
ABSTRACT
Recent advances have led to therapeutic options becoming available for people with inherited retinal disease. In particular, gene therapy has been shown to hold great promise for slowing vision loss from inherited retinal disease. Recent studies suggest that gene therapy is likely to be most effective when implemented early in the disease process, making consideration of paediatric populations important. It is therefore necessary to have a comprehensive understanding of retinal imaging in children with inherited retinal diseases, in order to monitor disease progression and to determine which early retinal biomarkers may be used as outcome measures in future clinical trials. In addition, as many optometrists will review children with an inherited retinal disease, an understanding of the expected imaging outcomes can improve clinical care. This review focuses on the most common imaging modality used in research assessment of paediatric inherited retinal diseases: optical coherence tomography. Optical coherence tomography findings can be used in both the clinical and research setting. In particular, the review discusses current knowledge of optical coherence tomography findings in eight paediatric inherited retinal diseases – Stargardt disease, Bests disease, Leber’s congenital amaurosis, choroideremia, RPGR related retinitis pigmentosa, Usher syndrome, X-linked retinoschisis and, Batten disease.
Introduction
Inherited retinal diseases are a heterogenous group of degenerative genetic eye diseases which can lead to profound vision impairment. Inherited retinal diseases can be caused by mutations in over 300 different genes (RetNet, https://sph.uth.edu/RetNet/), many of which result in similar clinical phenotypes. Whilst individually rare, the subtypes of inherited retinal diseases collectively are now the most common cause of legal blindness in working age Australians.Citation1 The disease processes start at a young age, this presents a significant challenge over a lifetime, increasing the socioeconomic impact of the condition.
The impact of inherited retinal diseases has led to a major effort to produce viable treatment options, which can either reverse disease pathology or halt its progression. Of these, retinal gene therapy has shown particular promise, leading to the regulatory approval of voretigene neparvovec-rzyl (Luxturna) for biallelic RPE65 mutations leading to Leber’s congenital amaurosis in December 2017 in the USA and November 2018 in Europe.Citation2–4 The clinical trials for this drug, as well as other inherited retinal diseases,Citation5 have shown that efficacy is often most pronounced in early disease stages.Citation3,Citation4,Citation6–8 In the case of Leber’s congenital amaurosis, an early onset inherited retinal disease, this means that children may be the best candidates for treatment.
For the success of gene therapy trials, the importance of natural history measures and appropriate outcome measures has been highlighted.Citation9,Citation10 Given that children are likely to be subjects for future gene therapy trials, a better understanding of the relevance of key outcome measures is required when applied to paediatric patients. Ocular imaging, especially optical coherence tomography (OCT), is a clear choice as an outcome measure in paediatric trials, as it is widely utilised in ophthalmologyCitation11 and enables fast image acquisition.Citation12
Whilst a number of reviews have been published on imaging in adult inherited retinal diseases,Citation13–16 a clinically-oriented summary of OCT features in preschool or school-aged children with inherited retinal diseases has apparently not been published previously. A recent review has detailed the use of OCT to evaluate various retinal pathologies in neonates and infants,Citation17 while noting that obtaining imaging in these younger children is in fact easier than children of preschool and school age. Given the increase of interest in this cohort, from both clinical and academic groups, a review of the current understanding of retinal pathology in children with inherited retinal diseases is presented, based on OCT imaging. The paediatric population is defined as all ages under 18 years.
Literature search
A literature search was conducted in July 2022, using the PubMed and Embase databases and the search terms ‘inherited retinal disease, optical coherence tomography and paediatric’ were used. A total of 247 papers were identified that incorporated some paediatric references and data. From this, papers were selected that focussed on key pathologies, as decided by those seen most commonly by one of the authors (JBR) in his paediatric practice, and those conditions currently in development for therapeutic treatment. One hundred and two papers were found which covered eight main pathologies – Stargardt disease, Leber’s congenital amaurosis, Bests disease, retinitis pigmentosa (RP), choroideremia, Usher syndrome, X-linked retinoschisis and Batten disease ().
Figure 1. Distribution of inherited retinal diseases (IRDs) in the 102 peer-reviewed papers included in this review. Publications identified through Pubmed and Embase literature review (to July 2022).
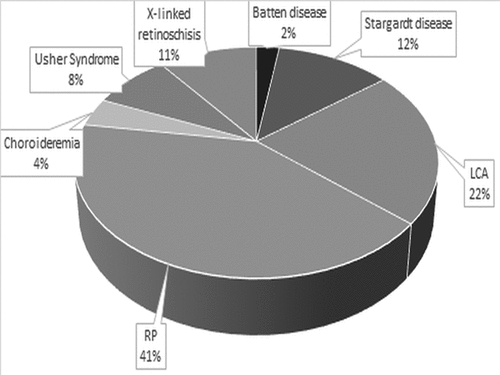
Given the broad nature of retinitis pigmentosa, what follows focuses on RPGR-related retinitis pigmentosa, due to current gene therapy advancements for this disease, and with it being the most common presentation of retinitis pigmentosa in a paediatric population. Further searches were conducted on these eight diseases for studies with adult cohorts, to further identify paediatric data within these papers. The other 102 papers, from the original search, contained a variety of other inherited retinal diseases. These papers were not included in the review, to allow the focus to be on the eight specific subtypes highlighted above.
Any references to retinal layers in this manuscript is per the ‘In.OCT’ consensus document.Citation18 The ‘ProgStar’ study reported that segmentation errors are common and need to be corrected in order to provide accurate thickness measurements, with good training of graders to ensure high agreement between different graders.Citation19 Several layers should be considered rather than total thickness to provide a picture of what is happening in the disease.Citation20 Further development of automatic accurate segmentation algorithms is required.Citation20,Citation21 Although this work was performed in Stargardt disease, it can be extrapolated to all inherited retinal diseases due to the degeneration caused in the retinal layers.Citation22
Of the 102 papers, 12 discussed Stargardt disease, 6 Bests disease, 19 Leber’s congenital amaurosis, 10 retinitis pigmentosa, 13 choroideremia, 11 Usher syndrome, 19 X-linked retinoschisis and, 12 Batten disease. Summaries of the findings in each disease subcategory are discussed below.
Stargardt disease
Autosomal recessive Stargardt disease is a maculopathy affecting around one in 8,000–10,000 people, and thereby the most common macular dystrophy.Citation23 This disease is caused by biallelic mutations in the cell membrane transporter gene, ATP Binding Cassette Subfamily A Member 4 (ABCA4). ABCA4 facilitates the clearance of all-trans-retinal from photoreceptor disc membranes following photoexcitation; loss of its function is toxic to the retinal pigment epithelium (RPE) and retina, leading to the degeneration of photoreceptors in the macula.Citation24
The disease commonly presents with central vision loss, macular atrophy and yellow-white flecks at the posterior pole.Citation25 The age of onset in Stargardt disease varies, and ABCA4-related macular atrophy and flecks have been observed in children as young as three,Citation25 as well as older adult-onset disease. The flecks appear on OCT as hyperreflective deposits that can traverse several layers. Disruption or loss of the ellipsoid zone occurs as a result of photoreceptor degeneration. A greater loss of ellipsoid zone is associated with an increased loss of visual acuity. Progressive thinning of the outer nuclear layer occurs. As the disease progresses, inner retinal thickening occurs indicating a process known as retinal remodelling.Citation26
Stargardt disease is the most common form of juvenile onset macular degeneration.Citation25 Consequently, this disease has allowed greater sample sizes for paediatric studies and is the best characterised inherited retinal disease in a paediatric population.
Tanna et al.Citation24 reported structural changes to the retina in 46 paediatric patients. This study used SD-OCT to longitudinally assess the ellipsoid zone of ABCA4 confirmed STGD1 patients with a mean age of 12.4 years. They found a mean annual rate of ellipsoid zone loss of 1.20 ± 1.29 mm2/year. Interestingly, the rate of ellipsoid zone loss in this study was found to be three times faster than that found in an older cohort (mean age of 33 years).Citation27
This finding was also supported by Fujinami et al., who identified that childhood-onset ABCA4 related Stargardt disease progresses faster and more severely in comparison to a later onset, their study included 42 patients who had onset of disease before the age of 17.Citation25 Foveal outer retinal disruption was found in all eyes whilst severe foveal thinning was found in both eyes of 18 out of the 21 patients who were assessed with SD-OCT.
Another common observation in paediatric Stargardt patients is a change to the external limiting membrane, with thickening and increased reflectivity notedCitation28–30 (see ). This was first observed by Burke et al.Citation28 in an asymptomatic five-year-old girl who had a family history of a pathogenic mutation in ABCA4 gene (splice mutation IVS 35 + 2, T>C). SD-OCT revealed external limiting membrane thickening, with no other abnormalities in other retinal layers. The authors postulated that the external limiting membrane thickening may be an early OCT feature of Stargardt disease.
