
Abstract
Purpose: Patients with essential hypertension who are receiving treatment with an angiotensin II receptor blocker and a calcium channel blocker often develop inadequate blood pressure (BP) control and require the addition of a diuretic. This study aimed to evaluate the long-term safety and efficacy of a triple combination therapy with 20 mg azilsartan (AZL), 5 mg amlodipine (AML) and 12.5 mg hydrochlorothiazide (HCTZ).
Materials and methods: The phase III, open-label, multicenter study (NCT02277691) comprised a 4-week run-in period and 52-week treatment period. Patients with inadequate BP control despite AZL/AML therapy (n = 341) received 4 weeks’ treatment with AZL/AML (combination tablet) + HCTZ (tablet) and 4 weeks’ treatment with AZL/AML/HCTZ (combination tablet) in a crossover manner, followed by AZL/AML/HCTZ (combination tablet) from Week 8 of the treatment period up to Week 52. The primary and secondary endpoints were long-term safety and BP (office and home), respectively.
Results: Most adverse events (AEs) were mild or moderate in intensity, and no deaths or treatment-related serious AEs were reported. The triple therapy provided consistent BP-lowering effects in both office and home measurements.
Conclusions: The triple combination therapy with AZL/AML/HCTZ was well tolerated and effective for 52 weeks in Japanese patients with essential hypertension.
Introduction
The Japanese Society of Hypertension Guidelines for the Management of Hypertension (JSH2014) [Citation1] recommend triple combination therapy of an angiotensin converting enzyme (ACE) inhibitor or angiotensin II receptor blocker (ARB) with a calcium channel blocker (CCB) and a diuretic in patients whose blood pressure (BP) cannot be adequately controlled with dual combination therapy. Initiation of treatment with a low dose of diuretic is also recommended [Citation1]. Such triple combination therapies may be administered either via co-administration of single tablets or as a fixed-dose combination (FDC) tablet if available.
Azilsartan (AZL) is an ARB that has been approved for the treatment of hypertension in Japan. In a randomized, double-blind phase III study, AZL (20–40 mg once daily) provided significantly greater reductions in sitting trough diastolic BP (DBP) and sitting trough systolic BP (SBP) compared with candesartan cilexetil (8–12 mg once daily) in Japanese patients with grade I–II essential hypertension [Citation2].
Amlodipine besylate (AML) is a long-acting CCB which has a longer half-life than second-generation agents and a slow onset, features widely held to be associated with a reduction in reflex sympathetic stimulation [Citation3]. The safety and efficacy of AML therapy has been well established [Citation4] and is, therefore, the most widely used CCB in Japan. The efficacy and safety of a dual combination therapy comprising AZL and AML has also been demonstrated in Japanese patients with grade I–II essential hypertension [Citation5].
Hydrochlorothiazide (HCTZ) is a thiazide diuretic that exhibits an antihypertensive effect by decreasing sodium chloride reabsorption at the distal tubule, increasing urinary excretion of sodium chloride and water loss [Citation6]. Co-administration of HCTZ with an ARB is expected to cancel out the side effects of each drug [Citation7].
We have previously demonstrated that triple combination therapy with AZL, AML, and HCTZ for 10 weeks has a substantial BP lowering effect and is well tolerated in Japanese patients with grade I–II essential hypertension (unpublished observations). However, the efficacy and safety of the long-term use of this triple combination therapy has not yet been investigated. Therefore, this phase III, open-label, multicenter study was designed to evaluate the safety and efficacy of long-term administration of once-daily AZL, AML, and HCTZ in Japanese patients with essential hypertension who had inadequate BP control with dual treatment of AZL and AML.
Materials and methods
Study design
The study comprised a 4-week run-in period and 52-week treatment period (56 weeks in total; ). Patients visited their study site every 2 weeks from the start of the run-in period (Week –4) to the start of the treatment period (Week 0), and every 4 weeks during the treatment period (16 visits in total). During the run-in period, all patients received one tablet daily, consisting of 20 mg AZL and 5 mg AML. At the end of the run-in period (Week 0), all patients were randomly assigned in a 1:1 ratio to either Group A or B based on the number of tablets per day of concomitant prescription drugs.
Figure 1. Study design. *RAS inhibitors (ACE inhibitors, ARBs, direct renin inhibitors), CCBs, thiazide diuretics, and diuretics similar to thiazides.
