
ABSTRACT
Background
The field of immunology has undoubtedly benefited from the in vitro use of cell lines for immunological studies; however, due to the “immortal” nature of many cell lines, they are not always the best model. Thus, direct collection and culture of primary cells from model organisms is a solution that many researchers utilize. To the best of our knowledge, there is not a singular protocol which encompasses the entire process of bone marrow cell collection through cryopreservation and resuscitation of cells from a murine model.
Methods
Bone marrow cells were collected from mice with a C57BL6 genetic background. Cells were differentiated using L929 conditioned media. Cells were assessed using a combination of microscopy, differential staining, immunocytochemistry, and trypan blue. Results: Primary murine BMDMs that underwent cryopreservation followed by resuscitation retained a high degree of viability. Furthermore, these BMDMs retained on overall ability to clear S. aureus.
Results
Primary murine BMDMs that underwent cryopreservation followed by resuscitation retained a high degree of viability. Furthermore, these BMDMs retained on overall ability to clear S. aureus.
Conclusion
Crypopreserved and resuscitated primary murine BMDMs were viable and retained their pverall S. aureus clearance ability.
Background
Macrophages are essential immune cells which have diverse functions. Under normal physiological conditions, macrophages are responsible for maintaining homeostasis via the clearance of cellular debris (Hirayama et al., Citation2017; Lendeckel et al., Citation2022). Under pathological conditions, however, macrophages become effector cells that are critical in the early defense against invading pathogens. During infection, macrophages play an important role in the secretion of pro-inflammatory cytokines as well as phagocytosis and clearance of both pathogens and infected/dead cells (Hirayama et al., Citation2017; Lendeckel et al., Citation2022). As a result, the in vitro study of macrophages using immortalized cell lines has played a pivotal role in aiding and furthering our understanding of how macrophages function (Cutolo et al., Citation2005; Khadaroo et al., Citation2003; Pearlman et al., Citation1988).
Although studies utilizing immortalized cell lines have been invaluable in helping to elucidate cellular functions, cell lines have limitations and, thus, are not always the best model. Because cell lines are transformed, their native responses and functions have strong potential to be altered (Castell et al., Citation2006; Parson et al., Citation2005). Additionally, extended culture and multiple passages of cell lines can result in genotypic and phenotypic drift (Castell et al., Citation2006; Parson et al., Citation2005). To omit these limitations, many researchers may instead choose to utilize primary cells for culture. Primary cells are extracted directly from tissues or organs of interest for in vitro culture (Philippeos et al., Citation2012). Herein, we present a comprehensive protocol for the collection/isolation, differentiation, cryopreservation, and resuscitation of primary murine bone marrow derived macrophages (BMDMs). Importantly, we found that primary BMDMs were both viable and functional following the cryopreservation and resuscitation protocols. However, we observed that cryopreserved/resuscitated primary BMDMs exhibited a reduced ability to clear a subsequent S. aureus infection compared to “freshly” isolated/differentiated primary BMDMs which did not undergo cryopreservation. However, despite this reduction in bacterial clearance, cryopreserved primary BMDMs retained an overall ability to clear S. aureus.
Methods
Materials
−Sterile surgical tools (tweezers, scissors)
−70% ethanol
−1X sterile PBS
−Dimethyl sulfoxide (DMSO)
−Fetal Bovine Serum (FBS)
−1X Roswell Park Memorial Institute 1640 (RPMI)
−ACK lysis buffer (150 mM NH4Cl, 10 mM KHCO3, .01 mM EDTA dissolved in deionized water and pH = 7.3, sterile filtered)
−Complete media (1X RPMI, 20% FBS, 30% L929 supernatant, 0.1 M Hepes 10,000 U/ml penicillin-streptomycin, 1.792 × 10−3 mM 2-Mercaptoethanol, and 4 mg/ml glutamine)
Mice
6–8-week-old female TNF-/- mice were purchased from Jackson Laboratory. 8–9-week-old female Elp+/LoxP and Wild Type (WT) C57BL6 mice were bred and housed at Montana State University’s Animal Resource Center. All mice were bred on a C57BL6 genetic background. Mice were maintained at the Montana State University (MSU) Animal Resource Center. All care and procedures were in accordance with the recommendations of the National Institutes of Health, the U.S. Department of Agriculture, and the Guide for the Care and Use of Laboratory Animals (8th ed) (National Academies Press, Citation2011). Animal protocols were reviewed and approved by the MSU Institutional Animal Care and Use Committee (IACUC).
