Abstract
Hypertension is a major risk factor for atherosclerosis. We tested the hypothesis whether high salt intake aggravates endothelial dysfunction and promotes atherosclerosis in apolipoprotein E‐deficient mice (ApoE−/− mice) and their littermate controls (C57Bl/6 mice). The role of increased oxidative stress was also examined. A high‐salt diet (NaCl 7%) for 12 weeks increased blood pressure and induced cardiac hypertrophy and albuminuria more pronouncedly in ApoE−/− mice compared with C57Bl/6. Endothelium‐dependent vascular relaxation in response to acetylcholine was almost maximally impaired in ApoE−/− mice during a normal sodium diet. A high‐salt diet did not further impair NO‐mediated vascular relaxation. A high‐salt diet also markedly attenuated endothelium‐dependent relaxation in C57Bl/6 mice. Preincubation with the superoxide scavenger Tiron normalized endothelial function almost completely in both mice strains indicating the central role of increased oxidative stress in the pathogenesis. Aortic superoxide production and the extent of atherosclerotic lesions were greater in ApoE−/− mice on a normal‐salt diet compared with C57Bl/6. The high‐salt diet increased vascular superoxide formation and promoted atherosclerosis in ApoE−/− mice. Changes in dietary salt intake did not influence serum lipids in either mouse strains. Our findings suggest a detrimental role for high salt intake in the development of atherosclerosis and underscore the importance of increased oxidative stress in the pathogenesis salt‐induced vascular damage.
Introduction
High blood pressure is one of the most important risk factors in the development of atherosclerosis and its related cardiovascular diseases. Compelling evidence suggests a pivotal role for high sodium intake in the pathogenesis of essential hypertension (for review see ref. 1). Although the molecular mechanisms of salt‐induced hypertension are only partially understood, several studies support the notion that disturbances in the regulatory systems altering renal pressure‐natriuresis, and thus renal net sodium excretion, are involved Citation[1]. Interestingly, recent studies by us Citation[2] and others have revealed that high sodium intake may induce vascular damage even by blood‐pressure independent mechanisms Citation[3–5].
Chronic high salt (NaCl) intake exerts harmful effects on cardiovasculature in addition to increasing arterial pressure Citation[6]. The major contributing factor to the adverse effects of a high‐salt diet has been suggested to be via hypertension‐induced oxidative stress with concomitant local vascular expression of angiotensin II and endothelin‐1 Citation[7], Citation[8]. Hypertension plays a pivotal role in the pathogenesis of atherosclerosis by accelerating endothelial dysfunction, which has been established in man and animal models Citation[9], Citation[10]. Murine studies have also demonstrated the detrimental role of a high‐salt diet on blood pressure and vascular function, which occur within a few days Citation[11], Citation[12]. Treatment with antioxidant apocynin or the lipid‐lowering drug atorvastatin has been shown to normalize salt‐induced albuminuria, vascular‐derived oxidative stress and endothelial dysfunction but without a strong effect on blood pressure Citation[13]. Thus, a high‐salt diet may affect directly the formation of vascular‐derived superoxide anions (O2−), independent of blood pressure. In addition to this, a recent in vitro study in renal cell culture has provided information that DNA repair is markedly impaired after exposition to high NaCl concentration Citation[14]. The superoxide anion is an oxygen‐derived radical naturally formed predominantly by NADPH oxidase and it can rapidly react with nitric oxide (NO) to scavenge it and further produce peroxynitrite and peroxynitrous acid, which is toxic to the vascular endothelial cell Citation[15].
Endothelial nitric oxide synthase (eNOS) is the main housekeeping enzyme secreting NO, which is continuously activated by laminar blood flow‐induced shear stress. NO is the main regulatory anti‐adhesive and anti‐inflammatory molecule and the changes in its production or half‐life are associated to smooth muscle hypertrophy and endothelial dysfunction. Genetically deficient mice for eNOS or those treated with N‐nitro‐l‐arginine exhibit endothelial dysfunction and develop hypertension along with increased atherosclerosis Citation[9]. They also show increased cardiac oxidative stress and upregulated vascular angiotensin‐converting enzyme activity (ACE) Citation[16]. Interestingly, high‐salt diet may decrease NO bioavailability by directly uncoupling eNOS, thus resulting in sustained arterial contraction and tissue hypoxia Citation[13].
Apolipoprotein E‐deficient (ApoE−/−) mice is a well‐established model for atherosclerosis. The ApoE−/− mouse develops spontaneous hypercholesterolemia, renal dysfunction and human‐like atherosclerotic lesions throughout the aortic tree Citation[17–20]. The C57Bl/6 mouse in turn is a susceptible model for high‐fat high‐cholesterol diet‐induced type II diabetes and salt‐sensitive hypertension Citation[21]. Furthermore, the results from the ApoE−/− mouse model have demonstrated the connection between impaired NO bioavailability, oxidative stress and endothelial dysfunction. Additionally, the mouse aortic tree is highly regulated by endothelial‐derived NO Citation[22], which further highlights this animal model being a rational option to investigate the interaction between oxidative stress and NO metabolism. Among the environmental factors, the role of high salt intake in the pathogenesis of atherosclerosis is only poorly understood. To the best of our knowledge, the potential link between salt‐induced hypertension and atherosclerosis has not been fully established. Accordingly, the present study was designed to further elucidate the role of dietary salt intake on atherosclerosis and endothelial function in the ApoE−/− mouse. We now show that dietary salt is a remarkable factor for the development of cardiovascular pathologies in the ApoE−/− knockout mouse.
