Abstract
The RecQ family of DNA helicases consists of specialized DNA unwinding enzymes that promote genomic stability through their participation in a number of cellular processes, including DNA replication, recombination, DNA damage signaling, and DNA repair pathways. Mutations resulting in the inactivation of some but not all members of the RecQ helicase family can lead to human syndromes which are characterized by marked chromosomal instability and an increased predisposition to cancer. An evolutionarily conserved interaction between RecQ helicases and topoisomerase 3s has been established, and this interaction is important in the regulation of recombination and genomic stability. Topoisomerases are critical in the cell because they relieve helical stress that arises when DNA is unwound. Topoisomerases function by breaking and rejoining DNA. By inhibition of the rejoining function, topoisomerase inhibitors are potent chemotherapeutic agents that have been used successfully in the treatment of hematologic malignancies and other cancers. This review discusses the roles of RecQ helicases in genomic stability, the interplay between RecQ helicases and topoisomerase 3s, and current and future prospects for targeting these interactions to develop novel anticancer therapies.
| Abbreviations | ||
| ATM | = | ataxia‐telangiectasia mutated |
| ATP | = | adenosine triphosphate |
| ATR | = | ataxia‐telangiectasia related |
| BRCA1 | = | breast cancer gene 1 |
| FANCD2 | = | Fanconi anemia D2 |
| MLL | = | myeloid/lymphoid or mixed lineage leukemia |
| MRN | = | Mre11/Rad50/Nbs1 |
| PML | = | promyelocytic leukemia |
| RecQ | = | recombination‐deficient mutant in Escherichia coli |
| UGT | = | UDP‐glucuronosyl transferase |
| UV | = | ultraviolet |
Introduction
Genomic instability is a cellular condition in which mutations accumulate at higher rates than normal. Genomic instability is the central feature of a sizeable group of human syndromes in which a defect results in one of the various processes that respond to DNA damage. Although the clinical manifestations of the genomic instability syndromes vary widely, cancer predisposition is a common diathesis. An acquired genomic instability is also a common feature of sporadic cancers, underscoring the importance of genomic instability in carcinogenesis.
The RecQ family of DNA helicases is highly conserved from bacteria to man. These enzymes harness the energy from the hydrolysis of adenosine triphosphate (ATP) to separate complementary strands of DNA. This is a necessary component of a number of cellular processes, including DNA replication, recombination, and repair. There are five distinct RecQ helicase genes in mammalian genomes, referred to as RECQL1, BLM, WRN, RECQL4, and RECQL5. Mutations in three of the five human RecQ helicases, BLM, WRN, and RECQL4, result in the rare, autosomal recessive disorders Bloom syndrome (BS), Werner syndrome (WS), and Rothmund‐Thomson syndrome (RTS), respectively Citation1. These disorders feature distinct forms of genomic instability that shed light on a group of important functions at the interface of DNA replication and recombination. While these genetic diseases are uncommon and affect only a small proportion of the population, they offer an excellent model in which to study the link between genomic stability and cancer. In this article, we focus on the role of the RecQ helicases in protection from cancer development and explore how RecQ helicase functions can be targeted in anticancer therapy.
Key messages
RecQ helicases are specialized DNA unwinding enzymes that play critical roles in maintaining the integrity of the genome through their participation in DNA replication, recombination, and repair pathways, and by responding to DNA damage.
Mutations in three human RecQ helicases, BLM, WRN, and RECQL4, result in Bloom syndrome, Werner syndrome, and Rothmund‐Thomson syndrome, respectively. All three syndromes feature striking genomic instability and cancer predisposition.
RecQ helicases and topoisomerase 3s physically and functionally interact. Topoisomerases control DNA topology in the cell, and inhibition of their functions causes DNA breaks; therefore these enzymes are useful targets for anticancer therapies.
RecQ helicases are specialized DNA unwinding enzymes that bind to recombination intermediates
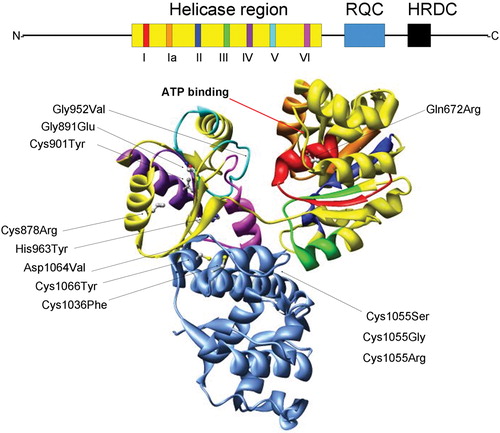
All RecQ helicases are defined by a helicase region that consists of seven highly conserved amino acid sequence motifs (I, Ia, II–VI) (). Helicase motifs I and II are homologous to the Walker boxes A and B that are common to many ATP‐binding proteins. Two additional regions are also found in most RecQ family members—the RecQ C‐terminal (RQC) region, which is required for DNA helicase activity and which also mediates specific protein‐protein interactions, and the Helicase and RNaseD C‐terminal (HRDC) domain, which participates in modulating specific protein‐nucleic acid interactions. Aside from these regions, RecQ helicases share little sequence homology. The highly variable N‐terminal regions present in many RecQ helicases mediate specific protein‐protein interactions that dictate unique functions of the respective family member. Most RecQ family members possess DNA‐dependent ATPase and ATP‐ and Mg2+‐dependent DNA unwinding activities. They unwind DNA in a 3′→ 5′ direction with respect to the strand on which the enzyme is bound. Their preferred substrates resemble DNA replication and recombination intermediates, including forked DNA structures, Holliday junctions, G‐quadruplex DNA (G4‐DNA) and D‐loops, suggesting that RecQ helicases play important roles in recombination.
