
Abstract
Introduction
Von Hippel-Lindau disease (e.g. VHL) is an autosomal dominant multi-organ cancer syndrome caused by a mutation in the VHL tumour suppressor gene. In this study, we introduce a novel genetic variant found in 11 family members diagnosed initially with isolated Pheochromocytoma. Subsequent findings revealed its association with VHL syndrome and corresponds to the Type 2 C phenotype.
Methods
The VHL gene was amplified through the utilisation of the polymerase chain reaction (PCR). PCR fragments were sequenced using bidirectional Sanger sequencing, using BigDye™ Terminator v3.1 Cycle Sequencing Kit, running on the 3500 genetic analyser. Results were assembled and analysed Using Software SeqA and chromas pro.
Results
A heterozygous in-frame duplication of three nucleotides, specifically ATG, c.377_379dup; p.Asp126dup in exon 2, was identified in all the patients tested within the pedigree.
Conclusion
In this study, we disclose the identification of a novel genetic variant in a Jordanian family, affecting eleven family members with pheochromocytoma associated with VHL disease. This finding underscores the importance of screening family members and contemplating genetic testing for individuals newly diagnosed with pheochromocytoma and could enhance our comprehension of the potential adverse consequences associated with VHL germline mutations.
PLAIN LANGUAGE SUMMARY
Goal: To study a novel gene change in a family with Von Hippel-Lindau (e.g. VHL) syndrome, which increases cancer chances.
Participants: 11 family members with Pheochromocytoma, a tumour linked to VHL.
Methods:
Used PCR to copy the VHL gene.
Analysed the gene using Sanger sequencing.
Findings:
Found a novel gene change in all family members. This change, called an in-frame duplication, affects a protein.
It’s in a specific part of the gene.
Conclusion:
Stressing the importance of genetic testing for Pheochromocytoma patients to grasp VHL mutation risks.
Introduction
Von Hippel-Lindau disease (e.g. VHL) is an inherited multi-organ cancer syndrome caused by a mutation in the VHL tumour suppressor gene. It follows an autosomal dominant inheritance pattern, and its incidence is approximately one in 36,000 live births [Citation1]. Individuals with VHL are at a higher risk of developing both benign and malignant tumours in various organs, such as the central nervous system, kidneys, adrenal glands, and pancreas [Citation1,Citation2]. The prevalence of pheochromocytoma in individuals with VHL falls within the range of 7% to 20% [Citation2].
The VHL tumour suppressor gene’s germline mutations are located on chromosome 3 (3p25-p26) [Citation3]. Lately, there has been substantial progress in screening patients for germline mutations, especially with the introduction of swift genetic screening methods. This progress enhances our capacity to predict the development of bilateral and malignant diseases, as well as to conduct screenings for family members [Citation4,Citation5].
The VHL tumour suppressor gene consists of three exons, and it is responsible for encoding a 213-amino acid protein featuring two structural domains: the α-domain and the β-domain [Citation6,Citation7]. Exon 1 encodes amino acids from 1 to 114, exon 2 encodes amino acids from 114 to 155, and exon 3 encodes amino acids from 155 to 213 [Citation7].
The inactivation of the germline VHL gene results in the upregulation of hypoxia-inducible factors, which subsequently leads to the activation of subsequent targets such as Vascular Endothelial Growth Factor and Erythropoietin [Citation8]. VHL disease can be classified into two main groups: type 1, typically characterised by the absence of pheochromocytoma, and type 2, where pheochromocytoma is predominantly present [Citation8]. Within VHL type 2, there are further subdivisions: type 2 A, involving renal cancer; type 2B, which does not involve renal cancer; and type 2 C, where patients develop isolated pheochromocytomas [Citation8,Citation9].
We present a group of 11 Jordanian family members who initially exhibited drug-resistant hypertension. Initially, they received a diagnosis of pheochromocytoma; however, subsequent investigation revealed that they had VHL syndrome, and their genetic variant was identified as a novel one, corresponding to Type 2 C.
A heterozygous in-frame duplication of three nucleotides, specifically ATG, c.377_379dup; p.Asp126dup in exon 2, was identified in all the patients tested within the pedigree. This novel genetic mutation was documented in a patient on cBioPortal for Cancer Genomics under sample ID (TCGA-RW-A67X-01), classified as an In_Frame_Ins mutation type [Citation10].