Materials and methods
Experimental animals, diets and sample preparation
Mice of the C57BL/6J background carrying targeted deletions of the apolipoprotein E gene (ApoE−/−; Jackson, Bar Harbor, ME, USA) and wild‐type (C57Bl/6) controls were used in this study. In the beginning of the study, 10–12‐week‐old male ApoE−/− mice and C57Bl/6 were divided into four groups (n = 10 in each group) to receive different dietary regimens for 12 weeks: (i) C57Bl/6 mice on a normal‐salt diet ((NS), NaCl 0.8% wt/wt); (ii) C57Bl/6 mice on a high‐salt diet ((HS) NaCl 7% wt/wt)); (iii) ApoE−/− mice on a NS diet; and (iv) ApoE−/− mice on a HS diet. The animals were housed in an animal laboratory (illuminated from 06.30 to 18.30 h, room temperature 22–24°C) and had free access to food and tap water. At the end of the experimental period, urine samples were collected over 24 h in metabolic cages. The mice were anesthetized by gas containing CO2/O2 (70%/30%) and the blood samples were collected from orbital puncture into heparinized tubes, centrifuged and stored at −20°C for analysis. Then the mice were euthanized by cervical dislocation and tissues were harvested. The heart and kidneys were excised, washed with ice‐cold saline, blotted dry and weighed. Tissue samples for histology were snap‐frozen in liquid nitrogen (−70°C). All samples were stored at −80°C until were assayed. For measurement of vascular responses and for superoxide measurement, the aorta was carefully dissected from the adherent connective tissue. The aortic samples for the determination of endothelial function were taken 2 mm below the origin of left subclavian artery and samples for the superoxide measurement were taken from the descending abdominal aorta. The protocols were approved by the Animal Experimentation Committee of the University of Helsinki, Helsinki, Finland.
Blood pressure measurement
At the end of the study, systolic blood pressure was measured non‐invasively using a tail‐cuff method (blood pressure analyzer: Apollo‐2AB Blood Pressure Analyzer, model 179‐2AB; IITC Life Science, Woodland Hills, CA, USA) on conscious, restrained mice. Briefly, the mice were allowed to adapt to restrainers and blood pressure measurement for 10 days. At week 12, the mean systolic blood pressure was calculated from 5–10 successful recordings. Blood pressure was measured by the same technician without the knowledge of the mice identity.
Assessment of endothelial function
Aortic relaxation responses were analyzed from aortic samples as previously described Citation[23]. Briefly, aortic rings 2–3 mm wide were connected to an isometric force transducer (EMKA technologies, France, 2000), suspended in an organ chamber filled with a Krebs solution of the following composition (mmol/l): NaCl 119.0, NaHCO3 25.0, glucose 11.1, CaCl2 1.6, KCl 4.7, KH2PO4 1.2, MgSO4 1.2 (37°C, pH 7.4) and aerated continuously with 95% O2/5% CO2. Isometric tension was recorded continuously. After a 60‐min equilibration period, rings were gradually stretched to the optimal point of their length‐tension curve as being determined to 0.7 g. Organ chambers experiments were carried out using mouse aortic rings preconstricted with noradrenaline 1 µmol/l noradrenaline (NA). Endothelium‐dependent relaxation to acetylcholine (Ach) was recorded before and after preincubation with the superoxide scavenger Tiron (1 mmol/l; 15). Endothelium‐independent relaxations were studied in response to sodium nitroprusside (SNP). Curves were produced and values were calculated using the Graph Pad Prism version 4.02 software program.
Measurement of superoxide production
Aortic superoxide formation was determined as described previously Citation[24–26]. Briefly, aortic rings (4 mm in width) were equilibrated in Krebs solution (pH 7.4, 37°C) aerated with oxygen (95%/5%; O2/CO2) for 30 min and then transferred into vials filled with Krebs enriched with lucigenin (5 µmol/l). The aortic rings were incubated in a 96‐well plate for 30 min in the dark and at 37°C. Thereafter superoxide was calculated from chemiluminesce measurements (Liquid Scintillation Counter, 1450 MicroBeta, Wallac, Boston, MA, USA). After the measurements, the aortic samples were dried and weighed, and the results were reported as counts/mg/min. The specificity of lucigenin to assess aortic O2− generation was confirmed by preincubation of aortic rings with 4,5‐dihydroxy‐1,3‐benzenedisulfonic acid 10 mmol/l (Tiron) that resulted in a 90% reduction in O2− measurement (data not shown). Tiron is a cell‐permeant, non‐enzymatic scavenger of superoxide anion, which was added to quench all superoxide‐dependent chemiluminescence Citation[24], Citation[25].