Figure 2. A nine-year-old patient with STGD1 (A) with an observable thickening of the ELM compared to an age-matched control (B). STGD1 image courtesy of Dr Jonathan Ruddle, Melbourne Children’s Eye Clinic. Control image provided by Dr Marianne Coleman from the Australian College of Optometry.
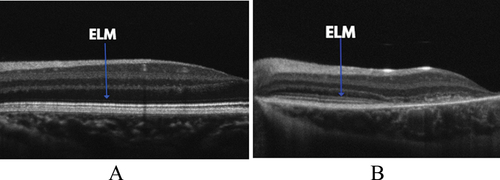
Khan et al. also found an increase in reflectivity in the external limiting membrane in four asymptomatic patients aged 8, 9, 11 and 13.Citation30 Following this initial finding, Lee et al.Citation29 analysed the external limiting membrane of 26 Stargardt patients under the age of 20 using SD-OCT. This study found thickening of the external limiting membrane was a consistent feature in all patients with Stargardt and was not found in age-matched healthy controls (mean thickness of 17.69 μm vs 11.45 μm in controls, p < 0.0001). There was also a mild downward trend of external limiting membrane thickness with age, suggesting that younger patients may show a more obvious thickening. These early changes to the external limiting membrane in Stargardt disease may provide clinicians with an early marker for Stargardt disease prior to the onset of other features, which can then be confirmed by genetic testing.
Stargardt disease can also be caused by autosomal dominant mutations in PROM1 or ELOVL4 genes. These mutations produce a similar phenotype to Stargardt and are distinguished based on the mode of inheritance.Citation31 Compared to Stargardt, there are fewer studies examining the OCT appearance of children with autosomal dominant Stargardt disease. Palejwala et al.Citation31 examined OCT images of PROM1 and ELOVL4 patients, which included four paediatric patients with known ELOVL4 mutations. They found thinning of the outer nuclear layer and disruption to the ellipsoid zone. Additionally, the youngest patient (aged 7) showed a thickening of the external limiting membrane similar to Stargardt disease.
Bests disease
Bests disease or Best Vitelliform Macular Dystrophy results from mutations in the BEST1 gene and can be inherited in an autosomal dominant pattern with rarer phenotypes inherited with recessive inheritance. BEST1 codes for a calcium channel protein in the RPE. The typical appearance is a yellow lesion, described as a vitelliform disc in the macula, and can resemble an egg yolk.Citation32,Citation33 This is formed from an accumulation of lipofuscin and photoreceptor debris.Citation33 Traditionally, the key diagnostic test has been an abnormal electrooculogram with a reduced Arden Index of less than 1.5, but this can be difficult for children to perform as it requires good concentration and co-operation to obtain high-quality eye movements. This is where imaging can play a part in the diagnosis and management of Bests Disease as the outer nuclear layer defects present in childhood.
Vitelliform lesions frequently present with normal vision, which decreases as the yolk appearance ‘scrambles’ and partially reabsorbs. As atrophy occurs, this can be accompanied by choroidal neovascularisation. The vitelliform lesion appears as hyperfluorescent on short wavelength fundus autofluorescence imaging, whilst atrophic areas appear as hypofluorescent.Citation33,Citation34 Fibrosis and neovascularisation can occur at any age, with active neovascularisation being differentiated from quiescent using multimodal imaging.Citation35
The earliest changes visible on OCT are a thickening of the interdigitation zone of cones and RPE apical processes centrally. Once the vitelliform lesion develops, a dome-shaped separation between the RPE and photoreceptors is visible at the fovea with hyperreflective borders and filled with subretinal fluid. Inner retinal layer morphology remains unaffected despite photoreceptor thinning.Citation33 Changes were seen at a variety of levels in the retina in different eyes, with a serious retinal detachment, choroidal changes, RPE elevation, subretinal hyperreflectivity above the RPE, intraretinal fluid, and fibrosis being noted.Citation35
Autosomal recessive bestrophinopathy is the rare recessive form resulting from biallelic mutations and results in a more severe in a more severe phenotype, often presenting earlier. Fundal appearance can appear normal or appear similar to Bests disease early on. This becomes a central serious detachment with a fibrous subretinal scar, with neovascularisation. Autofluorescence abnormalities cover a wider area than in Bests disease.Citation34,Citation36
The youngest presentation recorded in the literature is a child aged 10 months presenting with retinal pigment mottling. Circular lesions could be seen on colour negative fundus images.Citation37
Leber’s congenital amaurosis
Leber’s Congenital Amaurosis is one of the most severe forms of inherited retinal disease and usually causes visual impairment and associated nystagmus before the age of one.Citation38 Leber’s Congenital Amaurosis can be a result of a mutation in 13 different recessive genes,Citation8 and is relatively rare, with a prevalence of around 1:80,000 worldwide.Citation39–41
Clinical presentation is initially normal fundi but abnormal electrodiagnostic results (both electroretinogram and visual evoked potential), and behaviours such as eye poking and rubbing. Children are photophobic and often have a high refractive error, particularly hypermetropia. Leber’s Congenital Amaurosis can be associated with a range of neurological syndromes.Citation42
Phenotypes can vary widely. Fundus appearance can appear normal, pigmented such as seen in retinitis pigmentosa, oedematous, retinal vasculitis, or with nummular lesions of the RPE. A macular coloboma may be present. Astrocytoma or secondary angiomatosis may also be present.Citation42
The nystagmus associated with Leber’s Congenital Amaurosis presents challenges to clinicians when looking to obtain high-quality OCT images of the retina. Yang et al.Citation43 describe some steps taken to improve image quality in children with nystagmus, including the use of linear scans (rather than 3D volume scans) and the need for full pupil dilation.
The success and translation into clinical care of retinal gene therapy for Leber’s Congenital Amaurosis caused by biallelic mutations in RPE65 (voretigene neparvovec-rzyl)Citation3,Citation44 has meant that the disease has been relatively well studied in both children and adults.
Jacobson et al.Citation45,Citation46 analysed 11 RPE65 patients under the age of 18. Only two of these patients showed a decrease in foveal thickness. However, as eccentricity increased, all patients displayed a reduction in outer nuclear layer thickness, with increasing outer nuclear layer loss in the mid-periphery. This represents increasing loss of photoreceptors and increased disruption in the connectivity between the photoreceptors and the neural circuitry.Citation18 In the paediatric age group (3–14), outer nuclear layer thickness was on average 61% of the age matched controls, whilst the older age group (31–52) averaged an outer nuclear layer thickness of just 37% of the age matched controls. A second study by this group showed all subjects (aged 6–17 years) presented with abnormal outer retinal topography, but outer nuclear layer loss was variable, and did not correlate with age.Citation47
Studies have also investigated other genotypes linked with Leber’s Congenital Amaurosis. Miyamichi et al.Citation48 looked at four young patients with Leber’s Congenital Amaurosis caused by RPGRIP1 mutations. All subjects (aged 7–17) showed a reduced outer nuclear layer upon assessment. A single case report of a five-year old with the same gene mutation by Wang et al.Citation49 reported greater outer nuclear layer reduction than seen in the younger subjects of Miyamichi (1-year-old and three-month-old). This suggests outer nuclear layer loss occurs later in disease progression in RPGRIP1-related Leber’s Congenital Amaurosis, however, larger sample sizes would be required to investigate this hypothesis.
Jacobson et al.Citation46 studied 10 different genotypes across 24 patients with Leber’s Congenital Amaurosis. This cohort included three subjects with Leber’s Congenital Amaurosis caused by mutations in the GUCY2D gene (ages 11–14). Interestingly, this genotype was associated with a relatively normal appearance on OCT imaging, showing close-to-normal outer nuclear layer and retinal thickness across all eccentricities. Other studies have found paediatric patients to have a continuous or intact ellipsoid zone.Citation50 details the different OCT findings in LCA, based on the genotype.
Table 1. Summary of paediatric OCT findings in different LCA genotypes.