Figure 1. Structural features of the RecQ helicase catalytic core. The BLM helicase was modeled using the crystal structure of the catalytic core fromE. Coli RecQ (49–51) which only includes the helicase (yellow) and RQC (light blue) regions. Additionally, the seven helicase motifs and RQC domain are each displayed in a different color which corresponds to the schematic diagram (top): motif I (red), Ia (orange), II (dark blue), III (green), IV (purple), V (cyan), VI (fuchsia). The N and C termini and conserved HRDC domain (all shown in black) are not included in the structural representation of BLM. Diagram consisting of α‐helices (cylinders), β‐sheets (arrows), and variable regions (ribbons). The ATP binding site is indicated with an arrow (red). The E. coli RecQ structure was used to map 10 corresponding positions of 12 characterized BS missense mutations, shown with their original amino acid side chains. The mutations are indicated with an arrow (black) and listed as follows with the corresponding E. coli RecQ residue in parentheses. Helicase region: Gln672Arg (Gln30), Gly891Glu (Gly239), Cys901Tyr (Val249), Gly952Val (Gly299), His963Tyr (His310), Cys878Arg (Leu227); RQC region: Cys1036Phe (Cys380), Cys1055Ser (Gly392), Cys1055Gly (Gly392), 1055Arg (Gly392), Asp1064Val (Asp401), Cys1066Tyr (Cys402).
In addition to heterogeneity in protein‐protein interactions, there is catalytic heterogeneity, as WRN possesses an exonuclease activity in its N‐terminal region, whereas RECQL4 lacks detectable DNA unwinding ability but possesses ATPase and ssDNA annealing activities Citation2.
Functional homology between the microbial RecQ helicases and BLM
The RecQ family is named after the single Escherichia coli recQ gene, originally isolated in a screen for mutants resistant to thymineless death Citation3. RecQ is a component of the RecF recombination pathway, which was initially characterized in plasmid recombination assays Citation4. However, its main function is in DNA replication where recombination can be used to restore damaged replication forks. For example, RecQ acts to ameliorate replication fork stalling at sites of ultraviolet (UV) damage Citation5. Certain DNA substrates generated during replication can promote inappropriate recombination, and RecQ suppresses inappropriate replication‐associated recombination Citation6,7.
In yeast, there are single RecQ homologues—SGS1 in Saccharomyces cerevisiae (budding yeast) and rqh1+ in Schizosaccharomyces pombe (fission yeast). sgs1 and rqh1 null mutants exhibit an increased rate of illegitimate, homologous, and homeologous recombination Citation8,9, retarded proliferation rates, chromosome instability, and hypersensitivity to a number of DNA damaging agents, including hydroxyurea (HU), methyl methanesulfonate (MMS), UV irradiation, and the topoisomerase I inhibitor, camptothecin. This group of phenotypes closely resembles the cellular phenotypes of cells deficient for the RecQ helicase BLM (see below). Indeed, expression of the human BLM gene in a yeast sgs1 null cell can substitute for the mutant gene and suppress some of the sgs1 hyper‐recombination phenotypes Citation10. Hence, we will focus on the BLM helicase as the paradigmatic RecQ helicase in the remainder of this review.
BLM helicase deficiency results in genomic instability and cancer
Homozygous or compound heterozygous mutations in the BLM gene are associated with BS. BS is characterized by small but proportional size, a sun‐sensitive facial erythema, hypo‐ and hyperpigmented skin lesions, immune deficiency, infertility in males and sub‐fertility in females, a paucity of subcutaneous fat and defects in sugar metabolism, susceptibility to type 2 diabetes, and, most prominently, a predisposition to cancers Citation11. The cancer predisposition in BS is notable for its high incidence, broad spectrum (including leukemia, lymphoma and carcinomas), early diagnosis relative to the same cancer in the general population, and the development of multiple cancers in single individuals .
Table I. The first 100 cancers documented in the Bloom's Syndrome Registry.
At the cellular level, BS somatic cells exhibit striking chromosome instability. The most characteristic chromosomal abnormalities of BS cells are the quadriradial (a symmetrical, four‐armed configuration, composed of a pair of homologous chromosomes that apparently have undergone somatic crossing‐over) and the sister‐chromatid exchange (SCE)—an exchange event between the sister chromatids Citation12. In metaphases prepared from normal fibroblasts, the average number of SCEs is approximately 8; in metaphases from BS fibroblasts, the average is approximately 55 SCEs, which is a 7‐fold difference. Accordingly, high levels of SCEs are diagnostic of BS. In addition to this evidence of excessive recombination, BS cells display excesses of chromatid breaks and gaps, dicentric and ring chromosomes, acentric fragments, pulverized metaphases, telomere associations, and anaphase bridges. All of these abnormalities can be detected in cells from normal individuals, but the abnormalities are present at much higher levels in BS.
BS cells are also hypersensitive to certain genotoxic agents (e.g. ultraviolet light, mitomycin C, and topoisomerase inhibitors) Citation13. At the molecular level, BS somatic cells accumulate locus‐specific mutations at elevated rates Citation14. The increased rate of somatic mutations in concert with the increased rate of recombination between homologs in BS cells provides an explanation for the high incidence of cancers in persons with the syndrome. Consequently, BS is an important model for understanding how molecular mechanisms that suppress excessive recombination maintain genome integrity and how failure to suppress excessive recombination promotes carcinogenesis.
