
ABSTRACT
Chemorefractory ovarian cancer has limited therapeutic options. Hence, new types of treatment including neoantigen-specific immunotherapy need to be investigated. Neoantigens represent promising targets for personalized cancer immunotherapy. We here describe the clinical and immunological effects of a neoantigen peptide-loaded DC-based immunotherapy in a patient with recurrent and chemoresistant ovarian cancer. A 71-year-old female patient with chemorefractory ovarian cancer and malignant ascites received intranodal vaccination of DCs loaded with four neoantigen peptides that were predicted by our immunogenomic pipeline. Following four rounds of vaccinations with this therapy, CA-125 levels were remarkably declined and tumor cells in the ascites were also decreased. Concordantly, the tumor-related symptoms such as respiratory discomfort improved without any adverse reactions. The reactivity against one HLA-A2402-restricted neoantigen peptide derived from a mutated PPM1 F protein was detected in lymphocytes from peripheral blood by IFN-γ ELISPOT assay. Furthermore, the neoantigen (PPM1 F mutant)-specific TCRs were detected in the tumor-infiltrating T lymphocytes post-vaccination. Our results showed that vaccination with intranodal injection of neoantigen peptide-loaded DCs may have clinical and immunological impacts on cancer treatment.
Introduction
Recurrent ovarian cancer after surgery is generally a difficult disease to cure and patients usually succumb to this disease due to chemotherapy resistance (Pokhriyal et al. Citation2019). Although there is tremendous interest in immunotherapy for ovarian cancer, success in this field has been limited to date (Drerup et al. Citation2015).
Recent studies have revealed that neoantigens, which are produced in tumor cells as a result of non-synonymous, splicing, or frameshift mutations, have pivotal roles in the effectiveness of immune checkpoint inhibitors (ICIs) (McGranahan et al. Citation2016; Yarchoan et al. Citation2017). Furthermore, prediction of neoantigens through analysis of patients’ tumor cells using next-generation sequencing (NGS) and bioinformatic tools has become a reality (Durgeau et al. Citation2018; Liu and Mardis Citation2017).
Among neoantigens, human leukocyte antigen (HLA) class I-restricted antigens are important because these antigens are likely to be targets of tumor-specific cytotoxic T lymphocytes that can destroy tumor cells effectively (Durgeau et al. Citation2018). Recent studies have revealed that neoantigens presented on the surface of tumor cells, may be potent immunogenic tumor specific-antigens. In fact, neoantigen vaccines or neoantigen-targeting T cell therapy are demonstrated to be safe without serious adverse events (Ott et al. Citation2017; Sahin et al. Citation2017; Zacharakis et al. Citation2018).
Cancer genomic approaches in combination with next-generation sequencing (NGS) technologies and bioinformatics can predict possible tumor-specific neoantigens that are expected to have high binding affinity to HLA molecules and be recognized by anti-tumor CD8+ cytotoxic T lymphocytes (CTL) (Durgeau et al. Citation2018; Liu and Mardis Citation2017; Yarchoan et al. Citation2017). We previously described our neoantigen discovery pipeline for the design of personalized therapeutic vaccines (Kiyotani et al. Citation2018).
There is an increasing number of clinical applications for dendritic cell (DC)-based immunotherapies for cancer (Garg et al. Citation2017). With their powerful antigen-presenting capability, DCs have the potential to overcome tumor tolerance and reactivate anti-tumor immunity when they are loaded with tumor-specific antigens (Saxena and Bhardwaj Citation2018). DC vaccinations are often given intradermally in many studies, but only a few percent of the administered DCs actually reach the lymph nodes (Ridolfi et al. Citation2004). Moreover, it is reported that DCs did not reach lymph nodes at all by intravenous injection and subcutaneous injection (Morse et al. Citation1999). In addition, a recent report demonstrated that matured DCs may be beneficial, because matured DCs may expand beyond lymph nodes through stimulating fibroblastic reticular cells and T lymphocytes (Acton et al. Citation2014). Therefore, in this study, we used the techniques of intranodal administration using matured DCs.
In this clinical and immunological paper, we report a case that was successfully treated with intranodal neoantigen peptide-loaded DC vaccine. We further investigated TCR sequences of neoantigen-reactive T cell clones that were identified in the patient’s ascites fluid after vaccine therapy.
Materials and methods
Whole-exome and RNA sequencing
Both genomic DNA and total RNA were extracted from the patient’s malignant ascites cells using an AllPrep DNA/RNA mini kit (Qiagen), while normal control genomic DNA was extracted from the patient’s PBMCs. Whole-exome libraries were prepared from genomic DNA using SureSelect Human All Exon V6 kit (Agilent Technologies), according to the manufacturer’s instructions. RNAseq libraries were prepared using a TruSeq Stranded mRNA Library Prep kit (Illumina). The prepared whole-exome and RNAseq libraries were sequenced by 100 bp paired-end reads on a HiSeq sequencer (Illumina).