Adult patients with GUCY2D mutations also often maintain near-normal retinal structure, as reported by both Pasadhika et al.Citation51 and Jacobson et al.Citation52 However, despite the maintenance of retinal microstructure in these patients, visual acuity is still impaired. This has led to the conclusion that this form of Leber’s Congenital Amaurosis may present more biochemical dysfunction rather than structural.Citation51
An early diagnosis of Leber’s Congenital Amaurosis (particularly with the RPE65 genotype) is critical due to its severity and potential for treatment. If a patient is confirmed to have a biallelic RPE65 mutations, then voretigene neparvovec-rzyl gene therapy may be an option.Citation3
Choroideremia
Choroideremia is an X-linked recessive inherited retinal disease caused by mutations in the REP1 gene, and has a prevalence of approximately 1 in 50,000 people worldwide.Citation53 It is characterised by progressive degeneration of the choriocapillaris, RPE and retina. Nyctalopia is a characteristic early sign of disease and occurs in the first decade of life. This is followed by gradual peripheral field loss over the next three to five decades.Citation54
Fundus evaluation shows preservation of central and far peripheral islands of retina, visible on colour and autofluorescence imaging.Citation55 Outer nuclear thickness thinning occurs at the edge of the islands, with preservation of the ellipsoid zone within the islands. Outer retinal tubulations can be seen on the OCT in areas of marked degeneration. Choroidal thinning occurs independently to retinal thinning.Citation56 Central vision may be spared until later stages even until the sixth decade.Citation54,Citation57
Despite visual acuity changes usually occurring later in life, the central retina has noticeable changes in childhood. Aleman et al.Citation58 studied the natural history of 97 patients (aged 6–71 years) with choroideremia with SD-OCT, providing insight to both paediatric and adult patients. Outer nuclear layer and inner retinal thickness were normal in the first decade of life, with an inner retinal thickening and outer nuclear layer thinning observed in the second decade of life. Central retinal abnormalities were seen in all patients under the age of 15, including shortening of the photoreceptor outer segments and loss in the interdigitation zone between photoreceptor outer segments and the apical RPE.Citation58
Studying the central retina of choroideremia patients using OCT, all young patients maintained adequate central vision, despite having an increase in central retinal thickness (245 ± 21.2 µm vs 212 ± 8.01 µm in controls). Increased central retinal thickness has also been reported in other studies of paediatric choroideremia patients.Citation58–60 It is possible that this increase in central retinal thickness is a sign of early retinal remodelling due to Muller cells,Citation58 which likely extends to the second decade of life.Citation58
The relatively steady fixation in younger choroideremia patients (compared to cone-rod dystrophies, for example) allows OCT-angiography scanning, which has provided a means to examine the vasculature of affected patients. It is a non-invasive technique harnessing the reflectance of red blood cells to allow study of the retinal and choroidal vasculature. Murro et al.Citation61 used this tool to assess the vasculature of seven young choroideremia patients (five were <18 years). They found a decrease in both superficial and deep inner retinal capillary plexus (p < 0.01 for both), indicating a change in inner retinal blood flow in paediatric choroideremia patients. Similar changes have also been observed in studies with older age groups.Citation62,Citation63 This study also showed a significant reduction in choriocapillaris vessel density in young choroideremia patients compared to controls (51.62 ± 11.85% vs 74.53 ± 2.57%; p < 0.01). Gyrate atrophy is a condition that can be misdiagnosed as choroideremia. The OCTA findings in gyrate atrophy show the impact of cystic spaces, increasing the foveal avascular zone.Citation64
Choroidal assessment can be undertaken not only with OCT-A but with enhanced depth imaging or the chorioretinal mode on OCT. A method involving swept source OCT showed no impact of choroideremia on the choroidal vascular index, a metric related to vascular area. Thickness at different locations, however, proved to more a more sensitive marker of disease.Citation65 To fully understand disease changes, the normative range and choroidal changes caused by refractive error must be understood. A comprehensive study of schoolchildren showed there is topographical variation in choroidal thickness in control eyes with no significant refractive errorCitation66 and this did not change with age. Refractive error does impact the choroidal thickness with thickening seen in hyperopiaCitation67 and thinning in myopia.Citation68 This may be related to changes in ocular blood flow.Citation68
RPGR-related retinitis pigmentosa
RPGR-related retinitis pigmentosa is a severe form of retinitis pigmentosa which causes the degeneration of rod and cone photoreceptors at an early age, leading to severe visual deficiencies.Citation69 This disease has an X-linked mode of inheritance; hence, males are predominantly affected, although females demonstrate variable phenotypes.Citation70 Patients with RPGR retinitis pigmentosa present with common clinical hall marks of retinitis pigmentosa, such as pigmented peripheral regions on fundus images, poor night and peripheral vision, and reduced ERG responses.Citation71 It has also been shown that RPGR retinitis pigmentosa patients have loss of cone photoreceptors from early in disease course which affects visual acuityCitation72; hence, clinicians should consider this impact of visual function, including on education.
Menghini, Jolly et al.Citation72 utilised OCT to investigate foveal photoreceptor outer segments in a cohort which included a paediatric group (6 patients under the age of 16 years). OCT analysis found that RPGR retinitis pigmentosa patients had significantly thinner outer segments compared to controls (35.4 ± 2.6 µm vs 62.4 ± 0.7 µm in left eyes). When comparing the adult group of RPGR retinitis pigmentosa patients to the paediatric group, foveal photoreceptor outer segments thickness was similar (36.2 ± 4.1 µm adults vs 36.3 ± 2.1 µm paediatrics, in left eyes). This finding highlights early loss of cone photoreceptors in RPGR RP patients. Therefore, foveal outer segment thickness is an important area for clinicians to observe on OCT if RPGR retinitis pigmentosa is suspected, particularly with sub-clinically reduced visual acuity.
Birch et al.Citation73 explain that the point at which outer segment regions of photoreceptors disappear, indicates the edge of the healthiest region of the visual field. Their study used OCT to investigate the ellipsoid zone width in a cohort consisting predominantly of paediatric patients (44 patients with a mean age of 17.6 years). They showed that visual sensitivity rapidly declined just outside of the ellipsoid zone band. Hence, Birch et al. concluded that the anatomical ellipsoid zone measure can reflect a functional outcome. This may become an important outcome measure in monitoring the efficacy of gene therapy treatments and an important area to monitor for disease progression.
Despite this disease having an X-linked pattern, young symptomatic female carriers have also been identified.Citation74,Citation75 Females with mutations in the RPGR gene are generally asymptomatic or develop mild systems with a later onset.Citation76,Citation77 However, Shifera and KayCitation75 reported an eight-year-old female with RPGR RP, who displayed severe symptoms at the age of four. They report normal foveal contours on OCT with an intact RPE, however, given the obvious visual dysfunction, there are likely peripheral OCT abnormalities which have not been reported. Although rare, it is important for clinicians to consider that female carriers of X-linked conditions can present with severe symptoms.
Many of the hallmarks of RPGR retinitis pigmentosa will also be visible in other forms of retinitis pigmentosa.
Usher syndrome
Usher syndrome is syndromic form of retinitis pigmentosa associated with sensorineural hearing impairment.Citation78 The syndrome is an autosomal recessive disorder and has four clinical subtypes (1–4). Usher syndrome type 1, most commonly caused by MYO7A mutations, is responsible for around 30–40% of all Usher syndrome cases and causes visual impairment before the age of 10. Additionally, it causes deafness and vestibular dysfunction.Citation79,Citation80
Patients with Usher syndrome type 2 (mostly due to USH2A mutations) present with early moderate hearing loss and visual dysfunction that manifests in the second decade of life. However, vestibular function is normal.Citation80 Usher syndrome type 3 is the rarest form of disease and caused by CLRN1 mutations.Citation81 Patients present with severe visual defects and variable vestibular function, and progressive loss of hearing.Citation82 Usher syndrome type 4 is a recently identified autosomal recessive atypical form of Usher syndrome. It is characterised by late-onset retinitis pigmentosa and late-onset progressive sensorineural hearing loss without vestibular involvement.Citation83
The Usher syndrome associated genes can present as either Usher syndrome or as non-syndromic autosomal retinitis pigmentosa. When there is no associated hearing loss with the RP, the degeneration tends to present later, milder, and have less additional complications such as cystoid macular oedema.Citation81 In both cases, retinal presentation is as seen for retinitis pigmentosa and ellipsoid area is similar ().Citation84 Fundus autofluorescence shows a hyperfluorescent ring which is a marker of active degeneration. Alternatively, a focal increase in the autofluorescence signal can be seen at the fovea, and this was more likely in Ushers syndrome than non-syndromic variants. As degeneration progresses to atrophy, this will manifest as areas of hypofluorescence.Citation81,Citation85
Figure 3. Ellipsoid zone disruption shown on a 9-year-old USH1 patient (A) compared to control (B). Usher syndrome image provided by Dr Jonathan Ruddle, Melbourne Children’s Eye Clinic. Control image provided by Dr. Marianne Coleman from the Australian College of Optometry.
