
Abstract
Aim: 17 α-hydroxylase/17,20-lyase deficiency (17-OHD) is an extremely rare autosomal recessive disorder that typically causes hypertension, hypokalaemia, primary amenorrhoea, and the absence of secondary sex characteristics in 46,XX individuals. Partial 17-OHD is even rarer than complete 17-OHD and is prone to missed diagnosis due to its subtler symptoms. The aim of this study was to help early detection and diagnosis of partial 17-OHD.
Methods: We present a case of a 41-year-old female (46,XX) patient with partial 17-OHD caused by a novel missense CYP17A1 mutation, c.391 A > C (p.T131P). This patient experienced hypertension, hypokalaemia and adrenal hyperplasia, but did not present with primary amenorrhoea or absence of secondary sex characteristics. Initially, she was misdiagnosed and underwent right and left adrenalectomy, but the procedures were ineffective. Afterward, she received a one-month treatment of 0.5 mg dexamethasone, which greatly relieved her symptoms. Additionally, we reviewed reports of thirteen other patients with partial 17-OHD in 46,XX individuals from the literature, totalling fourteen probands.
Results: We found that primary amenorrhoea, hypertension, hypokalaemia, and ovarian cysts accounted for 15.4%, 42.9%, 38.5%, and 72.7% of these patients, respectively. In contrast, elevated serum progesterone was present in all patients.
Conclusion: Based on our literature review, the absence of primary amenorrhoea, hypertension or hypokalaemia cannot rule out suspicion for 17-OHD in 46,XX individuals. However, an elevation in serum progesterone levels is a highly sensitive indicator for diagnosing 17-OHD.
Plain language summary
17-OHD is a rare cause of secondary hypertension, often with hypokalaemia, primary amenorrhoea and absence of secondary sex characteristics.
Partial 17-OHD is an even rarer subtype of 17-OHD, with subtler symptoms.
There are few reports concerning partial 17-OHD, especially in 46,XX patients.
What is the context?
We reported a case of a 46,XX patient with partial 17-OHD caused by a novel missense CYP17A1 mutation, c.391 A > C (p.T131P).
We also conducted a literature review to summarise the clinical, hormonal and genetic characteristics of fourteen 46,XX probands with partial 17-OHD.
From the literature review, we found that:
Most 46,XX patients with partial 17-OHD presented with partial pubic hair, breast development, oligomenorrhea or secondary amenorrhoea, normotension, and/or normokalemia.
All 46,XX patients with partial 17-OHD presented with elevated serum progesterone.
However, the relationship between in vitro enzyme activities of the 17-hydroxylase and/or17,20-lyase and clinical severity is still unclear.
What is new?
The current study can help early detection and diagnosis of partial 17-OHD.
What is the impact?
Introduction
17 α-Hydroxylase/17,20-lyase deficiency (17-OHD) is an extremely rare autosomal recessive disorder caused by mutations in the CYP17A1 gene, which encodes the P450c17 enzyme. This enzyme plays a critical role in the production of both glucocorticoids and sex steroids, specifically by regulating the activities of 17 α-hydroxylase and 17,20-lyase, respectively [Citation1,Citation2]. The deficiency of 17 α-hydroxylase and 17,20-lyase has a significant impact on the patient’s health. The 17-hydroxylase deficiency results in decreased cortisol levels, which compensatively stimulates the secretion of adrenocorticotropic hormone (ACTH). It also increases the production of mineralocorticoid precursors, corticosterone, and 11-deoxycorticosterone (DOC), ultimately resulting in hypertension and hypokalaemia. In contrast, the 17,20-lyase deficiency leads to decreased sex steroid production, which compensatively stimulates Follicle-Stimulating Hormone (FSH) and luteinizing hormone (LH), resulting in primary amenorrhoea and a lack of secondary sex characteristics [Citation3]. Overall, patients with 17-OHD typically present with hypertension, hypokalaemia, primary amenorrhoea, and a lack of secondary sex characteristics.
There are two main types of 17-OHD: complete and partial, with the latter being much rarer than the former [Citation4]. Patients with complete 17-OHD typically exhibit the full range of symptoms associated with the disorder. On the other hand, patients with partial 17-OHD carry the CYP17A1 mutation but may only present with milder symptoms or only a subset of the full range of 17-OHD symptoms. For instance, some cases of partial 17-OHD have been reported to display hypertension and hypokalaemia while retaining some oestrogenic and androgenic functions [Citation5–10]. This can result in 46,XX patients with partial 17-OHD exhibiting partial pubic hair, breast development, oligomenorrhea or secondary amenorrhoea, and 46,XY patients presenting with ambiguous genitalia [Citation3,Citation6,Citation11]. Due to its rarity and atypical symptoms, 46,XX patients with partial 17-OHD are at risk of misdiagnosis and mistreatment, which can significantly impair their health and quality of life.