Isolation of murine bone marrow cells
NOTE: For anatomical pictures of murine bones described in this protocol, please reference Supplementary Figures .
Mice were euthanized according to IACUC protocol. After euthanasia, mice were sprayed down with 70% ethanol, and a small superficial incision was made midline at the abdomen, taking care to remove only the fur and fascia.
Taking hold of the fur to the top and bottom of the incision, the incision was widened and made to extend around the entire abdominal circumference of the mouse by pulling the fur-we refer to this as “depants-ing” because the separation of the skin/fur gives the appearance of pants.
From the midline, or “the top of the pants,” the fur was pulled downwards towards the tail. Scissors may be needed to make small incisions of the fur at the tail and feet in order to fully remove “pants.”
Once “pants” have been removed, scissors were used to cut away the muscle near the articulation point between the head of the femur and the pelvis. The articulation point can be felt/estimated by manipulating the hind leg while palpitating the pelvis area. It will feel like a bony protrusion that moves as the hind leg is manipulated. Take care not to accidently cut into the bone during this step.
Once the muscle surrounding this articulation point has been removed, the head of the femur articulating in the pelvic socket (acetabulum) should become visible and will appear as a small white ball.
There are three ligaments that attach the head of the femur to the acetabulum. All three ligaments need to be cut to detach the femur from the mouse. The ligaments will appear as small, taught white strands that go from the head of the femur into the acetabulum.
Repeat steps 5 and 6 for the other hind leg.
Remove foot by bending/breaking it in the opposite direction at the talus/calcaneus until it becomes loose.
Place kneecap on your thumb while gently grasping the femur between your index finger and thumb. With your other hand, pull broken foot downwards, in the direction that it was broken, while pushing up on the kneecap. Some ligaments may need to be cut if there is a lot of resistance (these will appear as white strands). This step will remove the foot and make proceeding muscle removal easier.
Gently grasp the femur between your thumb and index finger. With your other hand, break the kneecap by bending it in the opposite direction. Place your thumb at the distal end of the fibula/tibia, where the foot was broken, and push the fibula/tibia out towards the direction of the broken kneecap. This step should cleanly free the fibula/tibia and remove the majority of the muscle surrounding these bones. The fibula is a very thin bone, it is likely that it will break during this step if it has not already. This is okay. Breakage of the fibula will not contaminate/affect collection of bone marrow from the tibia.
Using sterile scissors, carefully remove the muscle surrounding the femur.
Any remaining muscle can be removed using a paper towel that has been soaked in 70% ethanol and air dried prior to use.
Repeat steps 8–12 for the other hind leg.
Pour 70% ethanol into a petri dish (enough to submerge bones) and soak collected bones for 5 min. This step is critical for sterilization of the bones.
All proceeding steps following 70% ethanol soak of bones MUST be performed with new sterile tools that have also been UV-sterilized to mitigate the risk of contamination.
Pour ~30 ml of 1X Roswell Park Memorial Institute 1640 (RPMI) into a sterile petri dish.
Aspirate 10 ml of RPMI into a 10 ml syringe, attach a 26.5-gauge needle. Using a NEW pair of sterile scissors and tweezers to manipulate the bones, rinse the bones off with 1X RPMI.
Cut small portions of both ends of the bones off. Insert needle attached to syringe and flush out bone marrow into a sterile 50 ml conical. Repeat as needed until all bone marrow has been removed and the bones take on a translucent appearance. The needle may not go into the bone easily during the first insertion. Gently rotating/small circular motions can aid needle insertion into the bone.
Repeat this process for the remaining bones.
Place a sterile 100 μm filter strainer into a new sterile 50 ml conical tube.
After all of the bone marrow has been collected into a 50 ml conical, use a sterile 10 ml serological to pipette up and down and break up any clumps. Pipette bone marrow solution onto 100 μm filter strainer.