Of the 64 unique mutations in the BLM gene that have been characterized in persons with BS in the Bloom's Syndrome Registry, 54 cause premature termination of translation (nonsense, frameshift, exon‐deletion, and splice‐site mutations), and 10 are missense mutations Citation15. Northern and Western analyses indicate that the premature translation‐termination mutations are null mutations because the BLM mRNA and BLM protein products are missing in cells that harbor this type of mutation. However, BLM mRNA and protein are present in cells that harbor missense mutations. The ten missense mutations localize to highly conserved residues of the helicase and RQC regions of BLM (). Purification of recombinant BLM proteins that contain the specific BS missense mutations Gly672Arg and Cys1055Ser demonstrated that they result in catalytically inactive helicases. Moreover, expression of the Gly672Arg and Cys1055Ser mutant proteins in BLM‐deficient BS cells does not correct their high SCE phenotype, indicating that BLM helicase activity is required for its function in the maintenance of genome integrity Citation16.
Nuclear preparedness: BLM, PML nuclear bodies, and control of access to substrates
BLM is a nuclear protein the levels of which are regulated during the cell cycle, being expressed at its highest levels in the DNA synthesis phase (S phase) Citation17. BLM also adopts a characteristic protein distribution in the nucleus. In normal, undamaged cells, BLM is distributed throughout the nucleoplasm in fine granules and in focal concentrations recognized as the promyelocytic leukemia (PML) nuclear bodies (PML‐NBs, which are also known as nuclear domains 10 and PML oncogenic domains) Citation18. The PML‐NBs number from 10 to 30 per nucleus, and they range in size from 0.2 to 1 micron. The PML‐NBs contain a wide variety of proteins. Besides BLM, the PML‐NBs contain other DNA repair proteins, including topoisomerase IIIα, p53, and the MRN complex (MRE11/RAD50/NBS1). Mutations in the p53, MRE11, and NBS1 genes are associated with human syndromes that feature genomic instability and cancer predisposition. The PML‐NBs also contain proteins involved in RNA transcription, apoptosis, antiviral defense, and tumor suppression. It is thought that the PML‐NBs control the availability of protein components for various nuclear processes by regulating the dynamic shuttling of proteins from the PML‐NBs to the nucleoplasm and back again Citation19.
The PML protein is one of the major structural components of the PML‐NBs. Although PML knockout mice (PML−/−) are viable, they are highly susceptible to developing tumors after treatment with carcinogens Citation20. Proteins that are normally focally concentrated in the PML‐NBs, including BLM, are not concentrated in foci in PML−/− mice; however, ectopic expression of PML causes the re‐formation of PML‐NBs. Mouse embryonic fibroblasts (MEFs) from PML−/− mice exhibit a two‐fold higher frequency of SCEs than PML+/+ mice, consistent with a role for the PML‐NBs in maintenance of genome integrity. The two‐fold increase in SCEs compares to the >six‐fold increase detected in BS cells. Thus, although the increase in genomic instability in the PML knockout mouse is modest, it could explain the cancer susceptibility seen in this animal. Because BLM is mislocalized in PML−/− mice, the data also suggest that localization of BLM to the PML‐NBs is important in BLM's ability to properly regulate excessive recombination Citation21. Consistent with this observation, expression in BLM‐deficient BS cells of a deleted form of human BLM that is unable to concentrate focally in the PML‐NBs only reduces the frequency of SCEs by half Citation22, supporting the hypothesis that the PML‐NBs play a key role in BLM's access to SCE‐producing DNA substrates.
DNA repair proteins gather in DNA damage foci to effect damage signaling and repair
In cells treated with DNA‐damaging agents (e.g. γ irradiation, DNA cross‐linking agents, and ultraviolet irradiation) BLM migrates out of the PML‐NBs and focally concentrates with other proteins, including phosphorylated histone H2AX (γH2AX), and the tumor suppressor proteins BRCA1, FAND2, and the MRN complex at sites of DNA damage—forming DNA damage‐inducible (DDI) foci. The recruitment and accumulation of DNA repair proteins at the DDI foci are important steps in DNA repair and DNA damage signaling. γH2AX Citation23,24 is the prototypic DDI focus protein. Phosphorylation of H2AX occurs in a large chromatin domain adjacent to the site of DNA damage Citation25; the antibody that recognizes γH2AX is widely used to mark DNA lesions. Intriguingly, mouse H2AX knockouts have defects in recombinational repair of DNA double strand breaks and elevated SCEs Citation26.
Another protein that is important in the formation of DDI foci is the Fanconi anemia (FA) protein FANCD2. FA is an autosomal recessive, chromosome instability syndrome that features developmental defects, sensitivity to DNA cross‐linking agents, and cancer predisposition. There are at least ten genes that when mutated result in FA. Most of the FA gene products are components of a multi‐protein complex that controls the localization and function of the FANCD2 protein in response to DNA damage. One of the gene products in the complex, FANCL, modifies FANCD2 with a single moiety of ubiquitin. Ubiquitin modification of FANCD2 is an important step in the formation of DDI foci Citation27. FANCD2 is also a key player in the recruitment of the breast cancer gene products BRCA1 and BRCA2 to chromatin Citation27,28. Persons who are born with single BRCA2 null mutations have a high risk of breast cancer development, whereas persons who are born with two BRCA2 mutations—one null and the other a C‐terminal BRCA2 truncation mutation—develop Fanconi anemia. Thus, BRCA2 is not only a breast cancer susceptibility gene, but also the FA gene FANCD1. Conversely, the FA genes are implicated in breast cancer predisposition.
Taken together, the observation that mutations in many of the genes encoding proteins involved in the maintenance of genomic stability in association with BLM are linked to cancer or cancer predisposing syndromes underscores the direct link between genomic instability and cancer.