At present, reports pertaining to the partial 17-OHD are few. Here, we report a novel case of a 46,XX patient with partial 17-OHD, caused by a homozygous novel missense CYP17A1 mutation, c.391 A > C (p.T131P). Moreover, we conducted a comprehensive literature review of partial 17-OHD in 46,XX, which provides valuable insights into this rare condition.
Case report
This 41-year-old female was admitted to the hospital in 2021 complaining weakness in her limbs for 2 years. She had a history of hypertension, bilateral ovarian cysts, and right adrenal space-occupying lesion in 2000, but was initially misdiagnosed and underwent a right adrenalectomy and bilateral ovarian cystectomies. In 2019, she underwent a left adrenalectomy due to hypertension, hypokalaemia, and a large mass in her left adrenal gland. Despite regular treatment with antihypertensive drugs and sustained-release potassium chloride tablets, her condition remained unsatisfactory. The patient began menstruating at the age of 12, with cycles of 7 days/28–30 days, and went through menopause at age 39. She is an offspring of healthy nonconsanguineous Chinses parents. There is no history of hypertension and/or hypokalaemia in her family.
Physical examination showed weight of 58 kg, height of 150 cm, blood pressure of 166/92 mmHg, Tanner stage V breast development, absent axillary hair and stage II pubic hair. Laboratory investigations showed that serum potassium (K+), cortisol, oestradiol (E2) and plasma renin concentration (PRC) were lower than normal, whereas serum ACTH, FSH, LH, progesterone (P) and 24-hour urinary potassium were higher than normal (). Besides, the serum aldosterone (ALD) and 17α-hydroxyprogesterone (17-OHP) were in the normal low range ().
Table 1. Biochemical and hormonal levels.
Post-operative adrenal enhancement computed tomography (CT) revealed a 19 mm × 7 mm and a 16 mm × 33 mm soft-tissue image respectively in the right and the left adrenal areas (). B-type ultrasound showed a postmenopausal uterus and no mass was observed in the bilateral ovaries.
Figure 1. Adrenal enhancement CT showed, post-operative bilateral adrenal review, a soft tissue image of 19 mm × 7 mm in the adrenal areas on the right and a 16 mm × 33 mm on the left.
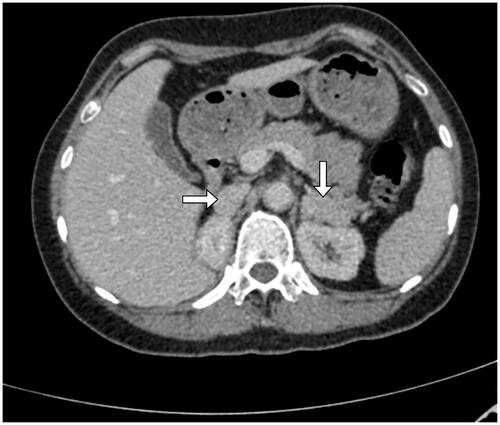
Based on the results of these biochemical and imaging assessments, the patient was diagnosed with a 17-OHD. As a treatment, the patient was given 0.5 mg of dexamethasone orally at night. One month later, the patient became normotensive, normokalemic, antihypertensives-free and potassium supplement-free.
To further determine the diagnosis of the disease, we performed whole exome sequencing with consent from the patient. The results showed that the patient carried a homozygous mutation in the exon 2 of CYP17A1 gene [c.391A > C] that predicts the aminoacid change p.T131P. This mutation is novel, which, to our best knowledge, has not been reported by previous research. Crucially, the PolyPhen-2 software (http://genetics.bwh.harvard.edu/pph2/) predicted that this mutation is potentially damaging.
Discussion
17-OHD is a rare form of congenital adrenal hyperplasia (CAH) that accounts for only 1% of CAH cases [Citation7]. It is classified into two main types: complete and partial. The partial type is much rarer than the complete type, especially in 46,XX patients. In this study, we present a case of a 41-year-old female (46,XX) patient carrying a novel homozygous missense CYP17A1 mutation, c.391 A > C (p.T131P), who was diagnosed with partial 17-OHD due to the presence of secondary sex characteristics and the lack of primary amenorrhoea.