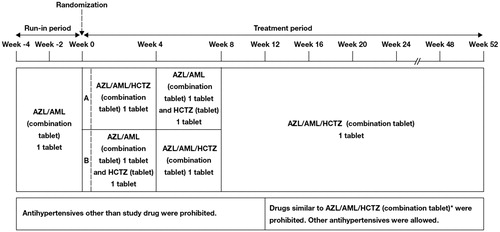
During the initial 8 weeks of the treatment period, patients received two different dosing regimens in a cross-over manner to conduct an exploratory survey concerning medication convenience of the triple FDC tablet therapy; thus, from Weeks 0 to 4 in the treatment period, Group A patients received one triple FDC tablet (containing 20 mg AZL, 5 mg AML and 12.5 mg HCTZ) once daily, while Group B patients received co-administration of one dual FDC tablet (containing 20 mg AZL and 5 mg AML) and one 12.5 mg HCTZ tablet once daily. From Weeks 4 to 8, patients in Group A received the co-administration and patients in Group B received the triple FDC tablet once daily (crossover). From Weeks 8 to 52, all patients in both groups received the triple FDC tablet once daily.
Use of additional antihypertensive drugs was not permitted from the start of the run-in period (Week –4) up to Week 12. However, if a patient did not achieve the target BP, as specified by the JSH2014 [Citation1], during the period between Week 12 and Week 52, the investigator was permitted to administer concomitant therapy with antihypertensive drugs, excluding any drugs similar to the study treatment (i.e. renin-angiotensin system [RAS] inhibitors [ACE inhibitors, ARBs, direct renin inhibitors], CCBs, thiazide diuretics, and diuretics similar to thiazides).
Patient eligibility criteria
Patients were eligible for enrolment into the study if they met all of the following main inclusion criteria: ≥20 years of age, essential hypertension, an office sitting SBP of <180 mmHg and office sitting DBP of <110 mmHg at the start of the run-in period, an office sitting SBP of <160 mmHg and an office sitting DBP of <100 mmHg at the start of the run-in period if they had previously received a triple combination therapy within 4 weeks prior to starting the run-in period, an office sitting SBP of ≥140 mmHg or office sitting DBP of ≥90 mmHg if they did not have concurrent diabetes mellitus or chronic kidney disease (CKD), or an office sitting SBP of ≥130 mmHg or office sitting DBP of ≥80 mmHg if they had concurrent diabetes mellitus or CKD at Week –2 and at the end of the run-in period (Week 0) according to the JSH2014 [Citation1]. Patients who met any of the following main exclusion criteria were excluded from the study: secondary or malignant hypertension, a history of hypersensitivity or allergy to any of the study drugs or related substances, a difference of ≥20 mmHg between left and right arms in office sitting SBP at the start of the run-in period, concurrent serious cardiovascular or cerebrovascular disorders (e.g. myocardial infarction, atrial fibrillation, congestive heart failure, transient ischemic attack), concurrent vascular disease or advanced hypertensive retinopathy, insufficient renal or hepatic function, gout or a history of gout within 24 weeks of the start of the run-in period or hyperuricaemia requiring drug treatment, abnormal levels of sodium or potassium, uncontrolled diabetes, or a malignant tumor.
This study was approved by the Institutional Review Board at each study site, and was conducted in accordance with the ethical principles of the Declaration of Helsinki, the International Conference on Harmonisation E6 (R1) Guidelines for Good Clinical Practice and all applicable local laws and regulations. All patients were required to provide written informed consent prior to the initiation of any study-related procedures. The study is registered in ClinicalTrials.gov (NCT02277691).
Endpoints and evaluations
The primary endpoint of the study was safety, including adverse events (AEs), vital signs (supine and standing BP, office sitting pulse rate), body weight, resting 12-lead electrocardiogram (ECG), and clinical laboratory tests. The secondary endpoints were office BP (i.e. sitting SBP and DBP at the time immediately before next dose) and home BP (i.e. morning and evening sitting SBP and DBP). Additional endpoints included the proportion of patients achieving target BP as defined by JSH2014 [Citation1], and patient-reported medication convenience during the initial 8 weeks of the treatment period. The target office BP was sitting SBP of <140 mmHg and sitting DBP of <90 mmHg for patients without diabetes mellitus or CKD, or sitting SBP of <130 mmHg and sitting DBP of <80 mmHg for patients with diabetes mellitus or CKD. Additionally, the target home BP was sitting SBP of <135 mmHg and sitting DBP of <85 mmHg for patients without diabetes mellitus or CKD, or sitting SBP of <125 mmHg and sitting DBP of <75 mmHg for patients with diabetes mellitus or CKD.