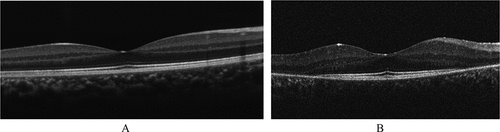
Where the hyperfluorescent ring is visible, the ellipsoid zone is preserved within the area encompassed within the ring. However, the eyes that show hyperfluorescence across the fovea, have widespread disruption of the ellipsoid zone visible on OCT imaging. Once atrophy occurs, this manifests as thinning of Bruch’s, RPE and the photoreceptor layers.Citation85
OCT studies in paediatric Usher syndrome patients have been relatively scarce, particularly in type 2,3 and 4. This is likely due to the later age of onset of visual impairment in Usher syndrome type 1, whereby Usher syndrome type 2 patients are generally diagnosed in early adulthood.
Pourova et al.Citation78 presented a case of a 4-year-old girl with congenital hearing loss. The authors used SD-OCT to analyse the retinal changes due to her MYO7A mutation (c.471-1G>T and c.4180C>T; p.Gln1394*) and a diagnosis of Usher syndrome type 1. SD-OCT revealed a loss of photoreceptors throughout the macula area, but not in the foveolar region, which correlated with preserved visual acuity. Consequently, the authors concluded that loss of outer nuclear layer on SD-OCT could be implemented as an early indication for Usher syndrome type 1, before the visual defects of the patient become symptomatic. Of course, other forms of retinal disease may show similar patterns of retinal loss, so this is not specific to Usher syndrome.
Subira et al.Citation86 were the first group to assess retinal abnormalities in paediatric patients with Usher syndrome type 1 using OCT. This group studied 16 patients with MYO7A mutations between the ages of 4 and 17. The authors identified several retinal changes in these patients, particularly in the macula. Of the 32 eyes examined, 84% had an absent external limiting membrane, 88% had a disrupted ellipsoid zone, 91% had absent cone outer segments and 94% had a loss of photoreceptors and RPE.
Comparatively, photoreceptor and RPE loss was 50% and 22% in perifoveal regions, highlighting that Usher syndrome type 1 retinal changes are primarily located in the fovea during childhood. This is supported by Malm et al.Citation87 who found foveal changes in two sisters (13 and 15) with USH1, whilst maintaining a relatively normal perifoveal region. Subira et al. also report that none of the paediatric patients presented with an epiretinal membrane. This is a common finding in adult patientsCitation88 and is a potential sign of disease progression.
X-linked retinoschisis
X-linked retinoschisis is a relatively common macular degeneration that affects males as early as infancy. It is caused by mutations in the retinoschisin gene (RS1). X-linked retinoschisis has a prevalence ranging from 1:5000 to 1:20,000 worldwide.Citation89 Patients present with a reduction in the b-wave amplitude, known as an ‘electronegative response’ on full field electroretinography.Citation90 The presence of pathology in the fovea of all affected patients leads to vision loss, with visual acuity generally stabilising at 6/15 to 6/36.Citation91,Citation92 There is an association between X-linked retinoschisis and high hyperopia.Citation93
Diagnosis for X-linked retinoschisis is often delayed until school age,Citation91 as this is when visual dysfunction is usually noticed.Citation91 However, there have been some cases described in infancy.Citation94,Citation95 Retinal appearance and function are often asymmetrical, albeit bilateral. Cavities appear in the retina which show up as a spoke-like pattern in fundus autofluorescence.Citation93 OCT has become the main tool used to diagnose X-linked retinoschisis, due to the obvious spoke-wheel pattern, cystic spaces, and schisis of the retinal layers (see ), which distinguishes the disease from others.Citation90 The breaks between the outer plexiform and nuclear layers increase the risk of retinal holes and retinal detachment.Citation93
Figure 4. Optical coherence tomography (OCT) image of a 7-year-old patient with XLRS due to an RS1 mutation. Image courtesy of Dr Jonathan Ruddle, Melbourne Children’s Eye Clinic.
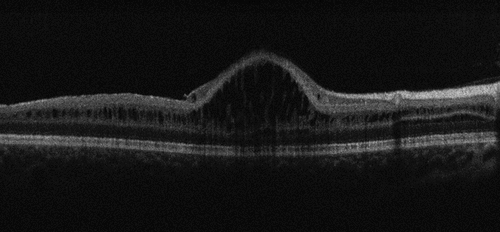
Imaging studies on patients with mild X-linked retinoschisis are common but are difficult to conduct on more severe forms of X-linked retinoschisis. However, inner nuclear layer schisis has been observed in children (aged 1–17 years) in several studies which have used OCT to analyse X-linked retinoschisis patients.Citation92,Citation96–102 The vast majority of these schisis are located primarily in the macula, with additional schitic pathology in the periphery. However, it has also been shown that RS1 mutations can cause a X-linked retinoschisis phenotype which spares the macula. Smith et al.Citation103 presented three such paediatric X-linked retinoschisis patients who had peripheral retinoschisis, with the absence of macula schisis. In addition, the three subjects showed normal electroretinogram responses. Consequently, it is important for clinicians to consider this diagnosis, and emphasises the importance of a thorough dilated peripheral retinal examination.
X-linked retinoschisis patients generally maintain a stable visual acuity until a secondary event such as retinal detachment occurs.Citation98,Citation104,Citation105 Despite the visual acuity remaining relatively constant, the central foveal thickness gradually reduces (below normal) over time, with adult X-linked retinoschisis patients usually presenting with foveal atrophy.Citation98,Citation105 This reduction in central foveal thickness is likely due to the reduction in the typical schisis type lesion and the formation of atrophic lesions.Citation106
Strupaite et al.Citation92 measured central foveal thickness in three patients which showed a similar trend with age. The oldest patient (17 years) had a central foveal thickness in the right eye of 373 μm, which was 197 μm less than any other patient. Hence, it is important for clinicians to consider the differences in foveal thickness between paediatric and adult patients, as it highlights the way in which this disease progresses.
The lack of correlation between central foveal thickness and visual acuity in X-linked retinoschisis could be due to the masking of pathological processes with this measurement. Central foveal thickness does not separate inner and outer retinal changes; thus, outer retinal degeneration can be masked by inner retinal thickening. Therefore, it is important for clinicians to consider both the inner and outer retina separately in X-linked retinoschisis patients.
Yang et al.Citation107 studied the correlation between SD-OCT findings and visual acuity in a X-linked retinoschisis cohort, which included 10 paediatric patients. Overall, the study included 20 X-linked retinoschisis patients with a mean age of 17.6 years. Photoreceptor outer segment length was found to be considerably shorter in X-linked retinoschisis patients compared to controls (29.3 vs 48.4 μm). This measurement was also found to be significantly correlated to visual acuity – shorter photoreceptor outer segments related to poorer visual acuity. This finding highlights that the photoreceptor outer segment measurements are a better indication for visual function than central foveal threshold, as foveal schisis height has not been found to correlate with visual function.
Batten disease
Batten disease, also known as neuronal ceroid lipofuscinosis, is a fatal-inherited disorder mainly affecting children. It is estimated to affect 1:100,000 people worldwide, but can have an incidence up to 1:14,500 in Scandinavia.Citation108 Juvenile Batten disease is most commonly due to mutations in the CLN3 gene, which causes an accumulation of autofluorescent waste material in the lysosome of cells.Citation109
Symptoms typically arise around the age of five or six years in the form of rapidly progressing vision loss due to macular degeneration.Citation110,Citation111 As the disease progresses, the peripheral retina degenerates, alongside cognitive issues and behavioural problems. The aggressive nature of the disease means that patients have an average life expectancy of around 15 years after onset.Citation112 This disease can also be caused by CLN2 mutations, inducing similar phenotype to CLN3 mutations but younger with presentation of neurological symptoms between the ages of two and four, with retinal structural changes visible on OCT as early as two years.Citation113
With CLN3-related Batten disease therapy available, an early diagnosis can lead to earlier treatment and slower motor dysfunction and decline. The retinal phenotype of Batten disease can be difficult to distinguish from early onset Stargardt disease. Kuper et al.Citation114 aimed to aid clinicians by highlighting key differences between the two diseases in visual acuity, colour vision, electroretinogram and OCT appearance. Paediatric patients with these two diseases have similar OCT presentations with macula atrophy and photoreceptor layer thinning in both. Differences can be observed in OCT images when analysing the inner retina: the retinal nerve fibre layer often presents with atrophy at this age in CNL3-related Batten disease, yet in Stargardt it is mostly regarded as normal. The study also reported that patients with CNL3-related disease presented with a more extensively and severely affected retina compared to Stargardt disease with more rapid loss of vision, absent dark-adapted ERG responses and severe colour vision deficiencies. As such, Batten disease should be considered in severe, fast-moving cases of suspected Stargardt disease and especially if there are co-existent seizures.