Assessment of atherosclerotic lesion
The cross‐sectional analysis of the extent of atherosclerosis was conducted from aortic sinus along the ascending aorta up to 5 mm according to previous study Citation[27]. The aortic samples were sectioned on a Leica cryostat (10 µm). Cross‐sectional serial sections were prepared and stained with hematoxylin and eosin to evaluate the atherosclerotic lesion area. The aortic lesions were quantified as percentage lesion area per total area of the cross‐sectional aorta. The percentage of area covered with fatty lesions were examined by light microscopy and analyzed by Leica imaging software.
Biochemical determinations
Albuminuria was measured by commercially available ELISA kit with mouse albumin used as a standard (CellTrend GmbH, Germany). Urinary nitrate plus nitrite (NOx) concentration was assessed with commercially available NOx colorimetric assay kit (Cayman Chemical Company, Ann Arbor, MI, USA). Serum lipids were analyzed by accredited laboratory (Helsinki University Hospital Laboratory, Finland; Hitachi 912 Automatic Analyzer, Hitachi, Tokyo, Japan). Serum total cholesterol was determined with an enzymatic method (Boehringer Mannheim CHOD‐PAP‐method), serum high‐density lipoprotein (HDL) cholesterol with an enzymatic direct method (Boehringer Mannheim HDL‐Chol Plus) and serum triglycerides with an enzymatic method (Boehringer Mannheim CHOD‐PAP‐method).
Drugs
Acetylcholine, eosin, hematoxylin, noradrenaline, sodium nitroprusside, tiron and chemical components of the Krebs solution: NaCl, NaHCO3, glucose, KCl, CaCl2, MgSO4,KH2PO4 were obtained from Sigma Chemical Company (St Louis, MO, USA). All concentrations used in organ chamber experiments are expressed as final molar concentration in the bath solutions.
Statistical analysis
All values are expressed as mean±SEM. Data were analyzed using one‐way analysis of variance (ANOVA) followed by the Tukey's test. The quantification of atherosclerotic lesion size was performed by unpaired Student's t‐test. Data were considered significantly different at p<0.05.
Results
Blood pressure, cardiac hypertrophy and albuminuria
There were no differences in blood pressure (BP) between mouse strains when kept on a NS diet. A HS diet increased BP in both mice strain. Salt‐induced increase in BP was more pronounced in ApoE−/− (Figure ).
Figure 1 Bar graphs showing the effects of high sodium diet on systolic blood pressure measured by non‐invasive tail‐cuff method (A), heart weight‐to‐body weight ratio (B) and albuminuria (C) in ApoE−/− and in C57Bl/6 mice at the end of the 12‐week‐treatment period. NS denotes normal‐salt and HS high‐salt diets. Means±SEM are given (n = 6 in each group). *p<0.05.
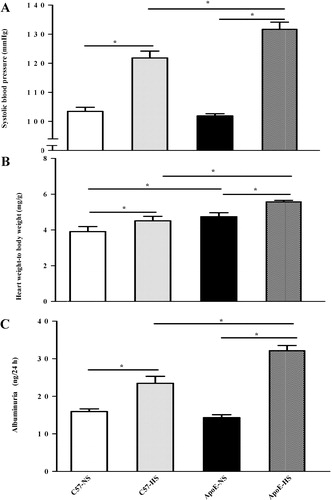
Opposite to this, there were no significant differences between mouse strains in heart rates (beats/min) in either HS or NS groups (ANOVA p = 0.67; ApoE−/− on HS and NS diets 402±10 vs 407±10 beats/min, respectively; C57Bl/6 on HS and NS diets 409±23 vs 461±11 beats/min, respectively).
During the NS diet, left ventricular hypertrophy (LVH), measured as heart weight‐to‐body weight ratio, was increased in ApoE−/− mice compared with C57Bl/6 mice. HS induced further LVH in both mice strains; however, the effect was more pronounced in ApoE−/− mice (Figure ).
There was no difference between the mice strains in 24‐h albuminuria during NS. HS increased albuminuria in both strains but more pronouncedly in ApoE−/− mice (Figure ).