Mutation calling was performed as described previously (Kiyotani et al. Citation2017) using the following parameters: (i) base quality of ≥15; (ii) sequence depth of ≥10; (iii) variant depth of ≥4; (iv) variant frequency in tumor of ≥10%; (v) variant frequency in normal samples of <2%; and (vi) Fisher P-value of <0.05.
Prediction of potential neoantigens
HLA class I genotypes of this patient were predicted from normal whole-exome sequencing data using the OptiType algorithm (Szolek et al. Citation2014). Neoantigens were predicted for each non-synonymous (SNV), and the binding affinities of all possible 8- to 11-mer peptides to HLA class I molecules (HLA-A, -B, and -C) were examined using NetMHC v3.4 software and NetMHCpanv2.8, as described previously (Kiyotani et al. Citation2018). Any candidate neoantigen peptides with predicted binding affinity IC50 values of less than or equal to 500 nM were selected for further consideration. Detection of mutated mRNA by RNAseq data was also considered for the selection of potential neoantigen candidates. All experiments were approved by the Ethics Committee (FGCC-EC009).
Generation of DC vaccine and intranodal injection methods
PBMCs were obtained using a leukapheresis procedure performed according to the manufacturer’s instructions (Haemonetics CCS). The leukapheresis product was diluted with RPMI-1640 (Kojin-Bio Inc. Japan) for isolation by Ficoll-Hypaque. After isolation, the cells underwent three washing cycles with RPMI.
DCs were generated from frozen PBMCs. PBMCs were thawed and then cultured in 6-well plates (BD FALCON) in complete medium containing 1% autologous serum for 30 min. After removing the floating cells and washing with RPMI, adherent cells were cultured in complete DC medium containing GM-CSF (Primmune Inc. Kobe, Japan) and interleukin-4 (Primmune). On day 6, the cells were stimulated with a maturation cytokine cocktail containing TNF-α (Pepro Tech Inc., NJ, USA) and interferon-α (Dainippon Pharma. Osaka, Japan) for 18 h.
Phenotypic DC changes were monitored by light microscopy and flow cytometric analysis. The cell-surface marker phenotype of monocyte-derived matured DCs was determined by single-color fluorescence analysis. Cells (2 × 105) were resuspended in 50 µl assay buffer (PBS, 2% FBS) and incubated for 30 min at 4°C with 10 µl appropriate FITC or PE-labeled mononuclear antibodies. After incubation, the cells were washed twice and resuspended in 500 µl assay buffer. Cellular fluorescence was analyzed by an FC-500 flow cytometer (Beckman Coulter, Marseille, France). Monoclonal antibodies specific for human CD14, CD40, CD86, HLA-Class I, control IgG1, control IgG2a (Beckman Coulter), and HLA-DR and CD11 c (BD Biosciences Pharmingen, San Diego, CA) were used.
DCs were pulsed with neoantigen peptides for 4 h at 37°C, then washed three times in saline and resuspended in a total volume of 0.5 ml saline in a 1 ml disposable syringe. The antigen-loaded DCs were immediately administered to the patient via intranodal injection under US guidance by a skilled medical doctor. The vaccination therapy was well-tolerated in the patient without any treatment-associated adverse events.
The cell processing, neoantigen examination, immunotherapy procedures, and immunological analysis were approved by the ethics committee of our institution with the patient’s written informed consent for the procedure, based on the Act on Securement of Safety of Regenerative Medicine in Japan.
ELISPOT assay
ELISPOT assay was performed using a Human IFN-γ ELISpotplus kit (MABTECH, Cincinnati, OH) according to the manufacturer’s instructions. Briefly, 96-well plates with nitrocellulose membranes (Millipore, Molshelm, France) precoated with primary anti-IFN-γ antibody were pretreated with RPMI medium containing 10% autologous serum at 4°C overnight. A total of 5 × 103 autologous immature DCs were added to each well, and then DC maturation cocktail was added to each well and incubated overnight. Then, each neoantigen peptide (at 25 µg/ml) or the mixtures were added to each well and incubated for 4 h. After washing three times with RPMI medium, 1.5 × 105 autologous lymphocytes were added to each well and then incubated for 48 h. The plate was washed three times with PBS, and then secondary antibody was added to each well and incubated for 2 h. The plates were incubated with HRP-reagent and stained with TNB (MABTECH). Positivity of the neoantigen-specific T cell response was quantitatively defined as specific spots. Spots were captured and analyzed by an automated ELISPOT reader 08 classic (AID GmbH, Strassberg, Germany). In some experiments using the most responsive peptide (PPM1 F mutant), we performed peptide-dose dependent assay in the lymphocyte IFN-γ response using PBMCs obtained at eight time points after the vaccination therapy was started.
Establishment of CTL clones
PBMCs were cultured with peptide and IL-2 (Novartis) for 2 weeks. Peptide was added to the culture once a week at final concentration of 10 μg/ml. Culture medium containing IL-2 was exchanged every three or four days (final concentration, 120 IU/ml).