Wright et al.Citation115 described the ocular phenotype of seven Batten disease patients (CNL3), with a mean age of onset of 5.3 years. All cases showed atrophy of the outer nuclear layer/outer plexiform layer and a complete loss of photoreceptors. There was also atrophy present in the inner retina; specifically, the retinal nerve fibre layer and the ganglion cell layer.Citation110,Citation116,Citation117 Mean choroidal thickness was shown to be within age-adjusted normal limits (~330 μm), suggesting the choroid was not affected in early-stage disease.
Other authors have correlated retinal degeneration with cognitive deficits in Batten disease. Orlin et al.Citation118 used an ophthalmic severity scale to grade the retinal phenotype in patients with Batten disease and compared these to a similar neurological scale (Weill Cornell LINCL neurological scale). This study had a cohort of 25 patients with a CLN2 mutation with a mean age of 4.9 years. The ophthalmic scale included OCT (score of 1 being normal retinal layers and a score of 5 including severe retinal thinning with photoreceptor/outer retina loss involving the entire macula). The ophthalmic rating was correlated strongly with the neurological rating – where a more severely affected retina was associated with worse cognitive function. Similar findings in a paediatric CLN3 cohort were made by Dulz et al.Citation119 This highlights that OCT may be a useful quantitative endpoint for neurological severity in CLN3-related Batten disease. It remains to be documented whether the impact of regular intraventricular enzyme therapy will decouple the association.
Discussion
With rapidly improving technology and the increasing chance of treatment or enrolment in a clinical trial, it is essential that researchers and clinicians are able to monitor structural changes to the retina of inherited retinal disease patients effectively. The paediatric retina is a strong candidate for retinal gene therapy, as less photoreceptor degeneration has occurred.
This review has summarised changes found in the retinae of paediatric inherited retinal disease patients using OCT imaging (). Most studies have focused on the outer retina, but inner retinal changes have also been noted in a number of conditions. Further study into the inner retinal changes in paediatric inherited retinal diseases may assist researchers in identifying eligibility, and monitoring success of retinal gene therapy, as well as providing a better understanding of adaptive processes.
Table 2. Summary of the key OCT findings from the seven IRDs discussed in this review.
OCT is a widely used technology in the optometry clinic, so it is often one of the first steps towards diagnosing the cause of visual dysfunction. Hence, it holds vital importance in the process of clinical care for paediatric patients. The aim of this review is to assist clinicians in understanding the paediatric retina, helping them to provide inherited retinal disease patients with proper clinical care. Due to the severity of some inherited retinal diseases, it is essential for clinicians to maintain an understanding of the rapidly progressing field of inherited retinal disease, particularly in paediatrics – which is less studied.
If clinicians observe any of the previously mentioned retinal changes on OCT, then the possibility of an inherited retinal disease should be considered. In particular, degeneration of photoreceptors is a common sign seen in paediatric patients with Stargardt disease,Citation24 Bests disease, Leber’s Congenital Amaurosis,Citation45 choroideremia,Citation58 Usher syndrome,Citation86 X-linked retinoshcisisCitation107 and Batten disease.Citation115 This is mostly observed through disruption or loss of ellipsoid zone and/or outer nuclear layer. These OCT changes can be paired with other functional changes such as a diminished ERG and pupillary light reflex in Leber’s Congenital AmaurosisCitation38 and scotopic visual defects in choroideremia.Citation54
Additionally, it has been shown that OCT can be used to unveil signs of IRD before patients have displayed any visual dysfunction.Citation28,Citation30,Citation58,Citation61 External limiting membrane thickening has been shown in Stargardt disease before the onset of symptoms,Citation28,Citation30 whilst an increase in central foveal thickness has been observed in choroideremia patients with normal visual acuity.Citation58,Citation61 Therefore, OCT scans become an important tool even in patients who do not present with severe visual impairment.
OCT can be incorporated as part of a routine optometric review to ensure a healthy retina in children, and to monitor any potential subtle retinal changes which may be caused by an inherited retinal disease. If optometrists suspect that a paediatric patient may be showing signs of an inherited retinal disease, they should be referred to an ophthalmologist to arrange further multimodal imaging, electroretinography and commence genetic counselling and testing (as per the Royal Australian and New Zealand College of Ophthalmologists inherited retinal disease guidelines).Citation120
The world of genetics is a rapidly evolving field, meaning that more genes are being identified due to cheaper genetic analysis and an increase in awareness. In line with this, more inherited retinal diseases are being genotyped and phenotyped, providing an exciting era of improved patient management and treatment options.Citation6 Given that it can often be difficult for clinicians to diagnose an inherited retinal disease based on only the phenotype of a patient,Citation121 more cost effective genetic testing has become a vital technique in the field. The more genetic testing is performed, the more not only definite gene variants, but also possibly causative variants are found.
The use of subtle OCT diagnostic signs in the affected family member and even younger siblings can be helpful confirming and upgrading certainty of genetic findings. The interlink between OCT and genetic test findings means that both lines of investigation provide feedback to each other, to allow greater insight into genes and their early manifestations. Thus, more accurate images and an increase in genetic testing is pivotal to the field moving forward.
With the progress of gene therapy and the evolution of treatments for inherited retinal diseases, imaging of inherited retinal disease patients is becoming increasingly important. The progression of early-stage structure/function correlation data in paediatrics will improve the overall understanding of these diseases. Additionally, it may provide information which can aid decision-making about the appropriate time to intervene with treatment options such as gene therapy, which is especially important when managing children.
Due to the limited number of studies assessing OCT in a paediatric population specifically, it is difficult to recommend biomarkers for clinical trials at this stage. Many techniques have not been systematically assessed, such as en-face imaging, in a paediatric population. This review is a starting point for discussion and further investigation of the best biomarkers to use as these may be different in children compared to adults.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Heath Jeffery RC, Mukhtar SA, McAllister IL et al. Inherited retinal diseases are the most common cause of blindness in the working-age population in Australia. Ophthalmic Genet 2021; 42: 431–439. doi:10.1080/13816810.2021.1913610.
- Boye SE, Boye SL, Lewin AS et al. A comprehensive review of retinal gene therapy. Mol Ther 2013; 21: 509–519. doi:10.1038/mt.2012.280.
- Jacobson SG, Cideciyan AV, Ratnakaram R et al. Gene therapy for Leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol 2012; 130: 9–24. doi:10.1001/archophthalmol.2011.298.
- MacLaren RE, Groppe M, Barnard AR et al. Retinal gene therapy in patients with choroideremia: initial findings from a phase 1/2 clinical trial. Lancet 2014; 383: 1129–1137. doi:10.1016/S0140-6736(13)62117-0.
- Britten-Jones AC, Jin R, Gocuk SA et al. The safety and efficacy of gene therapy treatment for monogenic retinal and optic nerve diseases: a systematic review. Genet Med 2022; 24: 521–534. doi:10.1016/j.gim.2021.10.013.
- Britten‐Jones AC, O’Hare F, Edwards TL et al. Victorian evolution of inherited retinal diseases natural history registry (VENTURE study): rationale, methodology and initial participant characteristics. Clin Exp Ophthalmol 2022; 50: 768–780. doi:10.1111/ceo.14110.
- Halioua-Haubold C-L, Jolly JK, Smith JA et al. Potential lifetime quality of life benefits of choroideremia gene therapy: projections from a clinically informed decision model. Eye 2019; 33: 1215–1223. doi:10.1038/s41433-019-0492-1.
- Maguire AM, High KA, Auricchio A et al. Age-dependent effects of RPE65 gene therapy for Leber’s congenital amaurosis: a phase 1 dose-escalation trial. Lancet 2009; 374: 1597–1605. doi:10.1016/S0140-6736(09)61836-5.
- Jolly JK, Bridge H, MacLaren RE. Outcome measures used in ocular gene therapy trials: a scoping review of current practice. Front Pharmacol 2019; 10: 509–519. doi:10.3389/fphar.2019.01076.
- Thompson DA, Ali RR, Banin E et al. Advancing therapeutic strategies for inherited retinal degeneration: recommendations from the monaciano symposium. Invest Ophthalmol Visual Sci 2015; 56: 918–931. doi:10.1167/iovs.14-16049.
- Ţălu Ş, Ţǎlu M, Giovanzana S et al. The history and use of optical coherence tomography in ophthalmology. Hum Vet Med 2011; 3: 29–32.
- Potsaid B, Gorczynska I, Srinivasan VJ et al. Ultrahigh speed spectral/fourier domain OCT ophthalmic imaging at 70,000 to 312,500 axial scans per second. Opt Express 2008; 16: 15149. doi:10.1364/OE.16.015149.
- Cicinelli MV, Marchese A, Bordato A et al. Reviewing the role of ultra-widefield imaging in inherited retinal dystrophies. Ophthalmol Ther 2020; 9: 249–263. doi:10.1007/s40123-020-00241-1.