Arterial function
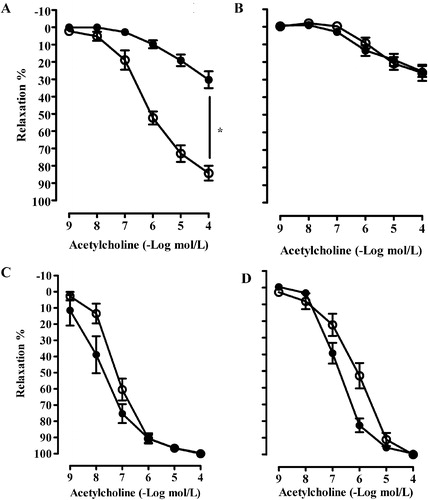
Endothelial function was impaired in ApoE−/− mice compared with C57Bl/6 during NS. The endothelium‐mediated aortic relaxation was aggravated by HS in C57Bl/6, but HS did not further impair the vascular function in ApoE−/− mice (Figure and ). Preincubation with superoxide scavenger Tiron (1 mmol/l) normalized the impaired relaxation response to acetylcholine in both strains (Figure and ). There were no changes in endothelium‐independent relaxations to sodium nitroprusside in both strains and dietary groups (data not shown).
Figure 2 Relaxations to acetylcholine after precontraction with 1 µmol/l noradrenaline in isolated intact aortic rings from C57Bl/6 mice (A) and ApoE−/− mice (B). Open circles denote normal‐salt (NS) diet and solid circles indicate high‐salt (HS) diet. Means±SEM are given (n = 5–6 in each group). *p<0.05.
Superoxide production
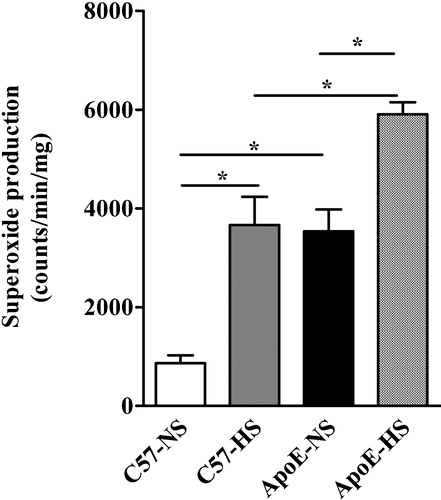
There was a marked elevation in aortic superoxide production in ApoE−/− mice during NS. HS increased aortic superoxide production in both mice strains but this effect was more pronounced in ApoE−/− mice (Figure ).
Figure 3 Determination of the vascular superoxide production from aortic rings in ApoE−/− and C57Bl/6 mice by lucigenin enhanced chemiluminescence assay. NS denotes normal salt and HS denotes high salt. Means±SEM are given (n = 5–6 in each group). *p<0.05.
Extent of atherosclerosis
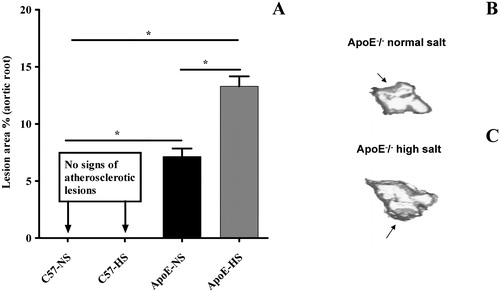
Atherosclerotic lesions were completely absent in C57Bl/6 mice fed either on NS or HS diets (Figure ) There were only modest fatty lesions in ApoE−/− mice fed on NS (Figure ) but these lesions of aortic area in ApoE−/− mice were significantly increased during HS (Figure ).
Figure 4 Bar graph showing the cross‐sectional analysis of aortic atherosclerotic lesions in C57Bl/6 and ApoE−/− mice (Panel A) after 12 weeks on different dietary regimens. High‐salt diet promoted atherosclerosis in ApoE−/− mice. Panels B and C show representative photomicrographs of the atherosclerotic aortic root from ApoE−/− mice receiving normal‐salt and high‐salt diet, respectively. Black arrows indicate the lesions area. There were no fatty streaks in C57Bl/6 mice fed either a normal‐salt (NS) or high‐salt (HS) diet. Means±SEM are given (n = 5–8 in each group). *p<0.05.
Tissue weights and biochemical determinations
Body weight, kidney weight, urine volume and biochemical markers from urine and serum from both mouse strains with dietary regimens are shown in Table . During the NS diet, there was a tendency to higher kidney weight‐to‐body weight ratio (kidney index) in ApoE−/− mice compared with controls. The HS diet increased relative kidney weight in both mice strains but the increase was more pronounced in ApoE−/− mice. As an indirect marker of sodium intake, HS lead to elevated urine volumes compared with NS in both mouse strains, but there were no statistical differences in urine volumes between ApoE−/− mice and C57Bl/6 mice during NS. Urinary nitric oxide metabolites nitrites/nitrates (NOx) remained similar independently of mouse strain or diet. Serum total cholesterol values were approximately three times higher in ApoE−/− mice compared with C57Bl/6 mice during NS. HS did not change the total cholesterol values in either of the mouse strains. During NS there were also a tendency toward higher HDL‐cholesterol concentrations in ApoE−/− mice compared with C57Bl/6, but HS did not have effect on these values. Finally, there were no changes in serum triglyceride concentrations between mouse strains in the presence or in the absence of an HS diet.