Limiting dilutions were performed to isolate CTL clones from PBMCs depleted of CD4 T cells by Dynabeads™ CD4 (Invitrogen) after in vitro stimulation. Peptide-specific CTL clones were screened using IFN-γ ELISPOT and expanded further using the rapid expansion method (Yoshimura et al. Citation2014) with some modifications. Briefly, the cells were cultured in T25 flasks containing AIM-V (GIBCO) supplemented with 5% human AB serum (MP Biomedicals), 40 ng/ml anti-CD3 antibody (clone UCHT1; BD Biosciences), 144 IU/ml IL-2, and feeder cells. EB-3 and Jiyoye were used as feeder cells (5 × 106 cells each) after treatment with mitomycin C (Kyowa Hakko Kirin). Culture medium was exchanged with fresh AIM-V supplemented with 5% human AB serum containing IL-2 every three or four days (final concentration of IL-2 was 36 IU/ml). EB-3 and Jiyoye were purchased from the American Type Culture Collection.
Determination of CTL response
CTL clones (Responder) were co-cultured overnight with TISI cells (stimulators) pulsed with serial dilutions of PPM1 F mutant or wild-type peptide. IFN-γ secretion was measured by ELISA in triplicate. OpEIA human IFN-γ ELISA set (BD Bioscience) was used to measure the secreted IFN-γ. Briefly, APCs were pulsed with mutant or wild-type PPMIF peptides, the lymphocytes were added to the culture for 16 h, and the supernatant was tested. Results are shown at a responder:stimulator ratio of 1.1:1 (CP008-1), 3.3:1 (CP008-2), 7.8:1 (CP008-4), and 1.7:1 (CP008-5), according to the available cell number of each CTL clone. TISI cells were purchased from the International Histocompatibility Working Group.
TCR sequencing and analysis
Total RNA was extracted from ascites pre- and post-DC vaccination using an RNeasy mini kit (QIAGEN). cDNAs with a common 5ʹ-RACE adapter were synthesized using SMARTScribe Reverse Transcriptase (Clontech). The cDNA libraries of TRA and TRB were acquired by modified protocol based on a previously described method (Choudhury et al. Citation2016). After adding barcodes to the Illumina index sequences using the Nextera XT index kit v2 (Illumina), the PCR products were sequenced by 300-bp paired-end reads on the MiSeq (Illumina). TRA and TRB sequencing reads were mapped to the reference sequences in IMGT/GENE-DB (Giudicelli et al. Citation2005) for identifying V-(D)-J segments including complementary determining region 3 (CDR3). Neoantigen-specific TCR sequences were identified from PPM1 F mutant peptide-specific CTL clones by Sanger sequencing, and then their frequency was tracked within TCR repertoire sequencing data acquired from T cells in ascites pre- and post-DC vaccination. TCR tracking was performed using TCR repertoire analysis software (ImmunoGrapher) developed by Cancer Precision Medicine, Inc.
Statistical analyses
The data are presented as the means ± standard deviation (SD). Student’s t-test was used to compare continuous variables between two groups. P-values of <0.05 were considered statistically significant.
Results
Case presentation
A 69-year-old woman (CP008) was diagnosed in July 2016 with advanced ovarian cancer with malignant ascites, stage IIIc (International Federation of Gynecology and Obstetrics classification). Her serum CA-125 levels were elevated at 1463 U/ml (normal, <35 U/ml). She had an abdominal paracentesis that showed malignant cells consistent with adenocarcinoma. A CT scan showed ascites and indicated a possible mesenteric mass. She had cytoreductive surgery with removal of the omentum, ovaries, uterus, and other visible disease. The pathology showed poorly differentiated serous adenocarcinoma in the peritoneum and ovaries. Postoperatively, a platinum- and taxane-based chemotherapy regimen was administered, and CA-125 levels were decreased to 200 U/ml. However, CA-125 increased to 418 U/ml in March 2017. Then, she received second-line chemotherapy with CPT-11 and CDDP. However, her CA-125 levels increased to 831 U/ml. Then, she received third-line chemotherapy with gemcitabine and bevacizumab. However, her CA-125 levels were continuously elevated to 3340 U/ml and her ascites volume was further increased.
In April 2018, she was admitted to our clinic to receive personalized immunotherapy. First, we inserted intraperitoneal tubing to drain the ascites fluid to relieve her symptoms. The CT findings before the immunotherapy revealed peritoneal thickness and ascites (). Cytological examination was performed before and after immunotherapy treatment. The cytology of the ascites fluid before the immunotherapy revealed a high number of tumor cell clusters (). We decided to predict the neoantigens expressed on her tumor cells for personalized neoantigen-specific immunotherapy using cancer cells in the ascites fluid.