BLM and DNA damage signaling during the S phase of the cell cycle
DDI foci can also be induced by treatment of cells with inhibitors of DNA replication such as hydroxyurea (HU), a ribonucleotide reductase inhibitor that stalls DNA polymerase on the DNA template by limiting it for nucleotides. After treatment of cells synchronized in S phase with DNA‐damaging agents or HU, BLM associates with sites of stalled or stressed replication forks within minutes of drug treatment Citation23. In normal cells, stalling replication forks with DNA damage or by HU treatment causes increased SCEs. Therefore, it is tempting to speculate that BLM normally regulates recombination events that are induced by stalling or stressing replication forks.
Damage recognition and signaling during S phase is clearly a highly regulated process, as many normal DNA substrates that are generated during DNA replication could be recognized as DNA damage. DDI foci that arise during replication stress contain many of the proteins that are components of the classic γ irradiation‐induced foci. Several lines of evidence indicate that BLM has important roles in regulating the assembly of S phase DDI foci. Accumulation of BRCA1 protein and the MRN complex in S phase DDI foci is partially dependent on BLM, because the recruitment of these proteins occurs with slower kinetics in BLM‐deficient BS cells Citation23,Citation29. BLM also regulates the recruitment of p53 to the DDI foci Citation30. If p53 is degraded in BS cells, the kinetic defect in the accumulation of BRCA1 and the MRN complex at DDI foci is alleviated Citation23. Because BLM is one of the earliest components to associate with S phase DDI foci, and because it is interacts with the major signaling factors known to be involved in DDI focus formation (FANCD2, BRCA1, p53, and the MRN complex), it is possible that BLM is involved both in sensing DNA damage and in the process of signaling the presence of DNA damage, specifically in S phase.
Over the years, a large number of proteins have been shown to interact physically with BLM. BLM interacts directly with the DNA damage response kinases ATM (ataxia‐telangiectasia mutated) and ATR (ataxia‐telangiectasia related) and the p53 gene. Some of these proteins with which BLM interacts can be isolated in large soluble complexes with BLM. One of these complexes is called BRAFT for the components BLM, the single stranded DNA binding protein RPA (p70, p32, and p14) Citation31, the FA gene product FANCA, and topoisomerase IIIα. BRAFT also includes the novel protein referred to as BLAP75 Citation32. BLM also interacts with the mismatch repair protein MLH1 Citation33, and the entire FA complex can be immunoprecipitated with BLM Citation31. The immunoprecipitation data suggest that BLM is associated with different groups of proteins at different times or under different cellular conditions. How these proteins and their activities are integrated into the biochemical pathways that regulate recombination is still unclear. It has been hypothesized that the BLM complex functions to disentangle interlinked DNA strands that are generated normally during DNA replication Citation34, but clearly BLM has additional roles in DNA damage response and signaling, and its part in the tangled network of protein interactions requires further study.
BLM gene dosage and mouse models of Bloom syndrome
Mouse models of BLM helicase deficiency have recapitulated the human disease phenotypes and provided novel insights into disease mechanisms. Several different mutated BLM alleles have been produced, and their effects on mouse development and the development of disease have been characterized. Phenotypic variation between mouse mutants is related to the type of mutation that was experimentally generated and the dosage of BLM protein. The null mutations that have been produced all result in the complete loss of normal BLM protein. A ‘hypomorphic’ mutation was produced that results in a quantitative reduction in the expression of normal protein.
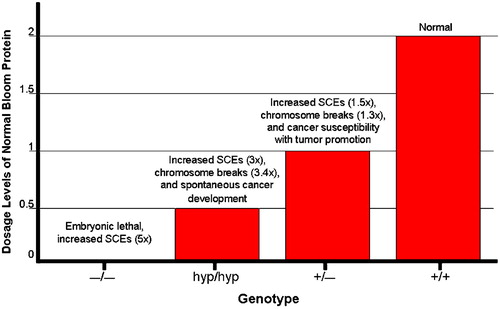
Three groups have produced multiple null alleles of the mouse BLM gene (a BLM− allele) and one of these groups also produced a hypomorphic BLM mutation (the BLMhyp allele). The phenotype of the homozygous null (BLM−/−) mouse is embryonic lethal with developmental delay, attributed to increased apoptosis in the epiblast, and anemia that leads to death by embryonic day 13.5 (). Similar to the human BS cells, homozygous null mouse cells exhibit a five‐fold increase in SCEs and chromosome breakage. The BLMhyp allele produces approximately 25% of normal BLM protein. BLMhyp/hyp or BLMhyp/− mice are viable and developmentally normal but have levels of SCE and chromosome breakage that are slightly lower than those in BLM−/− mice. A similar observation was made in BS cells, in which varying amounts of SCE reduction correlate with the amount of ectopic expression of BLM introduced experimentally on an expression vector. Cancer incidence in BLMhyp/hyp and BLMhyp/− mice is dramatically increased, with 40% and 76% of mice, respectively, developing a cancer by 2 years Citation35. A wide range of cancer types and sites were recorded, with diffuse lymphomas and adenomacarcinomas being the most common types. These observations confirm the classical connection made between excessive recombination and cancer predisposition in BS.