Table I. Body and kidney weights, urine volumes, urinary excretion of nitric oxide metabolites (NOx), and serum lipid profiles levels in C57Bl/6 (C57) mice and Apolipoprotein E‐deficient (ApoE−/−) mice after 12 weeks on different dietary regimens.
Discussion
Previous studies have provided evidence that high sodium intake may induce vascular damage by mechanisms independent of blood pressure Citation[2], Citation[12], Citation[28]. The present study aimed at exploring the influence of HS diet on the development of atherosclerosis and endothelial dysfunction in hyperlipidemic ApoE−/− mice and their normolipidemic controls, C57Bl/6 mice. Furthermore, the pathophysiological role of increased oxidative stress was also examined. The important finding was that HS diet increased vascular superoxide formation in both mice strains, which was associated with a pronounced endothelial dysfunction in C57Bl/6 mice and progression of atherosclerosis in ApoE−/− mice. We here also demonstrate for the first time that high salt intake increases blood pressure, cardiac hypertrophy and albuminuria more pronouncedly in ApoE−/− mice compared with C57/Bl6 mice, indicating a greater salt sensitivity of this mouse strain.
Salt‐sensitive hypertension is associated with impaired forearm vasorelaxation in hypertensive patients due to enhanced endothelium‐derived vasoconstrictors and impaired NO bioavailability Citation[29]. Endothelial dysfunction occurring both in clinical and experimental animal settings demonstrate a hallmark in atherosclerosis characterized by impaired vascular relaxation to acetylcholine via stimulation of the muscarinic receptors in the surface of endothelial cells Citation[10]. Clinically, endothelial dysfunction is also a first pertinent marker of salt‐sensitive hypertension before any end‐organ damage Citation[29]. Nitric oxide (NO) is the main vascular endothelial cell‐derived agent that promotes vasodilatation, inhibits platelets aggregation and prevents adhesion of inflammatory cells on the surface of large arteries Citation[30]. In the present study, we observed an almost complete absence of endothelium‐dependent vascular relaxation in atherosclerotic ApoE−/− mouse receiving the NS diet, whereas the HS diet did not further impair the function of the endothelium. However, the HS diet further increased vascular superoxide formation by 50%, and an additive effect of hyperlipidemia and high salt intake on vascular oxidative stress could be demonstrated. It is of particular interest that, in normolipidemic C57Bl/6 mice, HS diet induced a pronounced impairment of NO‐mediated vascular relaxation and increased the vascular superoxide formation to the same extent as hyperlipidemia in ApoE−/− mice. Furthermore, preincubation with the superoxide scavenger Tiron completely normalized endothelial dysfunction in C57Bl/6 mice. Our findings thus indicate that high salt intake exerts a direct effect on vascular oxidative stress and endothelial function in C57Bl/6 mice via mechanisms independent of serum cholesterol concentration. Due to limited amount of blood available for biochemical determinations, we were unable to confirm oxidative stress by serum 8‐isoprostane Citation[31] and MDA Citation[32] measurements, the two widely used indirect circulating markers of lipid peroxidation and free radical‐catalyzed peroxidation of arachidonic acid, respectively. Taken together, we report here an additive effect of high salt intake and genetic predisposition to atherosclerosis on vascular superoxide formation.
Nitric oxide is a labile species with a half‐life of only a few seconds in vivo. Therefore, the in vivo activity of NO must be monitored indirectly. Nitric oxide is rapidly degraded to unstable nitrite, and further converted to the more stable product nitrate (collectively referred to as NOx). However, the metabolic fate of NO is only partially understood, and some of the intermediate metabolites may also exert biological effects. Furthermore, it is known that NO reacts with superoxide anion to form peroxynitrite, a highly toxic agent to endothelial cell Citation[15]. Irrespective of some methodological limitations, measurement of NOx levels in plasma and urine has been used as indirect markers of whole body NO production Citation[33] and the 24‐h urinary NOx excretion during the highly controlled diet is considered a qualitative measure of total body (both systemic and renal) NO production Citation[33]. In the present study, we noticed a tendency towards lower 24‐h urinary NOx excretion in ApoE−/− mice fed high salt compared with their littermate controls, suggesting decreased total body NO production in response to salt loading. However, during normal salt intake, uninary NOx excretion in ApoE−/− was indistinguishable from C57BL6 mice, even though a fourfold increase vascular superoxide formation and pronounced endothelial dysfunction was found in ApoE−/− mice. Our findings thus indicate that 24‐h urinary NOx excretion cannot be considered a reliable marker of vascular oxidative stress and/or NO bioavailability.