Methodology
This study was approved by the University of Jordan Institutional Review Board committee (approval number 67/2019/7177) and was conducted in concordance with the Declaration of Helsinki’s latest report. It was conducted from January 2020 through March 2021 at Jordan University Hospital (JUH), a tertiary medical centre in Jordan. Informed consent was obtained from all participants.
Samples collection and DNA extraction
Samples were gathered in EDTA whole blood tubes, and Genomic DNA extraction was carried out using a commercial kit, specifically the Qiagen minielute kit from Germany, following the manufacturer’s instructions. The concentration of double-stranded DNA (dsDNA) was assessed using a spectrophotometer, namely the NanoDrop 2000 from the USA, and it exhibited an appropriate concentration of approximately 20 ng/μL with a purity ratio of approximately 1.8 at 260/280 nm.
Polymerase chain reaction (PCR)
The VHL gene was amplified through the utilisation of the polymerase chain reaction (PCR), employing newly designed Primers from IDT in the USA, as illustrated in .
Table 1. The specific forward and reverse primers utilised for sequencing the VHL gene.
Agarose gel electrophoresis
The samples were examined for a correctly sized PCR amplification using a 2% agarose gel and were viewed under a UV light using the BioRad Chemidoc System (Hercules, CA). The detection of a 500 bp band confirmed a successful PCR amplification for exon1, while exon2 and exon3 showed bands of 280 bp and 700 bp, respectively.
Sequencing
PCR fragments were sequenced using bidirectional Sanger sequencing, using BigDye™ Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, USA), running on the 3500 genetic analyser (Applied Biosystems, USA).
Analysis
Results were assembled and analysed Using Software SeqA (Applied Biosystems, USA) and chromas pro (Technelysium, au).
Results
A heterozygous in-frame duplication of three nucleotides, specifically ATG, c.377_379dup; p.Asp126dup in exon 2, was identified in all the patients tested within the pedigree. The clinical and genetic data of the study participants are presented in . and display the chromatogram results.
Figure 1. The forward primer designed for exon 2 of the VHL gene.
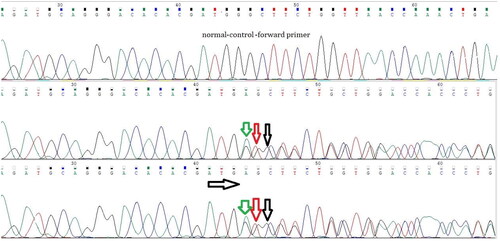
Figure 2. The reverse primer designed for exon 2 of the VHL gene.
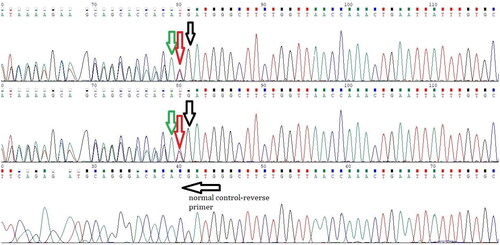
Table 2. The Clinical and genetic data of the study participants.
Discussion
VHL syndrome is a multi-organ disorder characterised by the development of benign and malignant tumours in various organs [Citation11]. These tumours include hemangioblastomas in the central nervous system and retina, clear cell renal cell carcinoma, pheochromocytoma, pancreatic cysts, and neuroendocrine tumours [Citation11].
The list of genes implicated in the development of pheochromocytoma and paraganglioma (e.g. PPGL) has steadily grown, now encompassing over 20 genes associated with either the hereditary or sporadic forms of the disease [Citation12]. It is estimated that around 40% of PPGL patients carry an autosomal dominant germline mutation in one of these 20 susceptibility genes [Citation12].
Approximately 35% of patients with VHL-associated pheochromocytoma display no symptoms, maintain normal blood pressure, and exhibit normal laboratory values for fractionated catecholamines and metanephrines [Citation13].
PPGL are regarded as the most heritable tumours, with an estimated 30–35% of patients possessing a germline mutation, and 40–45% having a somatic driver mutation in one of the more than 20 susceptibility genes identified thus far [Citation14]. The accuracy of genetic testing is crucial for precisely assessing tumour behaviour in affected individuals. This precision can facilitate earlier detection, personalised management, and ultimately improve outcomes [Citation14].