Heterozygous BLM+/− mouse cells have a small increase in SCEs and micronuclei formation (micronuclei formation is one indicator of DNA breakage). Increased cancer susceptibility in this mouse was demonstrated by crossing in a mutation in the APC gene (the so‐called min mouse); the resulting BLM+/−APC+/− mice develop adenomas at an increased rate. In addition, the time to death after infection with murine leukemia virus is faster in BLM+/− mice compared to the wild‐type mice. Human BLM heterozygotes do not exhibit increased SCEs, but heterozygotes have an increased susceptibility to colorectal cancer, suggesting that the dosage levels of BLM are also important in human cells. Thus, although the null mutation in mouse is more severe than the BS null mutations in humans, varying levels of BLM expression produce a gradient of effects on SCE levels and genomic instability, resulting in cancer susceptibility in both humans and mice. These data emphasize the importance of gene dosage in maintaining genome integrity for BLM, and similar observations have now been made for many other DNA repair proteins as well.
Figure 2. Distinct phenotypes observed in mice that express varying levels of Bloom protein: graphical depiction of the relationship between the level of normal BLM expression on the one hand and genomic instability and cancer susceptibility on the other. The null BLM−/− mutation which results in the complete absence of BLM protein causes embryonic lethality. Hypomorphic BLMhyp/hyp mice produce 25% of the normal Bloom protein level and exhibit increased SCEs (3‐fold higher than normal) and chromosome breakage (3.4‐fold higher), accompanied by spontaneous cancer development. Heterozygous BLM+/− mice produce 50% of the normal BLM protein levels and also exhibit increased SCEs (1.5‐fold higher than normal), chromosome breakage (1.3‐fold higher), and cancer susceptibility in the Apc min mouse.
Break and rejoin: topoisomerases and topoisomerase inhibitors in cancer
Topoisomerases are ubiquitous enzymes that are also conserved from bacteria to man. They function to maintain normal DNA topology, a critical requirement in any cellular pathway that involves separating the DNA strands, including DNA replication, recombination, and RNA transcription, and they are involved in chromosome mechanics (i.e. condensation and decondensation of the chromosomes, a process that takes place before and after both meiosis and mitosis) Citation36. The catalytic function of these enzymes is to relax positive or negative DNA supercoiling. There are three classes of topoisomerases: types IA, IB, and II. Type I topoisomerases include the bacterial and eukaryotic topoisomerases I and III. These enzymes possess a monomeric structure and are ATP‐independent in their ability to relax DNA by introducing a single‐strand break, passing a single DNA strand through the break, and subsequently rejoining the DNA. Type I topoisomerases have been further subdivided, based upon which side of the DNA break the enzyme binds as it forms its catalytic intermediate, referred to as the cleavage complex Citation37. The designation of type IA indicates that the enzyme is covalently bound to the 5′ end of the cleaved DNA, whereas type IB denotes that the enzyme is covalently bound to the 3′ end of cleaved DNA. Type II topoisomerases include bacterial DNA gyrase and topoisomerase IV and eukaryotic topoisomerase II. They are homodimeric proteins and function similarly to type I class except they introduce a double strand break in an ATP‐dependent manner Citation37. Moreover, this class of enzymes can catenate/decatenate, knot/unknot, and introduce supercoils into DNA. To date, seven topoisomerase genes have been identified in eukaryotes: the type I topoisomerases TOP1, TOP1mt, TOP3A, and TOP3B, and the type II topoisomerases TOP2A, TOP2B, and SPO11Citation38. These enzymes can have specialized functions. For instance, relaxation of DNA supercoiling that arises during RNA transcription in catalyzed by topoisomerase I, and DNA double strand breaks that initiate recombination during meiosis are catalyzed by SPO11. In mammals, the TOP1 and TOP3A genes are essential.
Given that the normal function of topoisomerases is to introduce breaks in DNA, an attractive hypothesis is that drugs that interfere with the normal function of topoisomerases by stabilizing the DNA damage introduced by the transient cleavage complexes should be potent anticancer agents. Indeed, it has been recently shown that this is the mechanism of action of agents long used as chemotherapeutics, such as camptothecin and the anthracyclines. Topoisomerase inhibitors are divided into two classes. Class I drugs are referred to as topoisomerase poisons, because, by trapping the enzyme as it cleaves DNA, they transform it into a potent apoptosis‐inducing cellular toxin. In contrast, class II drugs interfere with the normal enzymatic function of type II topoisomerases, but do not stabilize the enzyme‐DNA complex.
The prototypical inhibitor of type I topoisomerases is camptothecin Citation38. It and its related water‐soluble derivatives topotecan and irinotecan are unique in that their only known target is topoisomerase I. They rapidly permeate cells, and bind to the topoisomerase I cleavage complex. Because of the reversible nature of this interaction, however, prolonged infusions are required for them to maintain persistently the topoisomerase I cleavage complexes. These transient complexes are converted into irreversible topoisomerase I‐DNA covalent complexes and then into double strand breaks by replication that interact with the cleavage complexes when migrating replication forks collide with them. Hence, cells are only sensitive to camptothecin‐family‐mediated cytotoxicity during S phase Citation39. Clinically, irinotecan has been shown to be active in colon cancer, as well as breast, gastric, and small cell lung cancers, whereas topotecan is used primarily in the treatment of ovarian cancer, head and neck cancer, and malignant gliomas. The clinical use of the parental compound, camptothecin, has been limited by significant toxicities, primarily hemorrhagic cystitis and profound myelosuppression. These are significantly improved in its derivatives, whose primary toxicities are bone marrow suppression and diarrhea.