Previous studies have revealed that impaired activity of eNOS, increased formation of endothelial‐derived superoxide, and deficiency in cofactors L‐arginine or tetrahydrobiopterin (BH4), factors all leading to impaired NO bioavailability Citation[17], Citation[23], Citation[34], Citation[35] are involved in the pathogenesis of endothelial dysfunction in ApoE−/− mice. Additionally, previous studies in ApoE−/− mice with concomitant eNOS deficiency, induced either by genetic engineering or chemical inactivation by L‐NAME, have demonstrated marked progression of atherosclerosis and end‐organ damage supporting the crucial role of endothelium‐derived NO Citation[9], Citation[22]. Furthermore, endothelial dysfunction can be considered a reversible phenomenon Citation[13], Citation[27], Citation[35] with enhanced NO degradation by an HS diet. In hypertensive Dahl salt‐sensitive rats, salt‐induced endothelial dysfunction is associated with extracellular volume expansion, severe hypertension, increased formation of angiotensin II in the vascular wall, diminished levels of relaxing factors NO and prostacyclin, and reduction of the tissue glutathione concentration Citation[8], Citation[13].
Although reduction of dietary salt intake is recommended in the prevention and treatment of high blood pressure, it has also been claimed that severe salt restriction, at least acutely, may lead to unwanted side‐effects such activation of the sympathetic nervous system and renin–angiotensin–aldosterone system (RAAS), lipid disorders, and insulin resistance Citation[35]. In fact, recent experimental study by Ivanovski et al. Citation[37] revealed that, in ApoE−/− mice, severe dietary salt restriction (0.036% NaCl) for 16 weeks accelerated atherosclerotic lesions in the thoracix aorta of ApoE−/− mice. Aortic lesions were correlated with plasma angiotensin II and total cholesterol concentrations. However, severe salt restriction did not alter lesion area in the aortic root, suggesting the salt restriction may not affect the established and abundant plaque development that is present in the aortic root of ApoE−/− mice. However, it should be underlined that the dietary salt concentration used in the study by Ivanovski et al. Citation[37] was markedly below the nutritional requirement of mice (0.05% Na corresponding to 0.125% NaCl) Citation[38]. Interestingly, exposure to a less severe salt restriction (NaCl 0.15%) did not accelerate atherosclerotic lesions in low‐density lipoprotein (LDL) receptor deficient mice Citation[39]. Taken together, severe salt restriction may influence atherosclerosis via mechanisms linked to increased cholesterol concentration and angiotensin II formation; however, unlike the high salt intake used in the present study, moderate salt restriction seems to exert only a modest effect in the degree of atherosclerosis and does not influence myocardial or kidney morphology.
The ApoE−/− mouse was the first genetic engineered mouse model that spontaneously develops hypercholesterolemia independent of age and atherosclerotic lesions resembling those found in humans. The first atherosclerotic lesions are characterized by accumulation of foam cells and fatty streaks formation into the aortic root. With increasing age, the vascular lesions could also been found in the distal thoracic aorta Citation[18]. In good accordance with the previous studies we noticed modest atherosclerotic lesions from cross‐sectional samples of the aortic root in the 22–24‐week‐old ApoE−/− but not in the C57Bl/6 mice Citation[18]. The salt‐induced progression of atherosclerosis in ApoE−/− mice is likely to mediated by the cluster of well‐known risk factors for atherosclerosis such as hypertension Citation[17], kidney dysfunction Citation[9], Citation[20] and increase vascular‐derived superoxides Citation[34]. The dietary salt did not influence the plasma total cholesterol, HDL‐cholesterol or triglyceride concentrations; the present study cannot exclude the possibility that the detrimental effects of an HS diet were mediated, at least in part, by increased LDL‐cholesterol oxidation Citation[19].
In ApoE−/− mice, high salt intake for 12 weeks was associated with the salt‐sensitive hypertension and target organ damage such as cardiac hypertrophy and kidney damage. HS diet also increased blood pressure and induced target organ damage in normolipidemic C57Bl/6 mice; however, these effects were less pronounced compared with ApoE−/− mice. Previous studies have demonstrated that salt‐sensitive hypertension may manifest even after few days' dietary period both in mice Citation[11] and in rats Citation[12]. In the present study, an HS diet did not influence heart rate in either of the mice strains. Our findings are thus in agreement with previous studies Citation[11], Citation[21]. Interestingly, 13‐month‐old ApoE−/− mice fed on the NS diet show hemodynamic changes in terms of cardiac outflow velocities, pulse‐wave velocity and heart weight‐to‐body weight ratios compared with age‐matched normolipidemic C57Bl/6 mice; however, blood pressure and heart rate remained unaltered in this study Citation[40]. Consistent with these studies, we also found increased the heart weight‐to‐body weight ratio in ApoE−/− mice during the NS diet. Taking into account the growth promoting properties of angiotensin II and the important role of oxidative stress in the regulation of cellular growth and differentiation, it is tempting to speculate that the blood pressure‐independent cardiac hypertrophy ApoE−/− mice was mediated by increased local angiotensin II formation together with increased superoxide generation Citation[25], Citation[29].
In conclusion, using apolipoprotein E‐deficient mice as an experimental model of atherosclerosis, we here demonstrate that high salt intake promotes atherosclerosis and increases vascular superoxide formation. These salt‐induced changes were cholesterol‐independent. Our findings thus suggest a detrimental role for high salt intake in the development of atherosclerosis and underscore the importance of increased oxidative stress in the pathogenesis of salt‐induced vascular damage.