Figure 1. Computed tomography images (coronary view) before vaccine therapy, revealing diffuse peritoneal thickening (⇒) and ascites (→). ∆ indicates a port implanted in the abdominal wall and intraperitoneal tubing
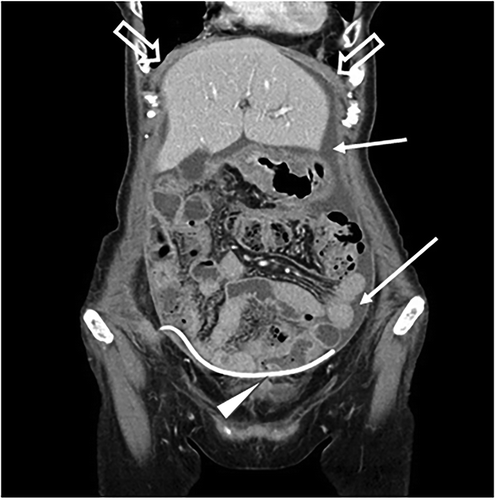
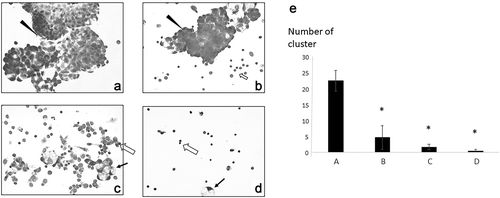
Figure 2. Cytological findings of ascitic cells. ➔ indicates macrophage ► indicates tumor cells. ⇦ are lymphocytes (a) May-Grünwald-Giemsa staining of ascitic cells before immunotherapy. A high number of tumor cells formed clusters. (b) Ascitic cells after four vaccinations. There was a significant decrease in tumor cells and an increase in immune cells. (c) Ascitic cells in December 2018: a small number of tumor cells and immune cells such as lymphocytes and macrophages were observed. (d) Ascitic cells in May 2019: almost all ascitic cells were lymphocytes and macrophages. (e) The number of tumor clusters formed by 10 or more tumor cells was counted under a microscope at x 100 visual field, and the average with standard deviation was calculated by EXCEL. * significant compared to the cluster number before vaccination
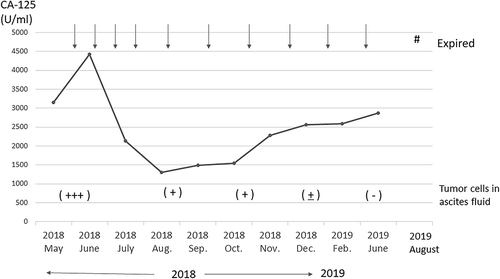
After cryopreserving the peripheral blood mononuclear cells (PBMCs) with leukapheresis, we prepared the neoantigen peptide-pulsed DC vaccine and started the intranodal injection therapy in May 2018. Leukopenia was not found at the start of DC vaccination. We chose co-stimulatory molecules (CD40 and CD86), HLA-class I/II, CD14, and CD11 c to define the DCs. DCs were defined as CD40+, CD86+, CD14−, CD11 c+, HLA-class I high, and HLA-DR high. The vaccine consisted of neoantigen peptide-loaded matured DCs (5–12 × 106 cells per vaccine) that were highly positive for CD40 and CD86 costimulatory molecules () and was administered into the groin lymph nodes (LNs) under ultrasonography guidance by a skilled physician every 2 weeks (). After four vaccinations, her symptoms such as abdominal fullness and respiratory discomfort were improved. At the same time, CA-125 levels decreased from 4470 U/ml before the second vaccine therapy to 1303 U/ml after the fourth vaccination. The cytology of the ascites fluid after four vaccinations revealed a significant decrease in tumor cells and an increase in immune cells such as lymphocytes, as shown in . We collected her PBMCs and ascites cells every 4 weeks to monitor the immunological parameters. The vaccine therapy was continued from May 2018 to June 2019. The clinical timeline is shown in . The cytology of the ascites fluid maintained a remarkable decrease in tumor cells and an increase in immune cells such as lymphocytes and macrophages throughout the treatment course (). Although the ascites volume and tumor marker levels were stable and did not show metastasis other than that to the peritoneal cavity, her bowel symptoms such as appetite loss from peritoneal thickness progressed and she died at the end of August 2019. shows tumor cluster numbers in the ascites fluid at various times during the clinical course, demonstrating a significant decrease in tumor cell clusters.