- Georgiou M, Kalitzeos A, Patterson EJ et al. Adaptive optics imaging of inherited retinal diseases. Br J Ophthalmol 2018; 102: 1028–1035. doi:10.1136/bjophthalmol-2017-311328.
- Muftuoglu IK, Al-Sheikh M, S J et al. Imaging in inherited retinal disorders. Eur J Ophthalmol 2021; 31: 1656–1676. doi:10.1177/1120672121990578.
- Sabbaghi H, Ahmadieh H, Jalili J et al. Choroidal thickness in different types of inherited retinal dystrophies. J Ophthalmic Vis Res 2020; 15: 351. doi:10.18502/jovr.v15i3.7454.
- Jayanna S, Jalali S, Padhi TR et al. OCT imaging in infants. Semin Ophthalmol 2022; 37: 358–372. doi:10.1080/08820538.2021.1970781.
- Staurenghi G, Sadda S, Chakravarthy U et al. Proposed lexicon for anatomic landmarks in normal posterior segment spectral-domain optical coherence tomography: the in OCT consensus. Ophthalmology 2014; 121: 1572–1578. doi:10.1016/j.ophtha.2014.02.023.
- Kong X, Ho A, Munoz B et al. Reproducibility of measurements of retinal structural parameters using optical coherence tomography in stargardt disease. Transl Vis Sci Technol 2019; 8: 46. doi:10.1167/tvst.8.3.46.
- Ervin AM, Strauss RW, Ahmed MI et al. A workshop on measuring the progression of atrophy secondary to Stargardt disease in the ProgStar Studies: findings and lessons learned. Transl Vis Sci Technol 2019; 8: 16–16. doi:10.1167/tvst.8.2.16.
- Velaga SB, Nittala MG, Jenkins D et al. Impact of segmentation density on spectral domain optical coherence tomography assessment in Stargardt disease. Graefes Arch Clin Exp Ophthalmol 2019; 257: 549–556. doi:10.1007/s00417-018-04229-3.
- Lang A, Carass A, Bittner AK et al. Improving graph-based OCT segmentation for severe pathology in retinitis pigmentosa patients. In: Krol A, and Gimi B, editors. Proceedings of SPIE–the International Society for Optical Engineering, San Diego, USA. NIH Public Access. p. 101371M.
- Tanna P, Strauss RW, Fujinami K et al. Stargardt disease: clinical features, molecular genetics, animal models and therapeutic options. Br J Ophthalmol 2017; 101: 25–30. doi:10.1136/bjophthalmol-2016-308823.
- Tanna P, Georgiou M, Strauss RW et al. Cross-sectional and longitudinal assessment of the ellipsoid zone in childhood-onset Stargardt disease. Transl Vis Sci Technol 2019; 8: 1. doi:10.1167/tvst.8.2.1.
- Fujinami K, Zernant J, Chana RK et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology 2015; 122: 326–334. doi:10.1016/j.ophtha.2014.08.012.
- Huang WC, Cideciyan AV, Roman AJ et al. Inner and outer retinal changes in retinal degenerations associated with ABCA4 mutations. Invest Ophthalmol Vis Sci 2014; 55: 1810. doi:10.1167/iovs.13-13768.
- Cai CX, Light JG, Handa JT. Quantifying the rate of ellipsoid zone loss in Stargardt disease. Am J Ophthalmol 2018; 186: 1–9. doi:10.1016/j.ajo.2017.10.032.
- Burke TR, Yzer S, Zernant J et al. Abnormality in the external limiting membrane in early Stargardt disease. Ophthalmic Genet 2013; 34: 75–77. doi:10.3109/13816810.2012.707271.
- Lee W, Nõupuu K, Oll M et al. The external limiting membrane in early-onset Stargardt disease. Invest Ophthalmol Vis Sci 2014; 55: 6139. doi:10.1167/iovs.14-15126.
- Khan KN, Kasilian M, Mahroo OAR et al. Early patterns of macular degeneration in ABCA4-associated retinopathy. Ophthalmology 2018; 125: 735–746. doi:10.1016/j.ophtha.2017.11.020.
- Palejwala NV, Gale MJ, Clark RF et al. Insights into autosomla dominant Stargardt-like macular dystrophy through multimodality diagnostic imaging. Retina 2016; 36: 119–130. doi:10.1097/IAE.0000000000000659.
- Spaide RF, Noble K, Morgan A et al. Vitelliform macular dystrophy. Ophthalmology 2006; 113: 1392–1400.e4. doi:10.1016/j.ophtha.2006.03.023.
- Bianco L, Arrigo A, Antropoli A et al. Multimodal imaging in Best vitelliform macular dystrophy: literature review and novel insights. Eur J Ophthalmol 2023: 112067212311664. doi:10.1177/11206721231166434.
- Johnson AA, Guziewicz KE, Lee CJ et al. Bestrophin 1 and retinal disease. Prog Retin Eye Res 2017; 58: 45–69. doi:10.1016/j.preteyeres.2017.01.006.
- Sayman Muslubas I, Arf S, Hocaoglu M et al. Best disease presenting as subretinal pigment epithelium hyperreflectivite lesion on spectral-domain optical coherence tomography: multimodal imaging features. Eur J Ophthalmol 2022; 32: 2702–2711. doi:10.1177/11206721211055961.
- Dhoble P, Robson AG, Webster AR et al. Typical best vitelliform dystrophy secondary to biallelic variants in BEST1. Ophthalmic Genet 2023: 1–6. doi:10.1080/13816810.2023.2188227.
- Griffith JF, Hess DJ, Berrocal AM. Best’s vitelliform macular dystrophy in 10- and 31-month-old siblings. J Pediatr Ophthalmol Strabismus 2014; 51: e8–e12. doi:10.3928/01913913-20140225-01.
- den Hollander AI, Roepman R, Koenekoop RK et al. Leber congenital amaurosis: genes, proteins and disease mechanisms. Prog Retin Eye Res 2008; 27: 391–419. doi:10.1016/j.preteyeres.2008.05.003.
- Aleman TS, Jacobson SG, Chico JD et al. Impairment of the transient pupillary light reflex in Rpe65−/− mice and humans with Leber congenital amaurosis. Invest Ophthalmol Visual Sci 2004; 45: 1259–1271. doi:10.1167/iovs.03-1230.
- Simonelli F, Ziviello C, Testa F et al. Clinical and molecular genetics of Leber’s congenital amaurosis: a multicenter study of Italian patients. Invest Ophthalmol Vis Sci 2007; 48: 4284. doi:10.1167/iovs.07-0068.
- Tsang SH, Sharma T. Leber congenital amaurosis. Adv Exp Med Biol 2018; 131–137.
- Fazzi E, Signorini SG, Scelsa B et al. Leber’s congenital amaurosis: an update. Eur J Paediatr Neuro 2003; 7: 13–22. doi:10.1016/S1090-3798(02)00135-6.
- Yang H, Yu T, Sun C et al. Spectral-domain optical coherence tomography in patients with congenital nystagmus. Int J Ophthalmol 2011; 4: 627–630. doi:10.3980/j.issn.2222-3959.2011.06.10.
- Maguire AM, Simonelli F, Pierce EA et al. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med 2008; 358: 2240–2248. doi:10.1056/NEJMoa0802315.
- Jacobson SG, Aleman TS, Cideciyan AV et al. Human cone photoreceptor dependence on RPE65 isomerase. Proc Natl Acad Sci USA 2007; 104: 15123–15128. doi:10.1073/pnas.0706367104.
- Jacobson SG, Cideciyan AV, Huang WC et al. Leber congenital amaurosis: genotypes and retinal structure phenotypes. Adv Exp Med Biol 2016; 169–175.
- Jacobson SG, Cideciyan AV, Aleman TS et al. Photoreceptor layer topography in children with Leber congenital amaurosis caused by RPE65 mutations. Invest Ophthalmol Vis Sci 2008; 49: 4573. doi:10.1167/iovs.08-2121.
- Miyamichi D, Nishina S, Hosono K et al. Retinal structure in Leber’s congenital amaurosis caused by RPGRIP1 mutations. Hum Genome Var 2019; 6: 32. doi:10.1038/s41439-019-0064-8.
- Wang S, Zhang Q, Zhang X et al. Clinical and genetic characteristics of Leber congenital amaurosis with novel mutations in known genes based on a Chinese eastern coast Han population. Graefes Arch Clin Exp Ophthalmol 2016; 254: 2227–2238. doi:10.1007/s00417-016-3428-5.
- Bouzia Z, Georgiou M, Hull S et al. GUCY2D-associated Leber congenital amaurosis: a retrospective natural history study in preparation for trials of novel therapies. Am J Ophthalmol 2020; 210: 59–70. doi:10.1016/j.ajo.2019.10.019.
