
Abstract
Purpose
Through describing the confusing misdiagnosis process of Liddle syndrome, we try to reveal the importance of accurate aldosterone-renin detection and a genetic test for Liddle syndrome.
Methods
We found a family of hypertension and hypokalaemia with the proband of a 21-year-old female who had been misdiagnosed as primary aldosteronism (PA). She presented with high aldosterone and low renin levels. Aldosterone is not suppressed in the saline infusion test and captopril challenge test. However, treatment with a standard dose of spironolactone has no blood pressure improvement effect. A heterozygous variant of SCNN1G was found with whole exome sequencing and Liddle syndrome is indicated. Treatment with amiloride was effective. We rechecked aldosterone-renin levels with two different aldosterone and renin test kits. Clinical features and the mutant gene SCNN1G of each family member were determined by the Sanger method.
Results
The two kits had nearly opposite results. Among those Liddle syndrome patients confirmed by a genetic test, for Test kit A all ARR were screened positive while for test kit B negative. It seems Test kit B is consistent with the diagnosis while test kit A misleads the diagnosis. A novel SCNN1G mutation, c.1729 C > T, was found in this family, which introduce a premature stop codon in the γ subunit in the epithelial Na+ channel (ENaC) and resulted in a deletion of 72 amino acids at the carboxyl end.
Conclusion
inaccurate ARR detection might misdiagnose Liddle syndrome. A Gene test is an important method for the diagnosis of Liddle syndrome. A novel SCNN1G missense mutation, c.1729 C > T, is found in a Chinese family.
Introduction
Hypertension combined with hypokalaemia is common in clinical practice. However, only a tiny part of it could be explained by Liddle syndrome. Liddle syndrome mimics the symptoms of mineralocorticoid excess characterised by early-onset hypertension, hypokalaemia, and metabolic alkalosis. Liddle syndrome is an autosomal dominant disease, first reported by Liddle in 1963 [Citation1] Mutations in the subunits of ENaC lead to over activation of the channel, which presents clinically as volume expansion and inhibition of aldosterone-renin levels. Treatments of salt restriction and the using ENaC inhibitors (amiloride, triamterene) are usually effective. There is no specific diagnosing process in any guideline for Liddle syndrome at present and endocrinologists usually apply the flow chart of hypertension and hypokalaemia to discriminate a cluster of endocrine hypertension [Citation2]. Accurate detection of aldosterone levels and aldosterone-rennin ratio (ARR) is important because it is the key point of the flow chart. Meanwhile, a genetic test is recommended for early-onset hypertension and familial hypertension to confirm hereditary hypertension [Citation3].
Method
Participants
A 21-year-old female was admitted to our endocrinology ward because of repeated hypokalaemia for 5 years and newly noticed high blood pressure (BP). Clinical features and medical history were collected. Laboratory tests were arranged to confirm secondary hypertension. Since some family members also have hypertension and hypokalaemia, 17 family members were further included in this study and a gene test was done on 11 family members (). This study was approved by the Committee of Shanghai Public Health Clinical Centre and carried out in accordance with the Declaration of Helsinki. Informed consent was obtained from all participants.
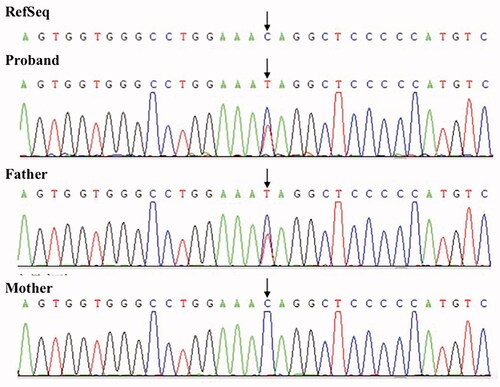
Figure 1. Sanger sequencing the results of the proband and her parents. RefSeq: National Centre for Biotechnology Information Reference Sequences.
Biochemical examination
For the test of aldosterone and renin levels, plasma is collected after two hours upright position. Aldosterone and renin levels were measured using chemiluminescence immunoassays by test kit A (China Antu) in our hospital and test kit B (Italy DiaSorin) in another hospital.