17-OHD is caused by mutations in CYP17A1 gene encoding P450c17, which results in deficient activity of 17 α-hydroxylase and 17,20-lyase [Citation1,Citation2]. 17 α-hydroxylase deficiency causes hypertension and hypokalaemia, while 17,20-lyase deficiency leads to primary amenorrhoea and absence of secondary sex characteristics. However, in rare cases of partial 17-OHD, this mutation elicits subtler symptoms or only some of the 17-OHD symptoms [Citation3,Citation5,Citation6,Citation11]. In some cases of partial 17-OHD, the symptoms caused by 17 α-hydroxylase deficiency are present, while the symptoms caused by 17,20-lyase deficiency are mild or even absent [Citation5–10] (see , patients No. 1, 4, 5, 11). Our patient had hypertension and hypokalaemia, but menstruation and development of secondary sex characteristics, indicating that a part of 17,20-lyase activities was retained. Because of the lack of such typical clinical manifestations, she was misdiagnosed and underwent right and left adrenalectomy successively. After bilateral adrenalectomy, the residual adrenal glands were unable to produce enough cortisol and sex hormones, resulting in decreased cortisol levels, significantly increased ACTH levels, uncontrollable hypertension and hypokalaemia, and amenorrhoea.
Table 2. Characteristics of partial 17-hydroxylase deficiency in 46, XX: literature review.
To improve understanding of partial 17-OHD in 46,XX and reduce misdiagnosis, we conducted a systematic literature review. We searched the PubMed database in July 2022 using the following keywords: ‘CYP17A1 AND partial’, ‘17-hydroxylase/17,20-lyase AND partial’, ‘17-hydroxylase and 17,20-lyase AND partial’, ‘17OH deficiency AND partial’, and ‘P450c17 AND partial’. From the literature, we identified 13 46,XX probands with genetically proven partial 17-OHD.
A cohort of fourteen 46,XX probands with partial 17-OHD (thirteen from the literature and one from our centre) were included for analysis. Characteristics of partial 17-OHD in 46,XX are shown in . Mean age of the patients at the time of first diagnosis was 31.9 ± 11.9 years (range: 18–67 years). The age of first diagnosis was much older for 46,XX patients with partial 17-OHD than that reported for 46, XY patients with partial 17-OHD [Citation11] and for complete 17-OHD [Citation7,Citation8,Citation12]. This is unsurprising, because, unlike complete 17-OHD, partial 17-OHD in 46,XX lack typical clinical manifestations, which can lead to delayed diagnosis. As to 46,XY patients with partial 17-OHD, they usually present with ambiguous genitalia in early childhood, so they can be diagnosed quite early [Citation13].
Literature review revealed that primary amenorrhoea, hypertension and hypokalaemia accounted for only 15.4% (2/13), 42.9% (6/14), 38.5% (5/13) of the cohort of probands, respectively, indicating that absence of primary amenorrhoea, hypertension or hypokalaemia cannot rule out suspicion for CYP17A1 deficiency. Meanwhile, we found that most 46,XX patients with partial 17-OHD manifested varied levels of breast development (91.67%) and/or pubic hair development (66.67%), which is different from the absence of secondary sex characteristics in complete 17-OHD, suggesting that presence of secondary sex characteristics cannot rule out suspicion for CYP17A1 deficiency. Importantly, 72.7% (8/11) of the patients in our study present with ovarian cysts. Similarly, another study has also reported that ovarian enlargement was found in 68.7% of the patients with 17-OHD and ovarian macrocysts in 62.5% [Citation14]. Additionally, recurrent ovarian cysts are considered to be a typical clinical manifestation of partial 17-OHD, which might be caused by the elevated gonadotropins or serum P [Citation6]. Taken together, we suggest that ovarian cysts, especially recurrent ovarian cysts, are important indicators for 17-OHD.