- Pasadhika S, Fishman GA, Stone EM et al. Differential macular morphology in patients with RPE65 -, CEP290 -, GUCY2D -, and AIPL1 -related leber congenital amaurosis. Invest Ophthalmol Vis Sci 2010; 51: 2608. doi:10.1167/iovs.09-3734.
- Jacobson SG, Cideciyan AV, Peshenko IV et al. Determining consequences of retinal membrane guanylyl cyclase (RetGC1) deficiency in human Leber congenital amaurosis en route to therapy: residual cone-photoreceptor vision correlates with biochemical properties of the mutants. Hum Mol Genet 2013; 22: 168–183. doi:10.1093/hmg/dds421.
- Sankila E-M, Tolvanen R, van den Hurk JAJM et al. Aberrant splicing of the CHM gene is a significant cause of choroideremia. Nat Genet 1992; 1: 109–113. doi:10.1038/ng0592-109.
- Tsang SH, Sharma T. X-linked choroideremia. Adv Exp Med Biol 2018: 37–42.
- Jolly JK, Edwards TL, Moules J et al. A qualitative and quantitative assessment of fundus autofluorescence patterns in patients with choroideremia. Invest Ophthalmol Vis Sci 2016; 57: 4498. doi:10.1167/iovs.15-18362.
- Xue K, Oldani M, Jolly JK et al. Correlation of optical coherence tomography and autofluorescence in the outer retina and choroid of patients with choroideremia. Invest Ophthalmol Vis Sci 2016; 57: 3674. doi:10.1167/iovs.15-18364.
- Foote KG, Roorda A, Duncan JL. Multimodal imaging in choroideremia. 2019. p. 139–143.
- Aleman TS, Han G, Serrano LW et al. Natural history of the central structural abnormalities in choroideremia. Ophthalmology 2017; 124: 359–373. doi:10.1016/j.ophtha.2016.10.022.
- Jacobson SG, Cideciyan AV, Sumaroka A et al. Remodeling of the human retina in choroideremia: Rab Escort Protein 1 (REP-1) mutations. Invest Ophthalmol Vis Sci 2006; 47: 4113. doi:10.1167/iovs.06-0424.
- Khan KN, Islam F, Moore AT et al. Clinical and genetic features of choroideremia in childhood. Ophthalmology 2016; 123: 2158–2165. doi:10.1016/j.ophtha.2016.06.051.
- Murro V, Mucciolo DP, Giorgio D et al. Optical coherence tomography angiography (OCT-A) in young choroideremia (CHM) patients. Ophthalmic Genet 2019; 40: 201–206. doi:10.1080/13816810.2019.1611880.
- Abbouda A, Dubis AM, Webster AR et al. Identifying characteristic features of the retinal and choroidal vasculature in choroideremia using optical coherence tomography angiography. Eye 2018; 32: 563–571. doi:10.1038/eye.2017.242.
- Battaglia Parodi M, Arrigo A, MacLaren RE et al. Vascular alterations revealed with optical coherence tomography angiography in patients with choroideremia. Retina 2019; 39: 1200–1205. doi:10.1097/IAE.0000000000002118.
- Mansour AM, Elnahry AG, Tripathy K et al. Analysis of optical coherence angiography in cystoid macular oedema associated with gyrate atrophy. Eye 2021; 35: 1766–1774. doi:10.1038/s41433-020-01166-6.
- Murro V, Mucciolo DP, Giorgio D et al. Choroidal vascularity index in young choroideremia patients. Retina 2021; 41: 1018–1025. doi:10.1097/IAE.0000000000002960.
- Read SA, Collins MJ, Vincent SJ et al. Choroidal thickness in childhood. Invest Ophthalmol Vis Sci 2013; 54: 3586. doi:10.1167/iovs.13-11732.
- Gerena Arévalo VA, Ruiz-Moreno JM. Choroidal thickness in a hyperopic pediatric population. Diagnostics 2022; 12: 2330. doi:10.3390/diagnostics12102330.
- Liu Y, Wang L, Xu Y et al. The influence of the choroid on the onset and development of myopia: from perspectives of choroidal thickness and blood flow. Acta Ophthalmol 2021; 99: 730–738. doi:10.1111/aos.14773.
- Talib M, van Schooneveld MJ, Thiadens AA et al. Clinical and genetic characteristics of male patients with RPGR-associated retinal dystrophies: a long term follow up study. Retina 2019; 39: 1186–1199. doi:10.1097/IAE.0000000000002125.
- Talib M, van Schooneveld MJ, Van Cauwenbergh C et al. The spectrum of structural and functional abnormalities in female carriers of pathogenic variants in the RPGR gene. Invest Ophthalmol Vis Sci 2018; 59: 4123. doi:10.1167/iovs.17-23453.
- Kortüm F, Kieninger S, Mazzola P et al. X-Linked retinitis pigmentosa caused by non-canonical splice site variants in RPGR. Int J Mol Sci 2021; 22: 850. doi:10.3390/ijms22020850.
- Menghini M, Jolly JK, Nanda A et al. Early cone photoreceptor outer segment length shortening in RPGR X-linked retinitis pigmentosa. Ophthalmologica 2021; 244: 281–290. doi:10.1159/000507484.
- Birch DG, Locke KG, Felius J et al. Rates of decline in regions of the visual field defined by frequency-domain optical coherence tomography in patients with RPGR-mediated X-linked retinitis pigmentosa. Ophthalmology 2015; 122: 833–839. doi:10.1016/j.ophtha.2014.11.005.
- Kousal B, Skalicka P, Valesova L et al. Severe retinal degeneration in women with a c.2543del mutation in ORF15 of the RPGR gene. Mol Vis 2014; 20: 1307–1317. doi:10.1111/j.1755-3768.2014.T070.x.
- Shifera AS, Kay CN. Early-onset X-linked retinitis pigmentosa in a heterozygous female harboring an intronic donor splice site mutation in the retinitis pigmentosa GTPase regulator gene. Ophthalmic Genet 2015; 36: 251–256. doi:10.3109/13816810.2013.879597.
- Neidhardt J, Glaus E, Lorenz B et al. Identification of novel mutations in X-linked retinitis pigmentosa families and implications for diagnostic testing. Mol Vis 2008; 14: 1081–1093.
- Souied E, Segues B, Ghazi I et al. Severe manifestations in carrier females in X linked retinitis pigmentosa. J Med Genet 1997; 34: 793–797. doi:10.1136/jmg.34.10.793.
- Kremlikova Pourova R, Paderova J, Copikova J et al. SD-OCT imaging as a valuable tool to support molecular genetic diagnostics of Usher syndrome type 1. J Am Assoc Pediatr Ophthalmol Strabismus 2018; 22: 312–314.e3. doi:10.1016/j.jaapos.2017.12.009.
- Mathur P, Yang J. Usher syndrome: hearing loss, retinal degeneration and associated abnormalities. Biochimic et Biophys Acta 2015; 1852: 406–420. doi:10.1016/j.bbadis.2014.11.020.
- Testa F, Melillo P, Bonnet C et al. Clinical presentation and disease course of Usher syndrome because of mutations in MYO7A or USH2A. Retina 2017; 37: 1581–1590. doi:10.1097/IAE.0000000000001389.
- Feenstra HM, Al-Khuzaei S, Shah M et al. Phenotypic and genetic characteristics in a cohort of patients with Usher genes. Genes (Basel) 2022; 13: 1423. doi:10.3390/genes13081423.
- Joensuu T, Hämäläinen R, Yuan B et al. Mutations in a novel gene with transmembrane domains underlie Usher syndrome type 3. Am J Hum Genet 2001; 69: 673–684. doi:10.1086/323610.
- Peter VG, Quinodoz M, Sadio S et al. New clinical and molecular evidence linking mutations in ARSG to Usher syndrome type IV. Hum Mutat 2021; 42: 261–271. doi:10.1002/humu.24150.
- Lad EM, Duncan JL, Liang W et al. Baseline microperimetry and OCT in the RUSH2A study: structure−function association and correlation with disease severity. Am J Ophthalmol 2022; 244: 98–116. doi:10.1016/j.ajo.2022.08.013.
- Lenassi E, Saihan Z, Cipriani V et al. Natural history and retinal structure in patients with Usher syndrome type 1 owing to MYO7A mutation. Ophthalmology 2014; 121: 580–587. doi:10.1016/j.ophtha.2013.09.017.
- Subirà O, Català-Mora J, Díaz-Cascajosa J et al. Retinal findings in pediatric patients with Usher syndrome type 1 due to mutations in MYO7A gene. Eye 2020; 34: 499–506. doi:10.1038/s41433-019-0536-6.