Genetic testing
DNA was extracted from peripheral blood leukocytes using the QIAamp DNA Blood Mini kit (QIAGEN, Hilden, Germany). The whole exome was sequenced by all exon capture chips (by IDT’s xGen Exome Research Panel, Integrated DNA Technologies) and by Illumina NovaSeq 6000 sequencer to identify candidate variants, which were selected by means of in silico analysis. The polymerase chain reaction products were sequenced using an ABI Prism 3730 DNA sequencer (Applied Biosystems, Foster City, CA, USA). Primers of the polymerase chain reaction were used according to Yang et al. [Citation4] to amplify Exon 13 of SCNN1G. Sanger sequencing on the ABI3730 platform was used to verify the target sequence (Applied Biosystems). Among the 17 family members of the proband invited to this study, a gene test was accepted in 11 family members.
Results
Clinical features
A 21-year-old female patient who complained of recurrent lower limb weakness for 5 years was admitted to the endocrinology ward. Two years ago, she visited a neurological clinic because of similar limb weakness. Her medical records then showed her BP was 130/70 mmHg, plasma potassium was 1.6 mmol/l (normal range, 3.5–5.3 mmol/l), aldosterone was 37.75 ng/dl (normal range, 4–31 ng/dl), renin was 0.73 ng/l (normal range 4–38 ng/dl) and aldosterone renin ratio (ARR) was 40.59 (positive for screening if >3.8). The aldosterone level was not inhibited in the captopril challenge test (data not available). She was diagnosed with primary aldosteronism and treated with spironolactone 20 mg bid and potassium chloride. However, the weakness of her limbs still emerged intermittently. She had no history of vomiting or diarrhoea, using diuretic or laxative, or using liquorice or glycyrrhetinic acid. There is no evidence of cortisol overabundance. Spironolactone had been stopped 1 month before admission and only a potassium chloride supplement was used. Her BP was 158/105 mmHg (not on anti-hypertension treatment) and hypertension was further confirmed after several repeated measurements. Plasma potassium was 3.2 mmol/l while synchronous 24 h urinary potassium was 96.2 mmol/day. Aldosterone was 20.85 ng/dl (normal range, 4–31 ng/dl), renin was 1.09 ng/l (normal range, 4–38 ng/dl) and ARR was 19.13 (positive for screening if >3.8). Saline infusion test showed aldosterone decreased from 28.78 to 22.86 ng/dl, indicating not suppressed. Enhanced CT scanning of the adrenal gland showed the medial branch of the left adrenal gland was slightly thickened, which was about 4 mm wide. Serum cortisol and plasm adrenocorticotropic hormone and 24 h urinary free cortisol were normal. Plasma-free or urinary-fractionated metanephrines were normal. Primary aldosteronism was diagnosed then. Considering other family members such as father, aunt and grandmother had hypertension and hypokalaemia, then we suspected the diagnose of glucocorticoid-remediable aldosteronism and carried out a dexamethasone inhibition test and gene test. However, the test was negative (aldosterone decreased from 38.53 ng/dL to 19.44 ng/dL after 4 mg dexamethasone was administrated in 48 h).
The patient’s family history was investigated, and 17 family members consented to participate in this study. Their clinical characteristics are shown in . Among these 17 family members, 12 had hypertension (8 on anti-hypertension treatment), 6 were diagnosed with hypertension at less than 30 years old, 6 had decreased blood potassium and 2 were on oral potassium treatment. Aldosterone-renin levels were tested using test kit A. Aldosterone was generally among the normal range while the renin levels were all below the normal range, all ARR is above 3.8 indicates the screening test for primary aldosteronism was positive for all the 17 family members whatever they have hypertension or not.
Table 1. Clinical and biochemical characters of the family members.
Genetic analysis
A heterozygous variant located in exon 13 of SCNN1G, c.1729 C > T, was identified in the 21-year-old female () while screening for hereditary hypertension. This variant was classified as “pathogenic” using the American College of Medical Genetics and Genomics (ACMG) criteria. Human genome variation software Provean and SIFT were used to predict the protein structure changes and revealed that this mutation changed the corresponding amino acid at codon 577 from glutamine to a stop codon and resulted in a deletion of 72 amino acids at the carboxyl end ofγsubunit of ENaC. This missense variant is predicted to be a pathogenic mutation, according to the guideline recommended by ACMG.
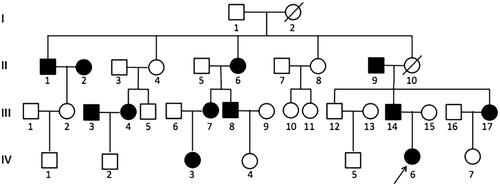
Among the 17 family members of the proband invited to this study, a gene test was accepted in 11 family members. Pedigree's investigation shows that this mutation is also presented in another 8 family members (, ).