For safety evaluation, investigators evaluated severity and causal relationship to the study drug of AEs during the study. Supine and standing BP were monitored at Weeks 0, 12, 24, 36, and 52, and office sitting pulse rate was monitored every visit. Weight measurement and resting 12-lead ECG recording were performed at Weeks –4 (body weight only), 0, 12, 24, 36, and 52. Blood samples for laboratory testing were obtained after patients had fasted for at least 10 hours, in Weeks –4, –2, 0, 4, 12, 24, 36 and 52. Hematology tests, serum chemistry tests, and urinalyses were conducted at the central laboratory (Bio Medical Laboratories, Inc., Tokyo, Japan). AEs were defined according to the Medical Dictionary for Regulatory Activities/Japanese edition (MedDRA/J) version 18.1.
Office sitting BP was measured during every visit to the study site with an automated BP monitor (HEM-907, Omron Healthcare Co., Ltd.) with a cuff (HEM-907-CR19, Omron Healthcare Co., Ltd.). In addition, patients were required to measure and record their home BP with the MedicalLINK® BP-monitoring service system and an automated telemetry home BP monitor (HEM-7251G, Omron Healthcare Co., Ltd.) with a cuff (HEM-CUFF-R22, Omron Healthcare Co., Ltd.). Home BP was measured on the non-dominant arm twice on awakening and twice before bed each day (total of four measurements per day) from Week –2 to Week 12; for the 8 days preceding the visits at Weeks 24, 36 and 52; and wherever possible outside these time periods.
Additionally, results of questionnaires for a survey on medication convenience were collected in this study to evaluate medication convenience of a triple FDC tablet. At Week 8, all patients undertook the survey during which they answered questions on the convenience of taking a single tablet or two tablets.
Statistical analysis
In accordance with the Extent of Population Exposure to Assess Clinical Safety for Drugs Intended for Long-Term Treatment of Non-Life-Threatening Conditions [Citation8], the target number of patients needed to complete 24 weeks of treatment was 300, while the target number of patients needed to complete 52 weeks of treatment was ≥100. Taking into account potential dropouts after randomization and for any other reason, the target number of randomized patients was set at 330. The safety endpoints were assessed in the safety analysis set, which was defined as all patients who received at least 1 dose of the study drug during the treatment period. AEs were displayed using frequency distribution. The efficacy endpoints were assessed in the full analysis set (FAS), which was defined as all patients who were randomized and received at least 1 dose of the study drug during the treatment period. A summary of key statistics (including mean, standard deviation [SD], maximum, minimum, and quartiles) and two-sided 95% confidence intervals (CIs) of the mean were calculated for the changes in trough office sitting SBP and DBP from baseline (the end of run-in period) to Week 12 (last observation carried forward [LOCF]), to Week 52 (LOCF), and to each timepoint during the treatment period. The same statistics and 95% CIs were calculated for morning and evening home sitting SBP and DBP.
Results
Study population
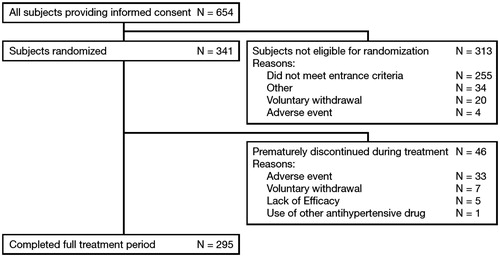
A total of 654 patients signed the informed consent form across 32 sites in Japan. At Week 0 (i.e. the end of the run-in period), 341 patients across 31 sites were determined to be eligible to enter the treatment period and were randomly assigned to Group A or B. The most common primary reason for not being eligible to enter the treatment period was ‘did not meet entrance criteria’, which was specified in 255 cases.
Of the 341 patients who received the study drug within the treatment period, 295 (86.5%) completed the full 52 weeks on study treatment, while 46 (13.5%) withdrew prematurely. The primary reasons for discontinuation of the study drug were ‘pre-treatment event/AE’ (n = 33), ‘voluntary withdrawal’ (n = 7), ‘lack of efficacy’ (n = 5), and ‘use of antihypertensive other than the study drug’ (n = 1) ().