Discard strainer, replace cap onto 50 ml conical containing bone marrow cells and centrifuge at 408 x g for 5 minutes at 4°C.
In a tissue culture cabinet, remove supernatant using a sterile 10 ml serological. Resuspend cells in 1 ml ACK lysis buffer for 5 minutes at room temperature.
Add 10 ml 1X RPMI and centrifuge cells at 408 x g for 5 minutes at 4°C.
In a tissue culture cabinet, remove supernatant and resuspend cells in 12 ml pre-warmed (37°C) complete media.
Add 17 ml complete media to six sterile petri dishes (100 mm×15 mm) and add 2 ml of re-suspended cells to each dish (4 bones = 6petri dishes with 2 ml of cell suspension each). If counting the cells, add ~ 5 to 8 million cells per petri dish.
For differentiation to macrophages, culture cells at 37°C with 5% CO2 for 6 days in complete media containing M-CSF. Change media on day 3. The source of M-CSF can be sourced from L929 cell supernatant (Boltz-Nitulescu et al., Citation1987).
On day 6, cells have completed differentiation and are now ready for use in assays. To lift cells for assay plating, wash cells with 5 ml ice-cold sterile 1X PBS two times and incubate on ice for 5 minutes with 5 ml sterile 1X PBS.
Using a pipette, gently wash cells off petri dish and collect in a 50 ml conical.
Centrifuge cells at 408 x g for 5 minutes at 4°C.
Count cells and re-suspend to desired cell density.We have found that a typical 6-9-week-old female mouse bred on a C57BL6 genetic background yields a total of ~ 30 million cells following 6-day differentiation period (assuming 2 femurs and 2 tibias were collected).
Once seeded into culture plates, cells will adhere within a few hours.
Cryopreservation of murine BMDMs
To cryopreserve extra cells, label sterile cryopreservation vials and keep on ice. DMSO reactions with water are extremely exothermic, so it is critical to pre-chill tubes and ensure that all reagents are ice cold.
Re-suspend cells in 90% FBS and 10% DMSO at 6 × 106 cells/ml.
Aliquot 1 ml cell suspension into each cryopreservation vial and place vials either in a Mr. Frosty (ThermoFisher) or sandwiched between Styrofoam and placed in −80°C for 24 hours.
Transfer vials to liquid nitrogen for long term storage.
Resuscitation of murine BMDMs
Using a water bath set to 37°C, warm up complete media.
Place cryopreservation vial in water bath until suspension has completely thawed.
Transfer 1 ml of thawed cell suspension into 15 ml conical tube and add 7 ml pre-warmed (37°C) complete media to conical tube containing cell suspension.
Centrifuge at room temperature for 7 minutes at 408 x g.
Pipette 17 ml of pre-warmed complete media into two petri dishes (100 mm×15 mm).
Re-suspend in 1 ml pre-warmed complete media and add 500 μl of cell suspension to each dish.
Culture at 37°C with 5% CO2 for 6 days in complete media containing M-CSF. Change media on day 3. The source of M-CSF can be sourced from L929 cell supernatant (Boltz-Nitulescu et al., Citation1987).
To lift cells for assay plating, wash cells with 5 ml ice-cold sterile 1X PBS two times and incubate on ice for 5 minutes with 5 ml sterile 1X PBS.
Using a pipette, gently wash cells off petri dish and collect in a 50 ml conical.
Centrifuge cells at 408 x g for 5 minutes at 4°C.
Count cells and re-suspend to desired cell density. We have found that a typical yield for cryopreserved/resuscitated primary murine BMDMs (frozen back at ~ 6million cells) yields a total of ~ 18 million cells following 6 day in resuscitation/culture.
Results
As described in the above protocol, primary bone marrow cells were cultured in differentiating media for 6 days and either used immediately (“fresh”) or cryopreserved. Cryopreserved bone marrow derived macrophages (BMDMs) were resuscitated and cultured in differentiating media again for 6 days prior to use (“frozen”). The cryopreservation recovery rate of “frozen” BMDMs on day 6 of culture was ~ 30%. This was determined by dividing the number of live cells (1.35×106 cells/ml) by the total number of cells frozen back (5×106cells/ml).