The most common VHL disease-associated genetic mutations encompass exonic deletions, in-frame insertions and deletions, truncating point mutations, missense mutations, splice-site mutations, and frameshift insertions and deletions [Citation15]. Among 804 germline VHL pathogenic variants documented in 945 VHL families, there were 491 missense mutations, 126 frameshift mutations, 106 nonsense mutations, 28 splice junction variants, 53 in-frame deletions/insertions, and 103 large deletion [Citation15].
We introduce a family comprising 11 members who initially displayed drug-resistant hypertension and were later diagnosed with pheochromocytoma linked to VHL syndrome. Their diagnosis of an isolated pheochromocytoma was established by the presence of elevated plasma fractionated metanephrines and/or 24-h urine levels of fractionated metanephrines and catecholamines, in conjunction with CT scans that revealed either unilateral or bilateral adrenal masses.
The majority of our patients presented with drug-resistant hypertension. Upon inquiry, some exhibited classic symptoms of pheochromocytoma, including recurrent headaches, palpitations, tremors, and anxiety attacks, while others were incidentally diagnosed with hypertension.
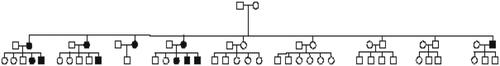
All of our patients underwent treatment, which included unilateral or bilateral adrenalectomies. Subsequently, it was revealed that all of them had VHL syndrome through genetic testing, with 9 out of the 11 patients consenting to undergo genetic testing. A heterozygous in-frame duplication of three nucleotides, specifically ATG, c.377_379dup; p.Asp126dup in exon 2, was identified in all the patients tested within the pedigree. displays the family pedigree of the participants.
Figure 3. The family pedigree of the participants, highlighting the 11 affected patients. Other family members either passed away or declined to participate, preventing us from ruling out their involvement.
Our patients predominantly exhibited elevated plasma normetanephrine concentrations while maintaining normal metanephrines levels, which is in line with prior studies suggesting that individuals with VHL disease typically exhibit elevated plasma normetanephrines levels exclusively, as indicated in previous research [Citation16,Citation17].
Out of the eleven patients, two presented with bilateral pheochromocytoma. Ophthalmologic examinations were conducted on all patients, and none of them exhibited retinal hemangioblastomas. Head CT scans revealed no signs of hemangioblastomas in any of the patients, and abdominal CT scans showed no presence of renal cell masses, pancreatic tumours, or any other abnormalities, except for the adrenal masses/pheochromocytomas.
Two of our patients experienced recurrent left adrenal pheochromocytoma. One recurrence occurred four years after the initial resection, while the other recurred within a year. This aligns with the earlier observations of an increased recurrence rate in patients with pheochromocytoma linked to genetic disorders [Citation18,Citation19].
Our study has some limitations. Despite making multiple attempts, asymptomatic non-hypertensive family members declined to participate in screening procedures. Their involvement could have been valuable in dispelling doubts regarding the pathogenicity of this novel VHL type 2 C genetic variant.
Conclusion
In this study, we disclose the identification of a novel genetic variant in a Jordanian family, affecting eleven family members with pheochromocytoma associated with VHL disease. This finding underscores the importance of screening family members and contemplating genetic testing for individuals newly diagnosed with pheochromocytoma and could enhance our comprehension of the potential adverse consequences associated with VHL germline mutations.
Consent form
We obtained consent for publication from all the participants.
Acknowledgements
We want to express our gratitude to all the participants who voluntarily enrolled in this study.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data supporting the results of this study can be requested from the corresponding author, Dr. Hussein Alhawari, MD.
Additional information
Funding
References
- Lonser RR, Glenn GM, Walther M, et al. Von Hippel-Lindau disease. Lancet. 2003;361(9374):1–7.
- Maher ER, Kaelin WGJr. Von Hippel-Lindau disease. Medicine. 1997;76(6):381–391. doi: 10.1097/00005792-199711000-00001.