Figure 2. Patient disposition throughout the study.
Baseline characteristics for the FAS are presented in . The mean age and body mass index (BMI) of all patients were 60.8 years and 26.20 kg/m2, respectively. Mean office sitting SBP and DBP were 143.7 mmHg and 86.2 mmHg, respectively.
Table 1. Baseline characteristics of all randomized patients.
Safety analysis
An overview of the AEs reported in this study is presented in . The overall incidence of AEs was 84.8% (289/341 patients) and the overall incidence of drug-related AEs was 38.4% (131/341 patients). Most of the AEs were mild or moderate in intensity.
Table 2. Summary of overall safety data.
The incidence of all-cause AEs occurring in >2% of patients is presented in . The most common AE was nasopharyngitis (31.1%). Of the remaining AEs, blood uric acid increased (25.2%) and hyperuricaemia (5.3%) were reported as AEs related to hyperuricaemia, but the majorities were mild in intensity and none led to study drug discontinuation.
Table 3. All-cause AEs occurring in >2% of patients.
Twenty-four serious AEs were reported in 20 patients but all of them were considered by the investigators as being not related to the study drug. No deaths were reported during the study. The incidence of AEs leading to study drug discontinuation was 9.4% (n = 32). Of these, the following events were considered to be related to the study drug: BP decreased, photosensitivity reaction, and hypotension (n = 4 each); anaemia, sinus bradycardia, hyponatraemia, dizziness postural, chronic kidney disease, drug eruption, eczema, purpura, and orthostatic hypotension (n = 1 each).
No clinically meaningful changes from baseline in mean serum chemistry test values were seen at any timepoint, with the exception of increases in blood uric acid. The increase was observed by Week 4, and then the mean change from baseline was maintained between 0.91 and 1.03 mg/dL until Week 52. For the hematology and urinalysis tests, no clinically meaningful change from baseline was seen in the test values at any timepoint.
Both supine BP and standing BP decreased from baseline after beginning administration of the study drug. The changes in pulse rate and weight from baseline were not clinically significant at any timepoint. Among those patients whose baseline (Week 0) ECG was interpreted as either ‘within normal limits’ or ‘abnormal, not clinically significant’, three patients had an ECG that was interpreted as ‘abnormal, clinically significant’ at Week 52.
Efficacy analysis
Decreases in mean office trough sitting SBP and DBP from baseline were observed at Week 4 and were maintained up to Week 52. At Week 12 (LOCF) and Week 52 (LOCF), the mean changes from baseline in office trough sitting SBP were –14.4 mmHg (SD 12.72 mmHg) and –13.9 mmHg (SD 12.14 mmHg), respectively, which were statistically significant (p < .0001, one-sample t-test). At Week 12 (LOCF) and Week 52 (LOCF), the mean changes from baseline in office trough sitting DBP were –8.6 mmHg (SD 8.97 mmHg) and –8.3 mmHg (SD 9.26 mmHg), respectively, and were also statistically significant (p < .0001, one-sample t-test). The statistically significant reductions in SBP and DBP were maintained throughout the study period, as shown in and .
Figure 3. Time profile of mean office trough sitting SBP and DBP. p < .0001 versus week 0 at all post-baseline timepoints.
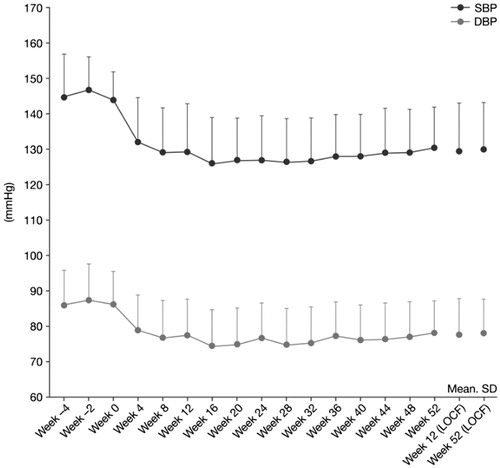
Table 4. Changes in office trough sitting SBP and DBP and morning home sitting SBP and DBP from baseline at each time point for the FAS.