Figure 3. Phenotype of mature dendritic cells (DCs). Flow cytometric analysis of DCs matured with tumor necrosis factor α and IFN-α for expression of DC surface markers. We performed single staining for all markers (CD40, CD86, HLA-Class I, HLA-DR, CD14 and CD11 c). DCs were highly positive for co-stimulatory molecule expression. NC: Negative Conrol. DC: Dendritic Cells
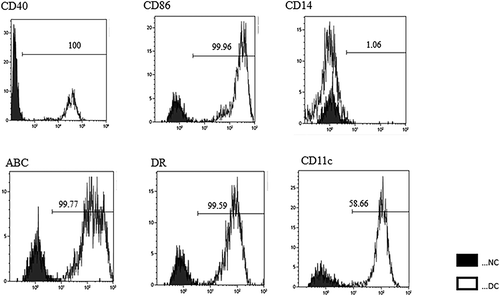
Figure 4. Intranodal administration of DC vaccine into unaffected groin lymph nodes (LNs) using direct ultrasound guidance provided by a skilled doctor. Left panel: the groin LNs before vaccine administration. Right panel: the same LNs after vaccine injection, showing swelling of the LNs
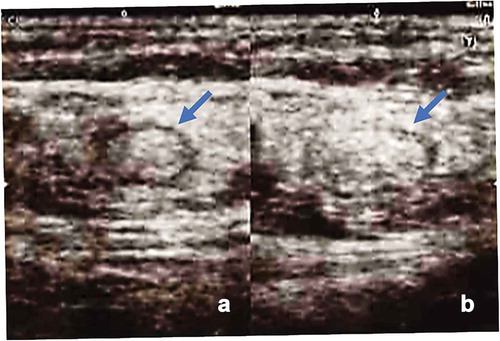
Figure 5. Timeline of CA-125 levels (U/ml) over the treatment course after vaccine therapy. Arrow indicates the vaccine therapy. After four rounds of vaccination, CA-125 levels decreased from 4470 U/ml to 1303 U/ml at the start of vaccine therapy
Prediction of neoantigens
Through whole-exome sequencing of genomic DNA from malignant ascites cells, which included ovarian cancer cells and the corresponding normal cells of patient CP008, we identified a total of 25 non-synonymous mutations (). We then predicted the binding affinity of possible neoantigen peptides to individual HLA-A, -B, and -C molecules and obtained the sequences of 57 candidate neoantigen epitope peptides that showed binding affinity IC50 values of less than or equal to 500 nM (). We also considered the expression of the neoantigen candidate epitopes by referring to the RNAseq data and then selected four potential neoantigen candidate epitopes derived from non-synonymous mutations in PPM1 F, PPOX, ARFGEF1, and TMEM161B genes ().
Table 1. List of non-synonymous SNVs
Table 2. List of predicted neoantigen
Table 3. The expression of the neoantigen candidate epitopes by referring to the RNAseq data
Immunological findings
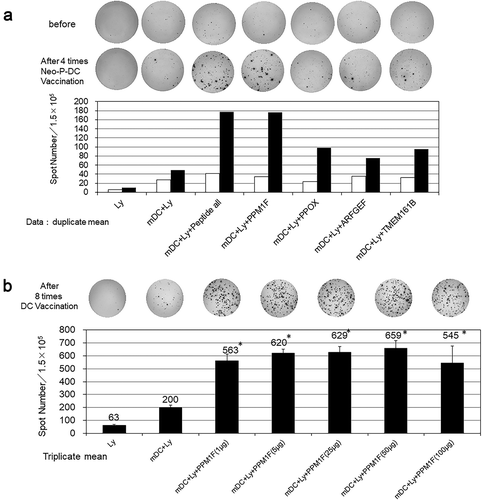
For vaccine-induced immune monitoring, we performed ELISPOT assays for the detection of neoantigen-specific T cell responses in the PBMCs after four vaccinations. At the same time, the tumor marker level decreased, as shown in . We detected high T cell responses against one peptide derived from the mutation in the PPM1 F mutant (). Further ELISPOT analysis of the IFN-γ response in lymphocyte samples obtained at eight time points after the vaccinations confirmed a specific immune response against neoantigen PPM1 F mutant peptide in a dose-dependent manner in the concentration range of 1 µg/ml to 50 µg/ml ().
Figure 6. a) Results of epitope-specific IFN-γ-secreting cells detected in ELISPOT assays. Four types of autologous-matured mutant peptide-loaded dendritic cells (5 × 103 cells/well) were co-incubated with or without the preserved lymphocytes before or after three rounds of vaccination (1.5 × 105 cells/well) for 48 h, and IFN-γ secretion spots were detected with an ELISPOT reader. Data indicate mean of duplicate assays. b) Dose-dependent IFN-γ secretion by lymphocytes incubated with the peptide-loaded DCs. Data show the mean of the triplicate assay. * significant compared to DCs without peptides. Ly; lymphocytes before DC stimulation, mDC; matured dendritic cells
Identification of neoantigen-specific CTLs
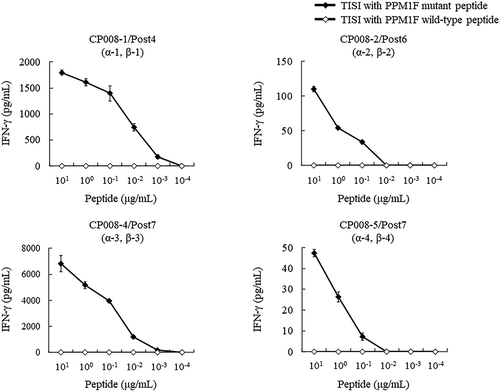
To further confirm cytotoxic T lymphocytes (CTLs) that recognize neoantigens, we attempted to establish CTL clones against the PPM1 F mutant peptide from PBMCs of the patient through in vitro stimulation after DC vaccination (post 4, 6, and 7). Consequently, we successfully established five CTL clones (CP008-1 to −5) and analyzed the TCR sequences of each clone. As shown in , four pairs of PPM1 F mutant peptide-reactive TCR were identified from CTL clones. Four CTL clones (CP008-1, -2, -4, -5) that express any one of these TCR-αβ pairs ().