Undifferentiated bone marrow cells (Day 0), Day 6 “fresh,” or Day 6 “frozen” BMDMs were placed into a funnel and centrifuged onto microscope slides (Cytospin) prior to DiffQuick staining. demonstrates the diverse cellularity of bone marrow cells immediately following isolation (Day 0) and the similarly uniform macrophage phenotype of Day 6 “fresh,” and Day 6 “frozen” BMDMs. demonstrates the cellular morphology of “fresh” and “frozen” BMDMs on days 0, 3, and 6 in culture.
Figure 1. Differential staining of primary cells obtained from bone marrow. Primary cells collected from bone marrowand subjected to centrifugation using cytospinand stained with diff quik immediately (day 0), after 6 days in macrophage differentiating media containing L929 (day 6 “fresh”), or following cryopreservation in liquid nitrogen, resuscitation, and culture for 6 days in macrophage differentiating media (day 6 “frozen”). Differential staining was performed on BMDMs isolated from three individual mice. Images at 40X magnification depict representative staining from one mouse.
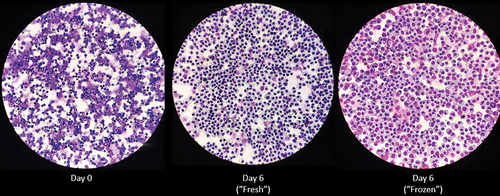
Figure 2. Similar morphology of “fresh” and “frozen” BMDMs. Freshly isolated bone marrow cells (a) or resuscitated cryopreserved BMDMs (b) were differentiated in media containing L929 for a total of 6 days. Microscopy images were taken at 40× magnification on day 0 (left), day 3 (middle), and day 6 (right) during culture. Images of BMDMs were taken from three individual mice. Data in this figure are representative of cells from one mouse.

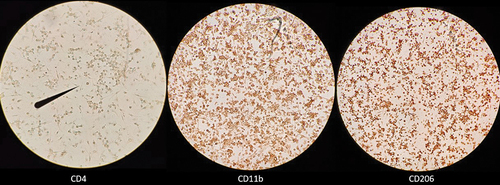
In line with the existing body of literature which describes macrophage differentiation from bone marrow cells, we used M-CSF sourced from L929 supernatant in our protocol (Haag & Murthy, Citation2021; Mendoza et al., Citation2022; Trouplin et al., Citation2013; Weischenfeldt & Porse, Citation2008). Following day 6 of culture in L929 differentiating media, both “fresh” and “frozen” primary BMDMs exhibited macrophage-like morphology (). However, to more directly characterize bone marrow cell differentiation to macrophages, we performed immunocytochemical staining of BMDMs for expression of classical macrophage differentiation markers CD11b and CD206 (Bisgaard et al., Citation2016; Zajd et al., Citation2020; Zhang et al., Citation2008). Being a classical T lymphocyte differentiation marker, CD4 is not expressed by murine macrophages and thus, served as a negative control (Moore et al., Citation1992). demonstrates BMDM positive staining (brown) for expression of the classical macrophage differentiation markers CD11b and CD206 and lack of staining for CD4.
Figure 3. BMDMs express CD11b and CD206. Day 6 “frozen” BMDMs were resuscitated and cultured in L929 containing media for 6 days. On day 6, BMDMs were lifted and seeded into 24-well plates containing collagen covers and subsequently incubated with biotinylated rat anti-mouse monoclonal IgG2 antibodies raised against CD4 (left; clone GK1.5), CD11b (middle; clone M1/70), or CD206 (right; clone MR5D3). All BMDMs received the same secondary avidin-HRP antibody and 3,3-diaminobenzidine chromogen. The presence of brown staining indicates expression of cell surface marker. Visualization was performed using standard inverted compound light microscope. Images depict staining from one representative experiment; however, two independent experiments were performed.