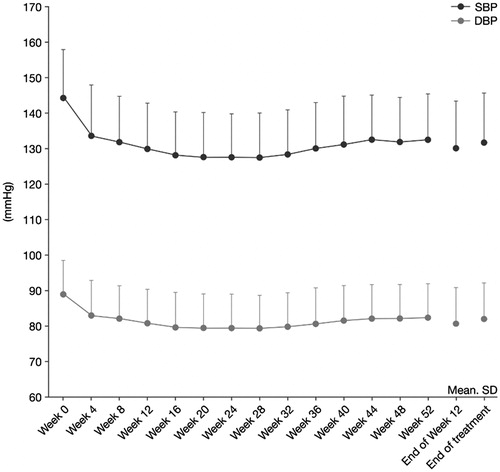
Decreases in mean morning home sitting SBP and DBP compared with baseline were also observed at Week 4 and maintained up to Week 52. At the end of Week 12 and at the end of treatment, the mean changes from baseline in morning home sitting SBP were –13.9 mmHg (SD 10.67 mmHg) and –12.4 mmHg (SD 11.75 mmHg), respectively, and were statistically significant (p < .0001, one-sample t-test). For morning home sitting DBP, the mean changes from baseline were –7.9 mmHg (SD 6.59 mmHg) and –6.9 mmHg (SD 7.23 mmHg), respectively, at the same two time points, and these were also statistically significant (p < 0.0001, one-sample t-test) as shown in and . A similar trend was observed when home sitting BP in the evening was measured.
Figure 4. Time profile of mean morning home sitting SBP and DBP. p < .0001 versus week 0 at all post-baseline timepoints.
The percentage of patients who achieved the target office or home BP outlined by the JSH2014 was 62.1% (n = 210) at Week 12 (LOCF), 62.1% (n = 210) at Week 52 (LOCF) for office BP, and 35.2% (n = 108) at the end of Week 12, 30.2% (n = 89) at the end of treatment for morning home BP (data were not shown).
Results from the medication convenience survey conducted at Week 8 are shown in Table S1. The majority of patients (77.1%; n = 256) considered that taking a single tablet was more convenient than taking two tablets. The most common reason for this answer was ‘easier to take out drug from sheet’ in both treatment groups.
Discussion
Overall, this phase III long-term study showed that triple combination therapy with 20 mg AZL, 5 mg AML and 12.5 mg HCTZ was well tolerated for 52 weeks in patients with essential hypertension whose BP was not adequately controlled by dual combination therapy with 20 mg AZL and 5 mg AML. Most of the reported AEs were mild or moderate in intensity and 5 reported severe AEs (in 4 patients), all of which were considered to be unrelated to the triple combination therapy. Additionally, no deaths or drug-related serious AEs were reported. Although blood uric acid increases were observed between Weeks 0 and 4 due to the pharmacological effects of HCTZ, most of the AEs related to hyperuricaemia were mild in intensity, with none being classed as serious or requiring drug discontinuation.
In terms of efficacy, it was shown that the BP-lowering effects of the triple therapy were maintained for 52 weeks, although it should be noted that the study design permitted the addition of other antihypertensive drugs (except for ACE inhibitors, ARBs, direct renin inhibitors, CCBs, thiazide diuretics, and diuretics similar to thiazides) after Week 12, if needed. The number of patients who needed additional antihypertensive drugs during the treatment period was only 14 out of 341 treated patients. The BP-lowering effects were observed consistently in both office and home BP readings.
In this study, we conducted an exploratory survey on medication convenience. Survey responses indicated that one FDC tablet was considered more convenient than co-administration of two separate tablets. It has been reported that an increase in the number of co-administrated drugs tends to reduce compliance with treatment [Citation9]. It has also been reported that inadequate medication adherence is related to inadequate BP control [Citation10–13] as well as increased risk of stroke, ischemic heart disease, and death [Citation14–18]. Thus, the triple FDC tablet with 20 mg AZL, 5 mg AML and 12.5 mg HCTZ is expected to contribute to improved medication adherence, better BP control and a reduction in the risk of vascular events.
The main limitation of this study was that the effectiveness of the triple therapy was not investigated in patients aged <20 years, or those with clinically evident hepatic or severe renal impairment. Further research would be required to confirm the long-term safety and efficacy of this treatment in those patient groups. In addition, this was a single-arm study and therefore has no comparator arm to allow direct comparison with the effectiveness of other treatment regimens.
In conclusion, triple combination therapy with a single tablet of 20 mg AZL, 5 mg AML and 12.5 mg HCTZ was well tolerated, and provided consistent BP-lowering effects for patients with essential hypertension whose BP was not adequately controlled by dual combination therapy with 20 mg AZL and 5 mg AML.