Table 4. PPM1 F mutant peptide-reactive TCRs
Figure 7. Response of CTL clones to PPM1 F mutant and wild-type peptides. CTL clone recognition of PPM1 F mutant and wild-type peptides was determined by IFN-γ ELISA. CTL clones (responders) were co-cultured overnight with IISI cells (stimulators) pulsed with PPM1 F mutant peptide or wild-type peptide. IFN-γ secretion was measured in triplicate. A pair of TCRα and TCRβ identified from CTL clones are shown by TCR ID (a-x, b-x), as listed in
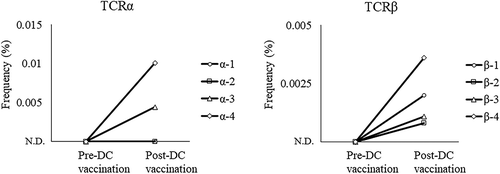
Next, we further examined whether the neoantigen-specific CTLs induced by DC vaccination infiltrated into tumor lesions and were present in malignant ascites. The frequencies of PPM1 F mutant peptide-reactive TCRα and TCRβ sequences were tracked in TCR repertoire sequencing data obtained from T cells in ascites pre-and post-DC vaccination. As shown in , three TCRαβ pairs (α-3 and β-3, α-4 and β-4, and α-5 and β-5), which were identified from PPM1 F mutant peptide-specific CTL clones (CP008-3, −4, and −5), were detected in ascites of post-DC vaccination but not in those of pre-DC vaccination. In addition, two TCRβ (β-1 and β-2) were also detected in ascites post-DC vaccination. These results strongly indicate the possibility that neoantigen DC vaccine-induced CTLs could infiltrate into tumor lesions and then be present in the malignant ascites.
Figure 8. Increase in PPM1 F mutant peptide-reactive TCRs after neoantigen DC vaccination. TCR repertoire analysis of T cells in the malignant ascites of the patient pre- and post-DC vaccination was performed by next-generation sequencing. The frequencies of PPM1 F mutant peptide-reactive TCRα and TCRβ sequences were tracked in TCR repertoire sequencing data obtained from ascites. ND, not detected
Discussion
In this study, we successfully characterized the neoantigen profile of chemorefractory ovarian cancer by NGS analysis and subsequently predicted neoantigen peptides that would possibly have the highest binding affinity to HLA-class I molecules of the patient. Using four neoantigen peptides and the patient’s monocyte-derived matured DCs, we prepared the neoantigen-peptide-loaded DC vaccine and injected the cells into the groin LNs guided by US. In this study, we used direct intranodal injection as the route of administration because of its certainty and its convenience of DC recruitment to lymph nodes. The CA-125 levels rapidly declined, and the abdominal fullness symptoms were relieved after four vaccinations without any adverse reactions. ELISPOT analysis revealed an increase in the number of neoantigen-specific lymphocytes in the PBMCs of the patient after vaccination. Moreover, TCR repertoire analysis demonstrated the appearance of clonal lymphocytes with neoantigen-specific TCR in the ascites fluid.
These results collectively indicate that MHC class-I-restricted neoantigen peptide-loaded DC vaccine could induce tumor-specific responses with an increase in the neoantigen-specific T cells in the peripheral blood and tumor site. However, we recognize the limitations of a single patient study, and further studies are needed to assess and monitor the anti-tumor effect of vaccine therapy. Nevertheless, the clinical and immunological effects in the patient with an even lower mutational burden will be meaningful, considering that neoantigen-targeting immunotherapy is an alternative approach to immune checkpoint inhibitors, and thus might be a promising therapy for chemorefractory cancer. Moreover, immunotherapy that targets neoantigens, such as neoantigen-peptide-loaded DC vaccines, are tumor-specific with minimal off-target effects, and are thus considered to be safe (Kissick Citation2018).
The patient died 15 months after the initial vaccine therapy was administered due to the progression of peritoneal carcinomatosis and malnutrition. Although the duration of the clinical effect of the neoantigen vaccine was approximately 12 months, the clinical benefit was obvious, considering that the patient was already chemoresistant and had massive ascites before the therapy.
Although Martin et al. reported that the low mutational burden in ovarian cancer may limit the utility of neoantigen-targeted vaccines, this conclusion was made in a single preclinical murine tumor model (Martin et al. Citation2016) and could have been overstated. In fact, recent clinical studies revealed that neoantigen vaccines were effective in medulloblastoma (Blaeschke et al. Citation2019) and pediatric leukemia (Zamora et al. Citation2019) that had low tumor mutational burden.