Figure 2. Family pedigree. The black arrow indicates the proband; Black filled symbols: subjects with hypertension. Empty symbols: without hypertension. Seventeen family members are further included in this study:II-1, II-2, II-5, II-6, II-9, III-2, III-3, III-4, III-6, III-7, III-8, III-14, III-15, III-17, IV-2, IV-3, IV-6; Eleven subjects accepted gene test: II-1, II-6, II-9, III-2, III-7, III-8, III-14, III-15, III-17, IV-3, IV-6; Slash indicates passed away due to cerebrovascular accident.
Treatment and effectiveness
This 21-year-old female presented with repeated hypokalaemia and latent hypertension, poor response to spironolactone, gene mutation located in SCNN1G, c.1729 C > T, leading to the diagnosis of Liddle syndrome instead of primary aldosteronism. Treatment was switched to amiloride accordingly. Since pure amiloride tablets are not available in the domestic market [Citation5]. compound amiloride hydrochloride one tablet daily was prescribed which contains amiloride hydrochloride 2.5 mg and hydrochlorothiazide 25 mg. Her BP decrease was noticed in 3 days, regularly measured 6 times daily, and her plasma potassium climbed to normal with 3.8 mmol/L. After 3 weeks’ treatment, the plasma potassium was 4.3 mmol/L without potassium supplement; BP decreased gradually and stabilised at 110–120/70–80mmHg.
Comparing of two different test kits
Although gene testing indicates this 21-year-old female and her 8 family members might be Liddle syndrome, none of their aldosterones were suppressed while the ARR is positive for PA Screen (). The proband aldosterone-renin level was rechecked in our hospital (Test kit A) and the reagent quality control standard also indicated that the detection was not abnormal. We sent thirteen family members’ samples to another hospital for verification where another kind of test kit (Test kit B) is used. Two sets of aldosterone-renin levels by different test kits are listed in .
Using Test kit A, all aldosterone levels are either among or above the normal range. All renin levels are far below the reference range. All ARRs are above 3.8, the positive cut-off point of the test kit for the PA screen ().
Figure 3. Aldosterone levels were tested by kits A (
 ) among family members. *Patient with Liddle syndrome diagnosed with a gene test.
) among family members. *Patient with Liddle syndrome diagnosed with a gene test.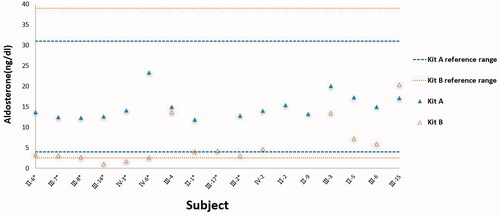
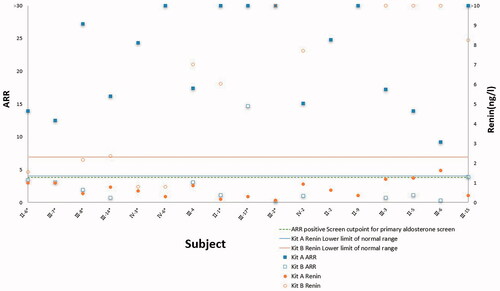
Using test kit B, aldosterone levels of those carrying the gene mutation are clustered near the lower limit of the reference range and two out of 9 subjects (III-14 and IV-3) are below the lower limit, which is both diagnosed as Liddle syndrome later. No renin levels are above the reference range; 8 out of 9 subjects carrying the gene mutation are below the reference range and the unrelated 4 subjects are among the reference range. All ARRs are below the positive cut-off point for the PA screen ().
Figure 4. ARR and renin levels were tested with different test kits among family members. Renin is detected below the lower limit of the detection range(less than 0.8 ng/l for patients IV-3 and IV-6) in kit B which is marked at the lower limit of the detection range, so the ARR ratio of patients IV-3 and IV-6 in kit B is not calculated.