Online_Supplement.docx
Download MS Word (14.6 KB)Acknowledgements
The authors would like to thank the patients and their families, as well as investigators and other staff members for their invaluable contributions to the study.
Disclosure statement
HR served as the medical expert for this study and received honoraria from Takeda Pharmaceutical Company Limited for lectures given during the study period. HR also declares the following conflicts of interest in relation to this presentation: lecture fees and research funding from various pharmaceutical companies in Japan that market antihypertensive drugs, including Takeda Pharmaceutical Company Limited.
KS, YN, YS and YU are all employees of Takeda Pharmaceutical Company Limited.
Additional information
Funding
Related Research Data
References
- Shimamoto K, Ando K, Fujita T, et al. The Japanese Society of Hypertension guidelines for the management of hypertension (JSH 2014). Hypertens Res. 2014;37:253–390.
- Rakugi H, Enya K, Sugiura K, et al. Comparison of the efficacy and safety of azilsartan with that of candesartan cilexetil in Japanese patients with grade I-II essential hypertension: a randomized, double-blind clinical study. Hypertens Res. 2012;35:552–558.
- Wang AL, Iadecola C, Wang G. New generations of dihydropyridines for treatment of hypertension. J Geriatr Cardiol. 2017;14:67–72.
- Derosa G, Maffioli P. Drug safety evaluation of amlodipine. Expert Opin Drug Saf. 2011;10:795–804.
- Rakugi H, Nakata E, Sasaki E, et al. Evaluation of the efficacy and tolerability of fixed-dose combination therapy of azilsartan and amlodipine besylate in Japanese patients with grade I to II essential hypertension. Clin Ther. 2014;36:711–721.
- Padilla MC, Armas-Hernández MJ, Hernández RH, et al. Update of diuretics in the treatment of hypertension. Am J Ther. 2007;14:154–160.
- Oshikawa J, Toya Y, Morita S, et al. Angiotensin receptor blocker (ARB)-diuretic versus ARB-calcium channel blocker combination therapy for hypertension uncontrolled by ARB monotherapy. Clin Exp Hypertens. 2014;36:244–250.
- Evaluation and Licensing Division, Pharmaceutical Affairs Bureau, Ministry of Health, Labour and Welfare. Extent of population exposure to assess clinical safety for drugs intended for long-term treatment of non-life-threatening conditions. PAB/ELD Notification No. 592 (May 24, 1995).
- Xie L, Frech-Tamas F, Marrett E, et al. A medication adherence and persistence comparison of hypertensive patients treated with single-, double- and triple-pill combination therapy. Curr Med Res Opin. 2014;30:2415–2422.
- Bramley TJ, Gerbino PP, Nightengale BS, et al. Relationship of blood pressure control to adherence with antihypertensive monotherapy in 13 managed care organizations. Jmcp. 2006;12:239–245.
- Fung V, Huang J, Brand R, et al. Hypertension treatment in a medicare population: adherence and systolic blood pressure control. Clin Ther. 2007;29:972–984.
- Ho PM, Magid DJ, Shetterly SM, et al. Importance of therapy intensification and medication nonadherence for blood pressure control in patients with coronary disease. Arch Intern Med. 2008;168:271–276.
- Yiannakopoulou ECh, Papadopulos JS, Cokkinos DV, et al. Adherence to antihypertensive treatment: a critical factor for blood pressure control. Eur J Cardiovasc Prev Rehabil. 2005;12:243–249.
- Bailey JE, Wan JY, Tang J, et al. Antihypertensive medication adherence, ambulatory visits, and risk of stroke and death. J Gen Intern Med. 2010;25:495–503.
- Cherry SB, Benner JS, Hussein MA, et al. The clinical and economic burden of nonadherence with antihypertensive and lipid-lowering therapy in hypertensive patients. Value Health. 2009;12:489–497.
- Dragomir A, Côté R, Roy L, et al. Impact of adherence to antihypertensive agents on clinical outcomes and hospitalization costs. Med Care. 2010;48:418–425.
- Kettani FZ, Dragomir A, Côté R, et al. Impact of a better adherence to antihypertensive agents on cerebrovascular disease for primary prevention. Stroke 2009;40:213–220.
- Perreault S, Dragomir A, White M, et al. Better adherence to antihypertensive agents and risk reduction of chronic heart failure. J Intern Med. 2009;266:207–218.