Acknowledgments
This study was supported by grants from the Academy of Finland, University's Research Funds and the Sigrid Jusélius Foundation. We are grateful to Ms Nada Bechara‐Hirvonen, Ms Anneli von Behr and Ms Sari Laakkonen for expert technical assistance.
References
- Lifton R. P., Gharavi A. G., Geller D. S. Molecular mechanisms of human hypertension. Cell 2001; 104: 545–556
- Cheng Z. J., Vaskonen T., Tikkanen I., Nurminen K., Ruskoaho H., Vapaatalo H., et al. Endothelial dysfunction and salt‐sensitive hypertension in spontaneously diabetic Goto‐Kakizaki rats. Hypertension 2001; 37: 433–439
- Messerli F. H., Schmieder R. E. Salt and hypertension: Going to the heart of the matter. Arch Intern Med 2001; 161: 505–506
- Messerli F. H., Schmieder R. E., Weir M. R. Salt. A perpetrator of hypertensive target organ disease?. Arch Intern Med 1997; 157: 2449–2452
- Simon G. Experimental evidence for blood pressure‐independent vascular effects of high sodium diet. Am J Hypertens 2003; 16: 1074–1078
- de Wardener H. E., MacGregor G. A. Harmful effects of dietary salt in addition to hypertension. J Hum Hypertens 2002; 16: 213–223
- Ferri C., Bellini C., Desideri G., Giuliani E., De Siati L., Cicogna S., et al. Clustering of endothelial markers of vascular damage in human salt‐sensitive hypertension: Influence of dietary sodium load and depletion. Hypertension 1998; 32: 862–868
- Zhou M. S., Adam A. G., Jaimes E. A., Raij L. In salt‐sensitive hypertension, increased superoxide production is linked to functional upregulation of angiotensin II. Hypertension 2003; 42: 945–951
- Knowles J. W., Reddick R. L., Jennette J. C., Shesely E. G., Smithies O., Maeda N. Enhanced atherosclerosis and kidney dysfunction in eNOS(−/−)apoe(−/−) mice are ameliorated by enalapril treatment. J Clin Invest 2000; 105: 451–458
- Kawashima S., Yokoyama M. Dysfunction of endothelial nitric oxide synthase and atherosclerosis. Arterioscler Thromb Vasc Biol 2004; 24: 998–1005
- Kennedy C. R., Zhang Y., Brandon S., Guan Y., Coffee K., Funk C. D., et al. Salt‐sensitive hypertension and reduced fertility in mice lacking the prostaglandin EP2 receptor. Nat Med 1999; 5: 217–220
- Zhu J., Mori T., Huang T., Lombard J. H. Effect of high‐salt diet on NO release and superoxide production in rat aorta. Am J Physiol Heart Circ Physiol 2004; 286: H575–H583
- Zhou M. S., Jaimes E. A., Raij L. Atorvastatin prevents end‐organ injury in salt‐sensitive hypertension: Role of eNOS and oxidant stress. Hypertension 2004; 44: 186–190
- Dmitrieva N. I., Cai Q., Burg M. B. Cells adapted to high NaCl have many DNA breaks and impaired DNA repair both in cell culture and in vivo. Proc Natl Acad Sci USA 2004; 101: 2317–2322
- Beckman J. S., Beckman T. W., Chen J., Marshall P. A., Freeman B. A. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc Natl Acad Sci U S A 1990; 87: 1620–1624
- Suda O., Tsutsui M., Morishita T., Tanimoto A., Horiuchi M., Tasaki H., et al. Long‐term treatment with N(omega)‐nitro‐l‐arginine methyl ester causes arteriosclerotic coronary lesions in endothelial nitric oxide synthase‐deficient mice. Circulation 2002; 106: 1729–1735
- d'Uscio L. V., Baker T. A., Mantilla C. B., Smith L., Weiler D., Sieck G. C., et al. Mechanism of endothelial dysfunction in apolipoprotein E‐deficient mice. Arterioscler Thromb Vasc Biol 2001; 21: 1017–1022
- Nakashima Y., Plump A. S., Raines E. W., Breslow J. L., Ross R. ApoE‐deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb 1994; 14: 133–140
- Meir K. S., Leitersdorf E. Atherosclerosis in the apolipoprotein‐E‐deficient mouse: A decade of progress. Arterioscler Thromb Vasc Biol 2004; 24: 1006–1014
- Wen M., Segerer S., Dantas M., Brown P. A., Hudkins K. L., Goodpaster T., et al. Renal injury in apolipoprotein E‐deficient mice. Lab Invest 2002; 82: 999–1006
- Yu Q., Larson D. F., Slayback D., Lundeen T. F., Baxter J. H., Watson R. R. Characterization of high‐salt and high‐fat diets on cardiac and vascular function in mice. Cardiovasc Toxicol 2004; 4: 37–46
