nived O, Truedsson L, et al. Genetically determined mannan-binding lectin deficiency is of minor importance in determining susceptibility to severe infections and vascular organ damage in systemic lupus erythematosus. Lupus. 2007; 16: 245–53
- Troelsen LN, Garred P, Madsen HO, Jacobsen S. Genetically determined high serum levels of mannose-binding lectin and agalactosyl IgG are associated with ischemic heart disease in rheumatoid arthritis. Arthritis Rheum. 2007; 56: 21–9