Hormonal characteristics of the thirteen patients with partial 17-OHD (one patient was not included due to lack hormonal data) are summarised in . Usually, the metabolic signature of 17-OHD in 46, XX includes decreased concentrations of oestradiol, cortisol and 17-hydroxyprogesterone together with elevated ACTH, FSH, LH and progesterone [Citation15]. However, we found low level of oestradiol, cortisol and 17-OHP in only 15.4% (2/13), 38.5% (5/13) and 20% (2/10) of the patients, respectively, and elevated ACTH, FSH and LH in only 76.9% (10/13), 38.5% (5/13) and 53.8% (7/13), respectively. These can lead to difficulty in early diagnosis of partial 17-OHD in 46,XX. But these biochemical parameters, especially the baseline serum cortisol, can be used to distinguish complete deficiency from partial deficiency [Citation11]. Importantly, we found serum progesterone was elevated in all patients (12/12). This was similar to a recent study about partial 17-OHD in 46, XY [Citation11], which reported that elevated serum progesterone had high sensitivity for diagnosis of partial 17-OHD (83.3%). Therefore, serum progesterone can be used as a simple and reliable test for the diagnosis of 17-OHD [Citation16].
Table 3. laboratory examination of partial 17-hydroxylase deficiency in 46, XX: literature review.
17-OHD usually causes low-renin, low-aldosterone hypertension. The low aldosterone levels in patients with 17-OHD was due to excessive accumulation of the mineralocorticoid precursors, and transcriptional downregulation of aldosterone synthase [Citation17]. However, in our study, aldosterone level was unsuppressed in six out of eight patients, with one patient’s aldosterone level even elevated. This is consistent with a few studies which also found unsuppressed aldosterone level in some of patients with 17-OHD [Citation9,Citation11,Citation18] and one recent study reported higher aldosterone levels in patients with 17-OHD than healthy controls (p = .003) [Citation13]. These might be related to a cross-reaction between ALD and DOC or the function of the residual 17 α-hydroxylase/17,20-lyase activity. In a word, normal or elevated aldosterone level cannot rule out suspicion for 17-OHD.
By reviewing the genetic characteristics of the fourteen probands (), we identified eighteen different CYP17A1 pathogenic mutations including missense mutations (n = 13), frameshift mutations (n = 4) and intronic mutations (n = 1). Among these fourteen patients, eight carries compound heterozygous mutations and six carries homozygous mutations. Three probands carries homozygous mutations p.Y201N, p.F54del and p.R496C, respectively. Previous in vitro research showed that homozygous mutations p.Y201N, p.F54del and p.R496C could exhibit residual enzymatic activities: residual 17 α-hydroxylase was 31.3% [Citation19], 10–23% [Citation11] and 18.4% [Citation20] of that of wild-type, respectively, and residual 17,20-lyase was 33.7% [Citation19], 5–12% [Citation11], and 6.6% [Citation20] of that of wild-type, respectively. This is consistent with the untypical clinical presentations and hormonal characteristics of the three probands carrying the above mutations. In contrast, the enzymatic activity of homozygous mutations p.R358Q and p.R347C is inconsistent with clinical presentation. Previous study has shown that these two mutations can result in isolated 17,20-lyase deficiency [Citation21,Citation22], which reduces sex steroid production to a minimum, and consequently impairs development secondary sex characteristics. But, in our reviewed papers, two probands carrying the homozygous mutations p.R358Q and p.R347C were reported to have secondary sex characteristics [Citation10,Citation23]. This implies that the same genotype can possibly cause different enzyme activities and clinical phenotypes. Similarly, Costa-Santos et al. [Citation7] reported that three 46, XY patients carrying the same homozygous mutation R362C presented with ambiguous genitalia, normokalemia, and complete 17-OHD syndrome, respectively. Furthermore, in vitro research showed that p.Y329fs, p.D487_F489del and p.F435S mutations cause complete loss of enzyme activities of 17 α-hydroxylase and 17,20-lyase [Citation8,Citation24–28]. However, in our reviewed papers, compound heterozygous mutations comprising theses mutations and other mutations causing partial loss of enzyme activity resulted in partial 17-OHD [Citation8,Citation20]. To sum up, the different combination of CYP17A1 mutations have different effects on the activities of the 17-hydroxylase and/or17,20-lyase, and the relationship between in vitro enzyme activity and clinical severity of 17-OHD is still unclear. We speculate that different phenotypes of individual partial 17-OHD might be influenced by other gene variations, which could be verified by high-throughput gene analysis.
Our results have some limitations. First, our data are taken from retrospective studies and the sample size is small, which can lead to unreliable results. Second, these data of clinical and biochemical phenotype and enzyme activity can be influenced by ages of patients and different laboratory protocols, which limited our ability to assess the relationship among phenotype, enzyme activity and genotype. Finally, the homozygous mutation p.T131P from our centre was a novel variant, but the enzymatic activity data was not available. Further studies are warranted to quantify the residual enzyme activity of this novel variant.