- Kauser K., da Cunha V., Fitch R., Mallari C., Rubanyi G. M. Role of endogenous nitric oxide in progression of atherosclerosis in apolipoprotein E‐deficient mice. Am J Physiol Heart Circ Physiol 2000; 278: H1679–H1685
- Jiang J., Valen G., Tokuno S., Thoren P., Pernow J. Endothelial dysfunction in atherosclerotic mice: Improved relaxation by combined supplementation with l‐arginine‐tetrahydrobiopterin and enhanced vasoconstriction by endothelin. Br J Pharmacol 2000; 131: 1255–1261
- Cifuentes M. E., Rey F. E., Carretero O. A., Pagano P. J. Upregulation of p67(phox) and gp91(phox) in aortas from angiotensin II‐infused mice. Am J Physiol Heart Circ Physiol 2000; 279: H2234–H2240
- Cheng Z. J., Grönholm T., Louhelainen M., Finckenberg P., Merasto S., Tikkanen I., et al. Vascular and renal effects of vasopeptidase inhibition and angiotensin‐converting enzyme blockade in spontaneously diabetic Goto‐Kakizaki rats. J Hypertens 2005; 23: 1757–1770
- Li Y., Zhu H., Kuppusamy P., Roubaud V., Zweir J. L., Trush M. A. Validation of lucigenin (bis‐N‐methylacridinium) as a chemilumigenic probe for detecting superoxide anion radical production by enzymatic and cellular systems. J Biol Chem 1998; 273: 2015–2023
- Wassmann S., Czech T., van Eickels M., Fleming I., Bohm M., Nickenig G. Inhibition of diet‐induced atherosclerosis and endothelial dysfunction in apolipoprotein E/angiotensin II type 1A receptor double‐knockout mice. Circulation 2004; 110: 3062–3067
- Lenda D. M., Sauls B. A., Boegehold M. A. Reactive oxygen species may contribute to reduced endothelium‐dependent dilation in rats fed high salt. Am J Physiol Heart Circ Physiol 2000; 279: H7–H14
- Bragulat E., de la Sierra A., Antonio M. T., Coca A. Endothelial dysfunction in salt‐sensitive essential hypertension. Hypertension 2001; 37: 444–448
- Wilkinson I. B., Franklin S. S., Cockcroft J. R. Nitric oxide and the regulation of large artery stiffness: From physiology to pharmacology. Hypertension 2004; 44: 112–116
- Morrow J. D. Quantification of isoprostanes as indices of oxidant stress and the risk of atherosclerosis in humans. Arterioscler Thromb Vasc Biol 2005; 25: 279–286
- Del Rio D., Stewart A. J., Pellegrini N. A review of recent studies on malondialdehyde as toxic molecule and biological marker of oxidative stress. Nutr Metab Cardiovasc Dis 2005; 15: 316–328
- Baylis C., Vallance P. Measurement of nitrite and nitrate levels in plasma and urine – what does this measure tell us about the activity of the endogenous nitric oxide system?. Curr Opin Nephrol Hypertens 1998; 7: 59–62
- Laursen J. B., Somers M., Kurz S., McCann L., Warnholtz A., Freeman B. A., Tarpey M., et al. Endothelial regulation of vasomotion in apoE‐deficient mice: Implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation 2001; 103: 1282–1288
- Deckert V., Lizard G., Duverger N., Athias A., Palleau V., Emmanuel F., et al. Impairment of endothelium‐dependent arterial relaxation by high‐fat feeding in ApoE‐deficient mice: Toward normalization by human ApoA‐I expression. Circulation 1999; 100: 1230–1235
- Matsumoto T., D'uscio L. V., Eguchi D., Akiyama M., Smith L. A., Katusic Z. S. Protective effect of chronic vitamin C treatment on endothelial function of apolipoprotein E‐deficient mouse carotid artery. J Pharmacol Exp Ther 2003; 306: 103–108
- Ivanovski O., Szumilak D., Nguyen‐Khoa T., Dechaux M., Massy Z. A., Phan O., et al. Dietary salt restriction accelerates atherosclerosis in apolipoprotein E‐deficient mice. Atherosclerosis 2005; 180: 271–276
- Subcommittee on Laboratory Animal Nutrition, Committee on Animal Nutrition, Board on Agriculture, National Research Council. Nutrient requirements of laboratory animals, fourth revised edition; 1995
- Catanozi S., Rocha J. C., Passarelli M., Guzzo M. L., Alves C., Furukawa L. N. S., et al. Dietary sodium chloride restriction enhances aortic wall lipid storage and raises plasma lipid concentration in LDL receptor knockout mice. J Lipid Res 2003; 44: 727–732
- Hartley C. J., Reddy A. K., Madala S., Martin‐McNulty B., Vergona R., Sullivan M. E., et al. Hemodynamic changes in apolipoprotein E‐knockout mice. Am J Physiol Heart Circ Physiol 2000; 279: H2326–H2334