Discussion
Liddle syndrome is not a common disease, and the clinical manifestations are insidious and atypical. Patients usually present with hypertension and/or hypokalaemia. When clinicians follow the diagnosing process of hypokalaemia and hypertension [Citation2], correct diagnosis depends greatly on the accuracy of biochemical tests. However, Liddle syndrome is often missed and misdiagnosed in our clinical practice. The proband was misdiagnosed as primary aldosteronism, which is closely related to the accuracy of aldosterone-renin levels tested in our hospital. To test this hypothesis, we compared two different aldosterone-renin test kits in this study. It is shown that test kits A and B yield obviously different aldosterone and ARR levels. This phenomenon is also verified in other studies that apparent differences exist between different aldosterone test methods [Citation6–8]. In one study, the test kits are identical to the test kits A and B used and the two chemiluminescence methods were compared with mass spectrometry, which is used as a golden standard, among healthy control, primary hypertension and primary aldosteronism patients. There was a good linear correlation between kit B and mass spectrometry (r = 0.776) while the correlation of kit A is poor (r = 0.058) [Citation7]. Most of the aldosterone levels of kit A are above 100 ng/dl and this might explain why the aldosterone could not be inhibited to 100 ng/dl below after saline infusion test and would lead to the false diagnosis of primary aldosteronism. Aldosterone levels of test kit B are obviously more accurate than kit A and it might not be a wise choice to diagnose Liddle syndrome based on the aldosterone levels of kit A.
Chemiluminescence detection is based on antigen-antibody reaction. Some substances with similar molecular weights and/or structures to aldosterone might interfere with the detection such as prednisolone, hydrocortisone, cortisone, 17 deoxycortisol and 18 hydroxycorticosterone [Citation9]. When plasma aldosterone is detected using the chemiluminescence method, the obtained value is usually higher than the actual value, especially when the plasma aldosterone concentrations are less than 7.2 ng/dl and the deviations usually are 50%–100% higher than the actual values [Citation6,Citation10]. The underlying reason might be the cross-reaction between aldosterone antibody and antigen such as aldosterone structural analogues and/or aldosterone metabolites [Citation9]. At present, there are several manufacturers providing chemiluminescent aldosterone test kits including Italy Diasorin, Zhengzhou Antu, Shenzhen Mindray, Changchun Dirui, Tianjin Boasais, Shenzhen New industry, etc in China. The results of the quality assessment of aldosterone in 2019 from 272 clinical laboratories in China showed that the mass spectrometry method was the most accurate, followed by the Italy Diasorin test kit; Other chemiluminescence methods had large deviations, which are usually negatively correlated with the sample concentration and the system deviation could be as high as 479% [Citation6,Citation10].
Unreliable aldosterone test contributes to the misdiagnosis of primary aldosteronism but there are still some clues worth exploring such as the patient’s reaction to spironolactone and the unusual family history. Gene results show the 21-year-old female carrying a heterozygous variant located in SCNN1G and the diagnosis of Liddle syndrome emerged to the surface finally. It is known that Liddle syndrome is caused by excessive activation of ENaC in renal tubular epithelial and ENaC is a heterotrimer comprised of three similar subunits: α, β, and γ, encoded by gene SCNN1A, SCNN1B and SCNN1G respectively. SCNN1A is in 12p13.31, while SCNN1B and SCNN1G are in 16p13-p12 [Citation11]. Autosomal dominant mutations in the subunits of the ENaC have been intermittently reported since Grant Liddle first reported Liddle syndrome in 1963. Up to now, only one gene mutation located in SCNN1A has been reported [Citation12], the remaining gene mutations are either located in SCNN1B or SCNN1G [Citation13]. Among SCNN1G mutations, a total of 11 Liddle syndrome gene mutations, including this study, have been reported until now. In this study, we find a novel heterozygous variant located in exon 13 of SCNN1G, c.1729 C > T, and this mutation changed the corresponding amino acid at codon 577 from glutamine to a stop codon, which resulted in a deletion of 72 amino acids at the carboxyl end ofγsubunit of ENaC. Similar to this study, four other studies reported mutations in different sites of SCNN1G resulting in different amino acid deletion at the C-terminal ofγsubunit and upregulation of sodium channel function. It is interesting that these deletion positions are limited to the 567–577 amino acids of the C-terminal of γsubunit [Citation14–17]. With the development of gene diagnosis, new gene mutations of Liddle syndrome have been reported in different populations. The incidence of Liddle syndrome is not rare [Citation18,Citation19] and Liddle syndrome might be a common cause of monogenic hypertension [Citation20], some scholars actively recommend gene tests as an important method of early diagnosis and differential diagnosis of Liddle syndrome.
Conclusion
An inaccurate aldosterone renin test might misdiagnose Liddle syndrome as primary aldosteronism. Gene detection is an important method of early diagnosis and differential diagnosis of Liddle syndrome. A novel SCNN1G missense mutation, c.1729 C > T, is found in a Chinese family.