In conclusion, we reported a partial 17-OHD case with a novel homozygous CYP17A1 mutation, c.391 A > C (p.T131P), which expanded the spectrum of identified CYP17A1 defects. Based on the literature review, presence of secondary sexual characteristics and/or normotension, normokalemia cannot rule out the suspicion for 17-OHD for 46,XX patients, and, more importantly, elevated serum progesterone is a critical clue for the diagnosis of 17-OHD. The current study can help early diagnosis of partial 17-OHD. But the relationship between enzymatic activities of the 17-hydroxylase and/or17,20-lyase and clinical severity of 17-OHD is still unclear, which needs further research, for example, analysing possible oligogenic effects of other gene variations.
Ethics approval and consent to participate
All procedures involving human subjects were approved by the ethics committee of Affiliated Jinhua hospital, Zhejiang University School of Medicine.
Authors’ contributions
HC drafted the manuscript; HC, HH and XL revised the manuscript; HC, YC and HM performed the clinical literature review. All authors read and approved the manuscript.
Consent for publication
The patient provided written informed consent for the publication of clinical details, and clinical images were obtained.
Acknowledgements
We are deeply grateful to the patient’s family for providing medical records and medical images for our research and to the clinicians for their contributions to this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The sequence datasets generated during the current study are not publicly available because it is possible that individual privacy could be compromised.
Additional information
Funding
References
- Kim YM, Kang M, Choi JH, et al. A review of the literature on common CYP17A1 mutations in adults with 17-hydroxylase/17,20-lyase deficiency, a case series of such mutations among Koreans and functional characteristics of a novel mutation. Metabolism. 2014;63(1):1–7.
- Miller WL. The syndrome of 17,20 lyase deficiency. J Clin Endocrinol Metab. 2012;97(1):59–67.
- Marsh CA, Auchus RJ. Fertility in patients with genetic deficiencies of cytochrome P450c17 (CYP17A1): combined 17-hydroxylase/17,20-lyase deficiency and isolated 17,20-lyase deficiency. Fertil Steril. 2014;101(2):317–322.
- Tian Q, Yao F, Zhang Y, et al. Molecular study of five Chinese patients with 46XX partial 17a-hydroxylase/17,20-lyase deficiency. Gynecol Endocrinol. 2012;28(3):234–238.
- Yanase T, Simpson ER, Waterman MR. 17 α-hydroxylase/17,20-lyase deficiency: from clinical investigation to molecular definition. Endocr Rev. 1991;12(1):91–108.
- Tian Q, Zhang Y, Lu Z. Partial 17alpha-hydroxylase/17,20-lyase deficiency-clinical report of five Chinese 46,XX cases. Gynecol Endocrinol. 2008;24(7):362–367.
- Costa-Santos M, Kater CE, Auchus RJ. Two prevalent CYP17 mutations and genotype-phenotype correlations in 24 Brazilian patients with 17-hydroxylase deficiency. J Clin Endocrinol Metab. 2004;89(1):49–60.
- Zhang M, Sun S, Liu Y, et al. New, recurrent, and prevalent mutations: clinical and molecular characterization of 26 CHINESE patients with 17alpha-hydroxylase/17,20-lyase deficiency. J Steroid Biochem Mol Biol. 2015;150:11–16.
- Wu C, Fan S, Qian Y, et al. 17alpha-hydroxylase/17, 20-lyase deficiency: clinical and molecular characterization of eight Chinese patients. Endocr Pract. 2017;23(5):576–582.
- Yamagata S, Kageyama K, Usui T, et al. Identification of a homozygous c.1039C > T (p.R347C) variant in CYP17A1 in a 67-year-old female patient with partial 17alpha-hydroxylase/17,20-lyase deficiency. Endocr J. 2022;69(2):115–120.
- Maheshwari M, Arya S, Lila AR, et al. 17α-hydroxylase/17,20-lyase deficiency in 46,XY: our experience and review of literature. J Endocr Soc. 2022;6(3):bvac011.
- Xia J, Liu F, Wu J, et al. Clinical and genetic characteristics of 17 α-Hydroxylase/17, 20-Lyase deficiency: c.985_987delTACinsAA mutation of CYP17A1 prevalent in the Chinese Han population. Endocr Pract. 2022;27(2):137–145.