Drugs that inhibit type II topoisomerases by stabilizing the cleavage complex are divided into two categories: DNA intercalating agents and nonintercalating agents Citation37. Intercalators include acridine compounds, anthracyclines, actinomycin antibiotics, and ellipticines. They are used in the treatment of a variety of cancers and are thought to act by inducing the unwinding of the DNA helix and concomitant generation of DNA strand breaks. Nonintercalators include the epipodophyllotoxins (etoposide) and the isoflavodins. While the mechanism by which the isoflavodins act remains controversial, the epipodophyllotoxins are comprised of two domains, one of which binds to type II topoisomerases, and the other of which binds DNA. Together, they act to inhibit the resealing of the type II topoisomerase‐induced double strand break. Class 2 drugs that inhibit the catalytic activity of type II topoisomerases but do not stabilize the enzyme‐DNA complex are also used as anticancer agents. These include the coumarins, fostriecin analogues, and other agents. Because the expression of type II topoisomerases is cell‐cycle‐dependent, it is expressed at higher levels in proliferating cells than in quiescent cells. Consequently, proliferating cells are more sensitive to these drugs than are quiescent cells.
A devastating complication of treatment with type II topoisomerase inhibitors, in particular the epipodophyllotoxins and anthracyclines, is therapy‐induced acute myelogenous leukemia (t‐AML) Citation40. Although t‐AML is induced by other agents as well, topoisomerase type II inhibitor‐induced t‐AML is characterized by chromosomal rearrangements involving the MLL or AML1 genes, a latency period of less than 2 years, and a dismal prognosis. Given that these drugs act reversibly to stabilize the cleavage complex, it is not surprising that, for etoposide, the risk is associated primarily with dosing schedule rather than cumulative dose.
Progress in the use of topoisomerase inhibitors to treat cancer will very likely proceed on several fronts. First, new topoisomerase I inhibitors unrelated to camptothecin are being developed, with a wider range of activity, a better pharmacokinetic profile, and fewer toxicities. Second, genetic biomarkers are being employed to identify patients at risk for the toxic side effects of these drugs, such as polymorphic variants in genes encoding UDP‐glucuronosyl transferase 1A1 isozymes that catalyze the inactivation of irinotecan Citation41. Third, rational drug combinations will be explored that maximize the synergistic effects of topoisomerase inhibitors with other DNA‐damaging agents. Finally, agents to overcome resistance to these drugs will be developed, one recent example of which is the use of imatinib mesylate to reverse irinotecan insensitivity Citation42.
DNA topology and genomic integrity: topoisomerase IIIα and BLM
Yeast cells that harbor a null mutation in their topoisomerase 3 gene (top3) grow slowly (due to partial lethality) and exhibit excessive recombination and chromosome loss Citation43,44. The sole recQ gene in budding yeast SGS1 was first identified as a suppressor of the slow growth phenotype characteristic of top3Δ cells Citation45, that is, cells that are double mutant sgs1Δ/top3Δ grow better than top3Δ cells. Although the phenotypes are not as extreme as in top3Δ cells, sgs1Δ cells also exhibit elevated levels of recombination and hypersensitivity to the DNA‐damaging agent MMS. These observations led to the discovery that the Sgs1 and Top3 proteins physically interact Citation45.
The importance of the RecQ and topoisomerase interaction is evidenced by its conservation in many species. In E. coli, RecQ functionally interacts with topoisomerase 3 to catenate and decatenate covalently closed plasmid DNA molecules, going through a hemicatenated DNA intermediate Citation46. In fission yeast, its sole RecQ helicase Rqh1 also interacts with Top3, and mutation of rqh1 suppresses the lethal phenotype of the top3 null mutation. How the RecQ mutation suppresses the top3 mutation is unclear, but hypothetically Top3 depends on the RecQ helicase for substrate, and generation of the substrate without resolution by Top3 produces an unresolvable DNA intermediate. Finally, human BLM interacts with topoisomerase IIIα Citation47.
In addition, there is conservation in sequences that mediate the interaction. In budding yeast, the region of Sgs1 that mediates the interaction with Top3 is in the N‐terminal 45 amino acids. In humans, it is in the N‐terminal 133 amino acids of BLM, and in fission yeast in the N‐terminal 322 amino acids of Rqh1. Amino acid homologies within these regions are observed. In yeasts, suppression of the top3 phenotypes occurs in the presence of an SGS1 gene that is deleted or mutated in its Top3‐interaction region.
As mentioned above, in human cells, both BLM and topoisomerase IIIα are focally concentrated in the PML‐NBs. However, in BLM‐deficient BS cells, topoisomerase IIIα is not in PML‐NBs. Ectopic expression in BS cells of BLM causes topoisomerase IIIα to relocalize to the PML‐NBs, but expression of an interaction incompetent form of BLM fails to cause relocalization, indicating that BLM recruits topoisomerase IIIα to the PML‐NBs. Furthermore, expression of the interaction incompetent BLM is unable to restore fully the number of SCEs to normal levels, suggesting that the interaction between the two proteins is important for efficient maintenance of genome integrity.
The BLM/topoisomerase IIIα complex has the ability to resolve substrates that resemble double Holliday junctions—an activity that neither protein has on its own Citation48. The double Holliday junction is an intermediate that is thought to arise during the recombinational repair of double strand breaks. Thus, one interpretation of the genetic and biochemical data is that the RecQ helicase‐topoisomerase 3 complex specifically processes the Holliday junctions that arise during the repair of stressed replication forks. In the absence of the RecQ helicase, as in BS or sgs1 cells, this substrate can be resolved by other proteins but it leads to crossing‐over (e.g., SCEs). Thus, based on the sensitivity of BS cells to damaging agents and the efficacy of topoisomerase inhibitors as anticancer therapeutic agents, we suggest that targeting the BLM‐topoisomerase IIIα interaction for pharmaceutical inhibition could enhance the efficacy of topoisomerase inhibitors in cancer treatment.