- Schreinemakers JM, Zonnenberg BA, Höppener JW, et al. A patient with bilateral pheochromocytoma as part of a von Hippel-Lindau (VHL) syndrome type 2C. World J Surg Oncol. 2007;5(1):112. doi: 10.1186/1477-7819-5-112.
- Sbardella E, Cranston T, Isidori AM, et al. Routine genetic screening with a multi-gene panel in patients with pheochromocytomas. Endocrine. 2018;59(1):175–182. doi: 10.1007/s12020-017-1310-9.
- Neumann HP, Bausch B, McWhinney SR, et al. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346(19):1459–1466. doi: 10.1056/NEJMoa020152.
- Sorrell AD, Lee S, Stolle C, et al. Clinical and functional properties of novel VHL mutation (X214L) consistent with type 2A phenotype and low risk of renal cell carcinoma. Clin Genet. 2011;79(6):539–545. doi: 10.1111/j.1399-0004.2010.01464.x.
- Dandanell M, Friis-Hansen L, Sunde L, et al. Identification of 3 novel VHL germ-line mutations in Danish VHL patients. BMC Med Genet. 2012;13(1):54. doi: 10.1186/1471-2350-13-54.
- Gläsker S, Neumann HP, Koch CA, et al. Von Hippel-Lindau disease. InEndotext [Internet] 2018 Sep 12. MDtext. com, Inc.
- Zbar B, Kishida T, Chen F, et al. Germline mutations in the Von Hippel‐Lindau disease (VHL) gene in families from North America, Europe, and Japan. Hum Mutat. 1996;8(4):348–357. doi: 10.1002/(SICI)1098-1004(1996)8:4<348::AID-HUMU8>3.0.CO;2-3.
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, Antipin Y. The cBio cancer genomics multidimensional cancer genomics data. Cancer discovery. 2012 May 1:2(5):401–4.
- Van Leeuwaarde RS, Ahmad S, Van Nesselrooij B, Zandee W, Giles RH. Von Hippel-Lindau Syndrome. In: GeneReviews. Uniuversity of Washington, Seattle (WA); 1993. PMID: 20301636
- Cascón A, Calsina B, Monteagudo M, et al. Genetic bases of pheochromocytoma and paraganglioma. J Mol Endocrinol. 2023;70(3):e220167. doi: 10.1530/JME-22-0167.
- Walther MM, Reiter R, Keiser HR, et al. Clinical and genetic characterization of pheochromocytoma in von Hippel-Lindau families: comparison with sporadic pheochromocytoma gives insight into natural history of pheochromocytoma. J Urol. 1999;162(3 Pt 1):659–664. doi: 10.1097/00005392-199909010-00004.
- Kiriakopoulos A, Giannakis P, Menenakos E. Pheochromocytoma: a changing perspective and current concepts. Ther Adv Endocrinol Metab. 2023;14:20420188231207544. doi: 10.1177/20420188231207544.
- Friedman E. Clinical implications of germline pathogenic variants in the VHL gene. In: Von Hippel-Lindau disease: a comprehensive guide to diagnosis, treatment, and management, Dhaval Thakor Patel (editor), Amit Tirosh (editor). Cham (Switzerland): Springer International Publishing; 2024. p. 1–26.
- Eisenhofer G, Lenders JW, Linehan WM, et al. Plasma normetanephrine and metanephrine for detecting pheochromocytoma in Von Hippel–Lindau disease and multiple endocrine neoplasia type 2. N Engl J Med. 1999;340(24):1872–1879. doi: 10.1056/NEJM199906173402404.
- Maranchie JK, Walther MM. Early identification of patients with von Hippel-Lindau disease at risk for pheochromocytoma. Curr Urol Rep. 2001;2(1):24–30. doi: 10.1007/s11934-001-0022-z.
- Plouin PF, Chatellier G, Fofol I, et al. Tumor recurrence and hypertension persistence after successful pheochromocytoma operation. Hypertension. 1997;29(5):1133–1139. doi: 10.1161/01.hyp.29.5.1133.
- Shirali AS, Clemente-Gutierrez U, Huang BL, et al. Pheochromocytoma recurrence in hereditary disease: does a cortical-sparing technique increase recurrence rate? Surgery. 2023;173(1):26–34. doi: 10.1016/j.surg.2022.05.003.