It is important to determine the optimal methods of vaccination to stimulate the most functional and rapid neoantigen-specific responses in patients. In the present case, the clinical and immunological effects were fast, showing a decline in CA-125 levels and the appearance of neoantigen-specific T cell responses after four DC vaccinations. We suppose that this rapid response could be due to the intranodal vaccination method. Intranodal DC vaccine therapy seems to be the most effective vaccine among various vaccines regarding the route of DC administration (Bedrosian et al. Citation2003; Gilliet et al. Citation2003; Lambert et al. Citation2001; Nestle et al. Citation1998). Although the intranodal administration of the vaccine is a skilled technique, US guidance is very supportive, as we demonstrated in this report.
Using TCR repertoire analysis, we provided evidence that neoantigen peptide-loaded DC vaccine induced infiltration of T cells with mutant-specific TCR into the tumor site. Recent reports by Keskin et al. and Johanns et al. also demonstrated that a neoantigen vaccine induced neoantigen-specific T cell infiltration into the tumor site in patients with glioblastoma, which usually has a relatively low mutational load (Johanns et al. Citation2019; Keskin et al. Citation2019).
However, there are some issues to be clarified in the future studies of this research. It remains to be resolved whether neoantigen-pulsed DC vaccine-induced CTLs are in fact directly involved in the antitumor effect, namely the recognition and destruction of tumor cells in patients. Moreover, it is unclear whether the neoantigen epitope is expressed on tumor cells. Further studies will confirm these observations.
Conclusions
Taken together, our findings in this report suggest that intranodal injection of neoantigen peptide-loaded DC vaccines is a promising cancer therapy, even in chemoresistant-advanced cancer patients. Further clinical studies of neoantigen peptide-loaded DC vaccine therapy are warranted.
Disclosure of potential conflicts of interest
Tetsuro Hikichi and Sachiko Yoshimura are employees of Cancer Precision Medicine, Inc.
Authors declare no conflict of interest.
Acknowledgments
We thank Dr. Hideichiro Kinoshita for clinical assistance. We also thank H. Nikki March, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
References
- Acton SE, Farrugia AJ, Astarita J, Mourão-Sá D, Jenkins RP, Nye E, Hooper S, van Blijswijk J, Rogers NC, Snelgrove KJ, et al. 2014. Dendritic cells control fibroblastic reticular network tension and lymph node expansion. Nature. 514(7523):498–502. doi:10.1038/nature13814.
- Bedrosian I, Mick R, Xu S, Nisenbaum H, Faries M, Zhang P, Cohen PA, Koski G, Czerniecki BJ. 2003. Intranodal administration of peptide-pulsed mature dendritic cell vaccines results in superior CD8 + T-cell function in melanoma patients. J Clin Oncol. 21(20):3826–35.
- Blaeschke F, Paul MC, Schuhmann MU, Rabsteyn A, Schroeder C, Casadei N, Matthes J, Mohr C, Lotfi R, Wagner B, et al. 2019. Low mutational load in pediatric medulloblastoma still translates into neoantigens as targets for specific T-cell immunotherapy. Cytotherapy. 21(9):973–86. doi:10.1016/j.jcyt.2019.06.009.
- Choudhury NJ, Kiyotani K, Yap KL, Campanile A, Antic T, Yew PY, Steinberg G, Park JH, Nakamura Y, O’Donnell PH, et al. 2016. Low T-cell receptor diversity, high somatic mutation burden, and high neoantigen load as predictors of clinical outcome in muscle-invasive bladder cancer. Eur Urol Focus. 2(4):445–52. doi:10.1016/j.euf.2015.09.007.
- Drerup JM, Liu Y, Padron AS, Murthy K, Hurez V, Zhang B, Curiel TJ. 2015. Immunotherapy for ovarian cancer. Curr Treat Options Oncol. 16(1):317. doi:10.1007/s11864-014-0317-1.
- Durgeau A, Virk Y, Corgnac S, Mami-Chouaib F. 2018. Recent advances in targeting CD8 T-cell immunity for more effective cancer immunotherapy. Front Immunol. 9:14. doi:10.1007/s11864-014-0317-1.
- Garg AD, Vara Perez M, Schaaf M, Agostinis P, Zitvogel L, Kroemer G, Galluzzi L. 2017. Trial watch: dendritic cell-based anticancer immunotherapy. Oncoimmunology. 6(7):e1328341.
- Gilliet M, Kleinhans M, Lantelme E, Schadendorf D, Burg G, Nestle FO. 2003. Intranodal injection of semimature monocyte-derived dendritic cells induces T helper type 1 responses to protein neoantigen. Blood. 102(1):36–42.
- Giudicelli V, Chaume D, Lefranc MP. 2005. IMGT/GENE-DB: a comprehensive database for human and mouse immunoglobulin and T cell receptor genes. Nucleic Acids Res. 33:D256–D261.