Concluding remarks
The association between genomic instability and cancer susceptibility has been amply established by the study of RecQ DNA helicases in human genetic syndromes in which these proteins are absent. RecQ helicases interact physically and functionally with many other DNA repair proteins at the sites of DNA damage, and they are important in DNA damage signaling. Reduced RecQ protein levels (for example, in heterozygotes) partially compromises repair functions and increases cancer susceptibility. One of RecQ's important functions in repair is in DNA replication‐associated recombination, which is a mechanism that can repair DNA double strand breaks and can restart stalled replication forks. RecQ's interactions with topoisomerase 3s, which are important in these repair functions, provide novel targets for chemotherapy.
Acknowledgements
The authors apologize to the many investigators whose work we did not appropriately reference in this review due to space limitations.
References
- Nakayama H. RecQ family helicases: roles as tumor suppressor proteins. Oncogene 2002; 21: 9008–21
- Macris M. A., Krejci L., Bussen W., Shimamoto A., Sung P. Biochemical characterization of the RECQ4 protein, mutated in Rothmund‐Thomson syndrome. DNA Repair (Amst) 2005; 5: 172–80
- Nakayama K., Irino N., Nakayama H. The recQ gene of Escherichia coli K12: molecular cloning and isolation of insertion mutants. Mol Gen Genet 1985; 200: 266–71
- Kolodner R., Fishel R. A., Howard M. Genetic recombination of bacterial plasmid DNA: effect of RecF pathway mutations on plasmid recombination in Escherichia coli. J Bacteriol 1985; 163: 1060–6
- Courcelle J., Hanawalt P. C. RecQ and RecJ process blocked replication forks prior to the resumption of replication in UV‐irradiated Escherichia coli. Mol Gen Genet 1999; 262: 543–51
- Hanada K., Ukita T., Kohno Y., Saito K., Kato J., Ikeda H. RecQ DNA helicase is a suppressor of illegitimate recombination in Escherichia coli. Proc Natl Acad Sci U S A 1997; 94: 3860–5
- Flores M. J., Sanchez N., Michel B. A fork‐clearing role for UvrD. Mol Microbiol 2005; 57: 1664–75
- Myung K., Datta A., Chen C., Kolodner R. D. SGS1, the Saccharomyces cerevisiae homologue of BLM and WRN, suppresses genome instability and homeologous recombination. Nat Genet 2001; 27: 113–6
- Stewart E., Chapman C. R., Al‐Khodairy F., Carr A. M., Enoch T. rqh1+, a fission yeast gene related to the Bloom's and Werner's syndrome genes, is required for reversible S phase arrest. EMBO J 1997; 16: 2682–92
- Yamagata K., Kato J., Shimamoto A., Goto M., Furuichi Y., Ikeda H. Bloom's and Werner's syndrome genes suppress hyperrecombination in yeast sgs1 mutant: implication for genomic instability in human diseases. Proc Natl Acad Sci U S A 1998; 95: 8733–8
- German J. Bloom's syndrome. Dermatol Clin 1995; 13: 7–18
- German J., Ellis N. A. Bloom Syndrome. The genetic basis of human cancer, B Vogelstein, K. W Kinzler. McGraw‐Hill, New York 2002; 267–88
- Imamura O., Fujita K., Shimamoto A., Tanabe H., Takeda S., Furuichi Y., et al. Bloom helicase is involved in DNA surveillance in early S phase in vertebrate cells. Oncogene 2001; 20: 1143–51
- Groden J., Nakamura Y., German J. Molecular evidence that homologous recombination occurs in proliferating human somatic cells. Proc Natl Acad Sci U S A 1990; 87: 4315–9
- German J., Sanz M. M., Ciocci S., Ye T. Z., Ellis N. A. Syndrome‐causing mutations in the BLM gene in persons in the Bloom's Syndrome Registry. Hum Mutat 2007, (in press)
- Neff N. F., Ellis N. A., Ye T. Z., Noonan J., Huang K., Sanz M., et al. The DNA helicase activity of BLM is necessary for the correction of the genomic instability of bloom syndrome cells. Mol Biol Cell 1999; 10: 665–76
- Dutertre S., Ababou M., Onclercq R., Delic J., Chatton B., Jaulin C., et al. Cell cycle regulation of the endogenous wild type Bloom's syndrome DNA helicase. Oncogene 2000; 19: 2731–8
- Dellaire G., Bazett‐Jones D. P. PML nuclear bodies: dynamic sensors of DNA damage and cellular stress. Bioessays 2004; 26: 963–77
- Matunis M., Zhang X. D., Ellis N. A. SUMO: The Glue that Binds. Dev Cell 2006; 11: 596–7
- Wang Z. G., Delva L., Gaboli M., Rivi R., Giorgio M., Cordon‐Cardo C., et al. Role of PML in cell growth and the retinoic acid pathway. Science 1998; 279: 1547–51
- Zhong S., Hu P., Ye T. Z., Stan R., Ellis N. A., Pandolfi P. P. A role for PML and the nuclear body in genomic stability. Oncogene 1999; 18: 7941–7
- Hu P., Beresten S. F., van Brabant A. J., Ye T. Z., Pandolfi P. P., Johnson F. B., et al. Evidence for BLM and Topoisomerase IIIalpha interaction in genomic stability. Hum Mol Genet 2001; 10: 1287–98
- Davalos A. R., Campisi J. Bloom syndrome cells undergo p53‐dependent apoptosis and delayed assembly of BRCA1 and NBS1 repair complexes at stalled replication forks. J Cell Biol 2003; 162: 1197–209