Author contributions
Yaling Yang participated in data analysis and writing. Yaling Yang, Chenwei Wu, Duoduo Qu, Xinyue Xu participated in data sorting, genetic analysing and article manuscript. Lili Chen and Quanya Sun participated in data and sample collection. Xiaolong Zhao designed the study, participated and guided all stages of the study, discussed and revised the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Botero-Velez M, Curtis JJ, Warnock DG. Brief report: Liddle's syndrome revisited-a disorder of sodium reabsorption in the distal tubule. N Engl J Med. 1994;330(3):178–181.
- Gardner D. Greenspan’s basic & clinical endocrinology. 9ed. New York City: Mcgraw-Hill Education; 2011.
- Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American college of medical genetics and genomics and the association for molecular pathology. Genet Med. 2015;17(5):405–424.
- Yang K-Q, Lu C-X, Xiao Y, et al. A novel frameshift mutation of epithelial sodium channel β-subunit leads to Liddle syndrome in an isolated case. Clin Endocrinol (Oxf). 2015;82(4):611–614.
- Fan P, Lu CX, Zhang D, et al. Liddle syndrome misdiagnosed as primary aldosteronism resulting from a novel frameshift mutation of SCNN1B. Endocr Connect. 2018;7(12):1528–1534.
- Weiyan Z, et al. Analysis on the performance of aldosterone testing and the results of EQA in China. Chin J Lab Med. 2020;43:267–273.
- Fengfan Z, et al. Comparison of the consistency of different methods in detecting serum aldosterone concentration. Chin J Endocrinol Metab. 2019;35:5.
- Wenbo L, Weiyan Z, Chuanbao Z. Laboratory measurement of aldosterone and its standardization. Chin J Clin Lab Sci. 2019;35:861–864.
- Ray JA, Kushnir MM, Palmer J, et al. Enhancement of specificity of aldosterone measurement in human serum and plasma using 2D-LC–MS/MS and comparison with commercial immunoassays. J Chromatogr B. 2014;970:102–107.
- Yin Y, Ma C, Yu S, et al. Comparison of three different chemiluminescence assays and a rapid liquid chromatography tandem mass spectrometry method for measuring serum aldosterone. Clin Chem Lab Med. 2019;58(1):95–102.
- Hanukoglu I, Hanukoglu A. Epithelial sodium channel (ENaC) family: phylogeny, structure-function, tissue distribution, and associated inherited diseases. Gene. 2016;579(2):95–132.
- Salih M, Gautschi I, van Bemmelen MX, et al. A missense mutation in the extracellular domain of αENaC causes liddle syndrome. J Am Soc Nephrol. 2017;28(11):3291–3299.
- Levanovich PE, Diaczok A, Rossi NF. Clinical and molecular perspectives of monogenic hypertension. Curr Hypertens Rev. 2020;16(2):91–107.
- Yamashita Y, Koga M, Takeda Y, et al. Two sporadic cases of Liddle’s syndrome caused by de novo ENaC mutations. Am J Kidney Dis. 2001;37(3):499–504.
- Hansson JH, Nelson-Williams C, Suzuki H, et al. Hypertension caused by a truncated epithelial sodium channel gamma subunit: genetic heterogeneity of Liddle syndrome. Nat Genet. 1995;11(1):76–82.
- Wang L-P, Yang K-Q, Jiang X-J, et al. Prevalence of Liddle syndrome among young hypertension patients of undetermined cause in a Chinese population. J Clin Hypertens. 2015;17(11):902–907.
- Wang Y, Zheng Y, Chen J, et al. A novel epithelial sodium channel gamma-subunit de novo frameshift mutation leads to Liddle syndrome. Clin Endocrinol. 2007;67(5):801–804.
- Liu K, Qin F, Sun X, et al. Analysis of the genes involved in mendelian forms of low-renin hypertension in Chinese early-onset hypertensive patients. J Hypertens. 2018;36(3):502–509.
- Tapolyai M, Uysal A, Dossabhoy NR, et al. High prevalence of Liddle syndrome phenotype among hypertensive US veterans in Northwest Louisiana. J Clin Hypertens. 2010;12(11):856–860.
- Padmanabhan S, Caulfield M, Dominiczak AF. Genetic and molecular aspects of hypertension. Circ Res. 2015;116(6):937–959.