- Malm E, Ponjavic V, Möller C et al. Alteration of rod and cone function in children with Usher syndrome. Eur J Ophthalmol 2011; 21: 30–38. doi:10.5301/EJO.2010.5433.
- Sliesoraityte I, Peto T, Mohand-Said S et al. Novel grading system for quantification of cystic macular lesions in Usher syndrome. Orphanet J Rare Dis 2015; 10: 157. doi:10.1186/s13023-015-0372-0.
- George ND, Yates JR, Moore AT. X linked retinoschisis. Br J Ophthalmol 1995; 79: 697–702. doi:10.1136/bjo.79.7.697.
- Molday RS, Kellner U, Weber BHF. X-linked juvenile retinoschisis: clinical diagnosis, genetic analysis, and molecular mechanisms. Prog Retin Eye Res 2012; 31: 195–212. doi:10.1016/j.preteyeres.2011.12.002.
- Murro V, Caputo R, Bacci GM et al. Case report of an atypical early onset X-linked retinoschisis in monozygotic twins. BMC Ophthalmol 2017; 17: 19. doi:10.1186/s12886-017-0406-6.
- Strupaitė R, Ambrozaitytė L, Cimbalistienė L et al. X-linked juvenile retinoschisis: phenotypic and genetic characterization. Int J Ophthalmol 2018; 11: 1875. doi:10.18240/ijo.2018.11.22.
- Altschwager P, Ambrosio L, Swanson EA et al. Juvenile macular degenerations. Semin Pediatr Neurol 2017; 24: 104–109. doi:10.1016/j.spen.2017.05.005.
- Lee JJ, Kim JH, Kim SY et al. Infantile vitreous hemorrhage as the initial presentation of X-linked juvenile retinoschisis. Korean J Ophthalmol 2009; 23: 118. doi:10.3341/kjo.2009.23.2.118.
- Prasad A, Wagner R, Bhagat N. Vitreous hemorrhage as the initial manifestation of X-linked retinoschisis in a 9-month-old infant. J Pediatr Ophthalmol Strabismus 2006; 43: 56.
- Gao F, Dong J, Wang D et al. Comprehensive analysis of genetic and clinical characteristics of 30 patients with X‐linked juvenile retinoschisis in China. Acta Ophthalmol 2021; 99: e470–e479. doi:10.1111/aos.14642.
- Han IC, Whitmore SS, Critser DB et al. Wide-field swept-source OCT and angiography in X-linked retinoschisis. Ophthalmol Retina 2019; 3: 178–185. doi:10.1016/j.oret.2018.09.006.
- Hu Q, Huang L, Chen X et al. X-linked retinoschisis in juveniles: follow-up by optical coherence tomography. Biomed Res Int 2017; 2017: 1–5. doi:10.1155/2017/1704623.
- Huang L, Sun L, Wang Z et al. Clinical manifestation and genetic analysis in Chinese early onset X‐linked retinoschisis. Molec Gen & Gen Med 2020; 8: e1421. doi:10.1002/mgg3.1421.
- Ling KP, Mangalesh S, Tran-Viet D et al. Handheld spectral domain optical coherence tomography findings of X-Linked retinoschisis in early childhood. Retina 2020; 40: 1996–2003. doi:10.1097/IAE.0000000000002688.
- Padrón-Pérez N, Català-Mora J, Díaz J et al. Swept-source and optical coherence tomography angiography in patients with X-linked retinoschisis. Eye 2018; 32: 707–715. doi:10.1038/eye.2017.281.
- Yu J, Ni Y, Keane PA et al. Foveomacular schisis in juvenile X-linked retinoschisis: an optical coherence tomography study. Am J Ophthalmol 2010; 149: 973–978.e2. doi:10.1016/j.ajo.2010.01.031.
- Smith LM, Cernichiaro-Espinosa LA, McKeown CA et al. X-linked peripheral retinoschisis without macular involvement: a case series with RS1 genetic confirmation. Ophthalmic Genet 2020; 41: 57–62. doi:10.1080/13816810.2020.1723115.
- Apushkin MA, Fishman GA, Janowicz MJ. Correlation of optical coherence tomography findings with visual acuity and macular lesions in patients with X-linked retinoschisis. Ophthalmology 2005; 112: 495–501. doi:10.1016/j.ophtha.2004.08.027.
- Kjellström S, Vijayasarathy C, Ponjavic V et al. Long-term 12 year follow-up of X-linked congenital retinoschisis. Ophthalmic Genet 2010; 31: 114–125. doi:10.3109/13816810.2010.482555.
- Takada Y, Fariss RN, Tanikawa A et al. A retinal neuronal developmental wave of retinoschisin expression begins in ganglion cells during layer formation. Invest Ophthalmol Vis Sci 2004; 45: 3302. doi:10.1167/iovs.04-0156.
- Yang HS, Lee JB, Yoon YH et al. Correlation between spectral-domain OCT findings and visual acuity in X-linked retinoschisis. Invest Ophthalmol Vis Sci 2014; 55: 3029. doi:10.1167/iovs.14-13955.
- Mole S, Williams R, Goebel H. The neuronal ceroid lipofuscinoses (Batten disease). Oxford, UK: Oxford University Press; 2011.
- Kleine Holthaus S-M, Aristorena M, Maswood R et al. Gene therapy targeting the inner retina rescues the retinal phenotype in a mouse model of CLN3 Batten disease. Hum Gene Ther 2020; 31: 709–718. doi:10.1089/hum.2020.038.
- Hansen MS, Hove MN, Jensen H et al. Optical coherence tomography in juvenile neuronal ceroid lipofuscinosis. Retin Cases Brief Rep 2016; 10: 137–139. doi:10.1097/ICB.0000000000000200.
- Lebrun A-H, Moll-Khosrawi P, Pohl S et al. Analysis of potential biomarkers and modifier genes affecting the clinical course of CLN3 disease. Mol Med 2011; 17: 1253–1261. doi:10.2119/molmed.2010.00241.
- Adams HR, Rose K, Augustine EF et al. Experience, knowledge, and opinions about childhood genetic testing in Batten disease. Mol Genet Metab 2014; 111: 197–202. doi:10.1016/j.ymgme.2013.10.017.
- Kovacs KD, Orlin A, Sondhi D et al. Automated retinal layer segmentation in CLN2-associated disease: commercially available software characterizing a progressive maculopathy. Transl Vis Sci Technol 2021; 10: 23. doi:10.1167/tvst.10.8.23.
- Kuper WFE, Talsma HE, van Schooneveld MJ et al. Recognizing differentiating clinical signs of CLN3 disease (Batten disease) at presentation. Acta Ophthalmol 2021; 99: 397–404. doi:10.1111/aos.14630.
- Wright GA, Georgiou M, Robson AG et al. Juvenile Batten disease (CLN3): detailed ocular phenotype, novel observations, delayed diagnosis, masquerades, and prospects for therapy. Ophthalmol Retina 2020; 4: 433–445. doi:10.1016/j.oret.2019.11.005.
- Preising MN, Abura M, Jäger M et al. Ocular morphology and function in juvenile neuronal ceroid lipofuscinosis (CLN3) in the first decade of life. Ophthalmic Genet 2017; 38: 252–259. doi:10.1080/13816810.2016.1210651.
- Kovacs KD, Patel S, Orlin A et al. Symmetric age association of retinal degeneration in patients with CLN2-associated Batten disease. Ophthalmol Retina 2020; 4: 728–736. doi:10.1016/j.oret.2020.01.011.
- Orlin A, Sondhi D, Witmer MT et al. Spectrum of ocular manifestations in CLN2-associated Batten (Jansky-Bielschowsky) disease correlate with advancing age and deteriorating neurological function. PloS ONE 2013; 8: e73128. doi:10.1371/journal.pone.0073128.
- Dulz S, Atiskova Y, Wibbeler E et al. An ophthalmic rating scale to assess ocular involvement in juvenile CLN3 disease. Am J Ophthalmol 2020; 220: 64–71. doi:10.1016/j.ajo.2020.07.015.
- Grigg J, Jamieson R, Chen F et al. Guidelines for the assessment and management of patients with inherited retinal diseases (IRD). 2020 [accessed 2023 Nov 7]. https://ranzco.edu/?s=Guidelines+for+the+assessment+and+management+of+patients+with+inherited+retinal+degenerations.
- Berger W, Kloeckener-Gruissem B, Neidhardt J. The molecular basis of human retinal and vitreoretinal diseases. Prog Retin Eye Res 2010; 29: 335–375. doi:10.1016/j.preteyeres.2010.03.004.
- Kumaran N, Georgiou M, Bainbridge JWB et al. Retinal structure in RPE65-associated retinal dystrophy. Invest Ophthalmol Vis Sci 2020; 61: 47. doi:10.1167/iovs.61.4.47.