- Johanns TM, Miller CA, Liu CJ, Perrin RJ, Bender D, Kobayashi DK, Campian JL, Chicoine MR, Dacey RG, Huang J, et al. 2019. Detection of neoantigen-specific T cells following a personalized vaccine in a patient with glioblastoma. Oncoimmunol. 8(4):e1561106. doi:10.1080/2162402X.2018.1561106.
- Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, Oliveira G, Giobbie-Hurder A, Felt K, Gjini E, et al. 2019. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial. Nature. 565(7738):234–39. doi:10.1038/s41586-018-0792-9.
- Kissick HT. 2018. Is it possible to develop cancer vaccines to neoantigens, what are the major challenges, and how can these be overcome? Neoantigens as vaccine targets for cancer. Cold Spring Harb Perspect Biol. 10(11):a033704.
- Kiyotani K, Chan HT, Nakamura Y. 2018. Immunopharmacogenomics towards personalized cancer immunotherapy targeting neoantigens. Cancer Sci. 109(3):542–49.
- Kiyotani K, Park J-H, Inoue H, Husain A, Olugbile S, Zewde M, Nakamura Y, Vigneswaran WT. 2017. Integrated analysis of somatic mutations and immune microenvironment in malignant pleural mesothelioma. Oncoimmunol. 6(2):e1278330.
- Lambert LA, Gibson GR, Maloney M, Durell B, Noelle RJ, Barth RJ. 2001. Intranodal immunization with tumor lysate-pulsed dendritic cells enhances protective antitumor immunity. Cancer Res. 61(2):641–46.
- Liu XS, Mardis ER. 2017. Applications of immunogenomics to cancer. Cell. 168(4):600–12. doi:10.1016/j.cell.2017.01.014.
- Martin SD, Brown SD, Wick DA, Nielsen JS, Kroeger DR, Twumasi-Boateng K, Holt RA, Nelson BH. 2016. Low mutation burden in ovarian cancer may limit the utility of neoantigen-targeted vaccines. PLoS One. 11(5):e0155189.
- McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal-Hanjani M, Wilson GA, Birkbak NJ, Hiley CT, et al. 2016. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science. 351(6280):1463–69. doi:10.1126/science.aaf1490.
- Morse MA, Coleman RE, Akabani G, Niehaus N, Coleman D, Lyerly HK. 1999. Migration of human dendritic cells after injection in patients with metastatic malignancies. Cancer Res. 59(1):56–58.
- Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R, Burg G, Schadendorf D. 1998. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med. 4(3):328–32.
- Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, Zhang W, Luoma A, Giobbie-Hurder A, Peter L, et al. 2017. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature. 547(7662):217–21. doi:10.1038/nature22991.
- Pokhriyal R, Hariprasad R, Kumar L, Hariprasad G. 2019. Chemotherapy resistance in advanced ovarian cancer patients. Biomark Cancer. 11:1179299X1986081.
- Ridolfi R, Riccobon A, Galassi R, Giorgetti G, Petrini M, Fiammenghi L, Stefanelli M, Ridolfi L, Moretti A, Migliori G, et al. 2004. Evaluation of in vivo labelled dendritic cell migration in cancer patients. J Transl Med. 2(1):27. doi:10.1186/1479-5876-2-27.
- Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, Bukur V, Tadmor AD, Luxemburger U, Schrörs B, et al. 2017. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature. 547(7662):222–26. doi:10.1038/nature23003.
- Saxena M, Bhardwaj N. 2018. Re-emergence of dendritic cell vaccines for cancer treatment. Trend Cancer. 4(2):119–37.
- Szolek A, Schubert B, Mohr C, Sturm M, Feldhahn M, Kohlbacher O. 2014. OptiType: precision HLA typing from next-generation sequencing data. Bioinformatics. 30(23):3310–16.
- Yarchoan M, Johnson BA III, Lutz ER, Laheru DA, Jaffee EM. 2017. Targeting neoantigens to augment antitumor immunity. Nat Rev Cancer. 17(4):209–22. doi:10.1038/nrc.2016.154.
- Yoshimura S, Tsunoda T, Osawa R, Harada M, Watanabe T, Hikichi T, Katsuda M, Miyazawa M, Tani M, Iwahashi M, et al. 2014. Identification of an HLA-A2-restricted epitope peptide derived from hypoxia-inducible protein 2 (HIG2). PLoS One. 9(1):e85267. doi:10.1371/journal.pone.0085267.
- Zacharakis N, Chinnasamy H, Black M, Xu H, Lu Y-C, Zheng Z, Pasetto A, Langhan M, Shelton T, Prickett T, et al. 2018. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer. Nat Med. 24(6):724–30. doi:10.1038/s41591-018-0040-8.
- Zamora AE, Crawford JC, Allen EK, Guo XZJ, Bakke J, Carter RA, Abdelsamed HA, Moustaki A, Li Y, Chang T-C, et al. 2019. Pediatric patients with acute lymphoblastic leukemia generate abundant and functional neoantigen-specific CD8 + T cell responses. Sci Transl Med. 11(498):eaat8549. doi:10.1126/scitranslmed.aat8549.