- Kurnaz E, Kartal Baykan E, Turkyilmaz A, et al. Genotypic sex and severity of the disease determine the time of clinical presentation in steroid 17α-hydroxylase/17,20-lyase deficiency. Horm Res Paediatr. 2020;93(9–10):558–566.
- Carvalho LC, Brito VN, Martin RM, et al. Clinical, hormonal, ovarian, and genetic aspects of 46,XX patients with congenital adrenal hyperplasia due to CYP17A1 defects. Fertil Steril. 2016;105(6):1612–1619.
- Auchus RJ. Steroid 17-hydroxylase and 17,20-lyase deficiencies, genetic and pharmacologic. J Steroid Biochem Mol Biol. 2017;165(Pt A):71–78.
- Martin RM, Lin CJ, Costa EM, et al. P450c17 deficiency in Brazilian patients: biochemical diagnosis through progesterone levels confirmed by CYP17 genotyping. J Clin Endocrinol Metab. 2003;88(12):5739–5746.
- Takeda Y, Yoneda T, Demura M, et al. Genetic analysis of the cytochrome P-450c17alpha (CYP17) and aldosterone synthase (CYP11B2) in Japanese patients with 17alpha-hydroxylase deficiency. Clin Endocrinol. 2001;54(6):751–758.
- Han B, Xue L, Fan M, et al. Clinical and molecular manifestation of fifteen 17OHD patients: a novel mutation and a founder effect. Endocrine. 2016;53(3):784–790.
- Taniyama M, Tanabe M, Fau- Saito H, et al. Subtle 17alpha-hydroxylase/17,20-lyase deficiency with homozygous Y201N mutation in an infertile woman. J Clin Endocrinol Metab. 2005;90(5):2508–2511.
- Xu Y, Jiang S, Yan Z, et al. Phenotypic heterogeneity and fertility potential of patients with 17-hydroxylase/17,20-lyase deficiency. J Clin Endocrinol Metab. 2022;107(6):e2610–e2618.
- Geller DH, Auchus RJ, Mendonca BB, et al. The genetic and functional basis of isolated 17,20-lyase deficiency. Nat Genet. 1997;17(2):201–205.
- Van Den Akker EL, Koper JW, Boehmer AL, et al. Differential inhibition of 17alpha-hydroxylase and 17,20-lyase activities by three novel missense CYP17 mutations identified in patients with P450c17 deficiency. J Clin Endocrinol Metab. 2002;87(12):5714–5721.
- Sousa Paredes SC, Marques O, Alves M. Partial deficiency of 17alpha-hydroxylase: a rare cause of congenital adrenal hyperplasia. BMJ Case Rep. 2019;12(12):e230778.
- Qiao J, Han B Fau- Liu B-L, Liu Bl Fau- Liu W, et al. A unique exonic splicing mutation in the CYP17A1 gene as the cause for steroid 17α-hydroxylase deficiency. Eur J Endocrinol. 2011;164(4):627–633.
- Chen H, Yuan K, Zhang B, et al. A Novel compound heterozygous CYP17A1 variant causes 17α-Hydroxylase/17, 20-Lyase deficiency. Front Genet. 2019;10:e0996.
- Fardella CE, Zhang LH, Mahachoklertwattana P, et al. Deletion of amino acids Asp487-Ser488-Phe489 in human cytochrome P450c17 causes severe 17 alpha-hydroxylase deficiency. J Clin Endocrinol Metab. 1993;77(2):489–493.
- Lam CW, Arlt W, Chan CK, et al. Mutation of proline 409 to arginine in the meander region of cytochrome p450c17 causes severe 17 alpha-hydroxylase deficiency. Mol Genet Metab. 2001;72(3):254–259.
- Qiao J, Chen X, Zuo CL, et al. Identification of steroid biosynthetic defects in genotype-proven heterozygous individuals for 17alpha-hydroxylase/17,20-lyase deficiency. Clin Endocrinol. 2010;72(3):312–319.
- Croxson M, Ogilvie CM, Milsom S, et al. Delayed puberty from partial 17-alpha hydroxylase enzyme deficiency. N Z Med J. 2012;125(1355):71–74.
- Xia Y, Shi P, Xia J, et al. Novel mutations of the CYP17A1 gene in four Chinese 46,XX cases with partial 17a-hydroxylase/17,20-lyase deficiency. Steroids. 2021;173:108873.