- Wu L., Bachrati C. Z., Ou J., Xu C., Yin J., Chang M., et al. BLAP75/RMI1 promotes the BLM‐dependent dissolution of homologous recombination intermediates. Proc Natl Acad Sci U S A 2006; 103: 4068–73
- Rogakou E. P., Boon C., Redon C., Bonner W. M. Megabase chromatin domains involved in DNA double‐strand breaks in vivo. J Cell Biol 1999; 146: 905–16
- Celeste A., Petersen S., Romanienko P. J., Fernandez‐Capetillo O., Chen H. T., Sedelnikova O. A., et al. Genomic instability in mice lacking histone H2AX. Science 2002; 296: 922–7
- Garcia‐Higuera I., Taniguchi T., Ganesan S., Meyn M. S., Timmers C., Hejna J., et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 2001; 7: 249–62
- Meetei A. R., de Winter J. P., Medhurst A. L., Wallisch M., Waisfisz Q., van de Vrugt H. J., et al. A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet 2003; 35: 165–70
- Davalos A. R., Kaminker P., Hansen R. K., Campisi J. ATR and ATM‐dependent movement of BLM helicase during replication stress ensures optimal ATM activation and 53BP1 focus formation. Cell Cycle 2004; 3: 1579–86
- Sengupta S., Linke S. P., Pedeux R., Yang Q., Farnsworth J., Garfield S. H., et al. BLM helicase‐dependent transport of p53 to sites of stalled DNA replication forks modulates homologous recombination. EMBO J 2003; 22: 1210–22
- Meetei A. R., Sechi S., Wallisch M., Yang D., Young M. K., Joenje H., et al. A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol 2003; 23: 3417–26
- Yin J., Sobeck A., Xu C., Meetei A. R., Hoatlin M., Li L., et al. BLAP75, an essential component of Bloom's syndrome protein complexes that maintain genome integrity. EMBO J 2005; 24: 1465–76
- Langland G., Kordich J., Creaney J., Goss K. H., Lillard‐Wetherell K., Bebenek K., et al. The Bloom's syndrome protein (BLM) interacts with MLH1 but is not required for DNA mismatch repair. J Biol Chem 2001; 276: 30031–5
- Liberi G., Maffioletti G., Lucca C., Chiolo I., Baryshnikova A., Cotta‐Ramusino C., et al. Rad51‐dependent DNA structures accumulate at damaged replication forks in sgs1 mutants defective in the yeast ortholog of BLM RecQ helicase. Genes Dev 2005; 19: 339–50
- McDaniel L. D., Chester N., Watson M., Borowsky A. D., Leder P., Schultz R. A. Chromosome instability and tumor predisposition inversely correlate with BLM protein levels. DNA Repair (Amst) 2003; 2: 1387–404
- Nitiss J. L. Investigating the biological functions of DNA topoisomerases in eukaryotic cells. Biochim Biophys Acta 1998; 1400: 63–81
- Topcu Z. DNA topoisomerases as targets for anticancer drugs. J Clin Pharm Ther 2001; 26: 405–16
- Pommier Y. Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer 2006; 6: 789–802
- Del Bino G., Lassota P., Darzynkiewicz Z. The S‐phase cytotoxicity of camptothecin. Exp Cell Res 1991; 193: 27–35
- Baldwin E. L., Osheroff N. Etoposide, topoisomerase II and cancer. Curr Med Chem Anticancer Agents 2005; 5: 363–72
- Maitland M. L., Vasisht K., Ratain M. J. TPMT, UGT1A1 and DPYD: genotyping to ensure safer cancer therapy?. Trends Pharmacol Sci 2006; 27: 432–7
- Johnson F. M., Krug L. M., Tran H. T., Shoaf S., Prieto V. G., Tamboli P., et al. Phase I studies of imatinib mesylate combined with cisplatin and irinotecan in patients with small cell lung carcinoma. Cancer 2006; 106: 366–74
- Wallis J. W., Chrebet G., Brodsky G., Rolfe M., Rothstein R. A hyper‐recombination mutation in S. cerevisiae identifies a novel eukaryotic topoisomerase. Cell 1989; 58: 409–19
- Bailis A. M., Arthur L., Rothstein R. Genome rearrangement in top3 mutants of Saccharomyces cerevisiae requires a functional RAD1 excision repair gene. Mol Cell Biol 1992; 12: 4988–93
- Gangloff S., McDonald J. P., Bendixen C., Arthur L., Rothstein R. The yeast type I topoisomerase Top3 interacts with Sgs1, a DNA helicase homolog: a potential eukaryotic reverse gyrase. Mol Cell Biol 1994; 14: 8391–8
- Harmon F. G., DiGate R. J., Kowalczykowski S. C. RecQ helicase and topoisomerase III comprise a novel DNA strand passage function: a conserved mechanism for control of DNA recombination. Mol Cell 1999; 3: 611–20
- Wu L., Davies S. L., North P. S., Goulaouic H., Riou J. F., Turley H., et al. The Bloom's syndrome gene product interacts with topoisomerase III. J Biol Chem 2000; 275: 9636–44
- Wu L., Hickson I. D. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature 2003; 426: 870–4
- Bernstein D. A., Zittel M. C., Keck J. L. High‐resolution structure of the E. coli RecQ helicase catalytic core. EMBO J 2003; 22: 4910–21
- Wu L., Chan K. L., Ralf C., Bernstein D. A., Garcia P. L., Bohr V. A., et al. The HRDC domain of BLM is required for the dissolution of double Holliday junctions. EMBO J 2005; 24: 2679–87
- Huber M. D., Duquette M. L., Shiels J. C., Maizels N. A conserved G4 DNA binding domain in RecQ family helicases. J Mol Biol 2006; 358: 1071–80