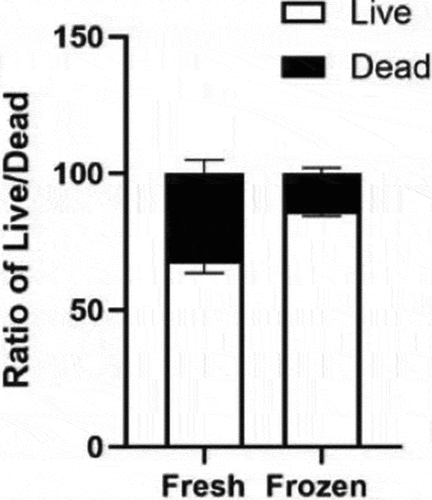
demonstrates that the viability of “frozen” and “fresh” BMDMs was comparable on day 6 of culture. In fact, the ratio of live/dead for “frozen” cells (6.4) was improved compared to the ratio of live/dead for “fresh” cells (2.2). Day 6 of culture for “fresh” BMDMs represents the completed differentiation period when BMDMs can be either cryopreserved or used in subsequent assays. As described in the above protocol, “fresh” and “frozen” BMDMs were lifted from the plate using ice cold PBS, centrifuged, and re-suspended in media. Cell density and viability was determined after trypan blue staining.
Figure 4. Similar viability of “fresh” and “frozen” BMDMs. Trypan blue staining assessing the viability of “fresh” BMDMs on day 6 of culture in L929 containing media (left) compared to viability of “frozen” BMDMs following resuscitation for 6 days in L929 containing media (right). Results show mean and SD of two independent experiments. Each experiment contained two technical replicates.
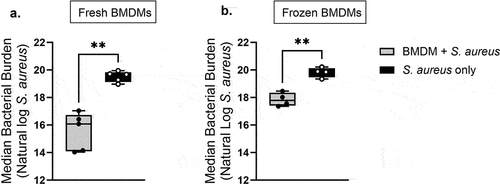
Having determined that primary BMDMs that had undergone cryopreservation followed by resuscitation retain a high degree of viability, we next sought to determine the functionality of “frozen” BMDMs. We compared the bacterial clearance ability of “fresh” BMDMs and “frozen” BMDMs. demonstratesthat “frozen” BMDMs had a reduced ability to clear S. aureus infection compared to “fresh” BMDMs. For “fresh” BMDMs, treatments that contained only S. aureus (no BMDMs), had a median bacterial burden (CFUs/ml) that was approximately 52.90 times greater than the median bacterial burden for treatments that contained “fresh” BMDMs in addition to S. aureus. For “frozen”BMDMs, treatments that contained only S. aureus (no BMDMs), had a median bacterial burden that was approximately 7.19 times greater than the median bacterial burden for treatments that contained “frozen” BMDMs in addition to S. aureus. These data demonstrated that in comparison to “fresh” BMDMs, “frozen” BMDMs display a significant decrease in S. aureus clearance efficiency. However, as evidenced by the decrease of S. aureus bacterial burden in treatments containing frozen/resuscitated BMDMs compared to S. aureus conditions alone, our results demonstrated that “frozen” BMDMs retained an overall ability for S. aureus bacterial clearance, albeit to a limited capacity.
Figure 5. Cryopreservation reduces S. aureus clearance capacity of primary BMDMs. (a) after 6 days in macrophage differentiating media, primary TNF-/- cells were treated with saline and infected with S. aureus for 5 hours at MOI 2:1. “Fresh” BMDM CFU data shown here were used in another manuscript (Luu et al, submitted). Shown is data from five independent experiments that contained two technical replicates each. (b) Primary TNF-/- BMDMs were cryopreserved in liquid nitrogen, thawed, and resuscitated in macrophage differentiating media for an additional 6 days. Cells were treated for either 12 hr or 1 hr with WT BMDMs fixed in 1% PFA before infection with S. aureus for 5 hr at MOI 2:1. Data were obtained from four independent experiments that contained two technical replicates each. (a, b) the average of two technical replicates per experiment is shown. Grey bars represent S. aureus bacterial burden (with BMDMs) compared to black bars which represent S. aureus uninhibited growth (without BMDMs) during a 5-hr period. Statistical analysis was performed using a one-sided paired t-test p-value = .0014 (a) p-value = .0061 (b) on log transformed data.
Conclusions
In vitro cell culture is a vital facet of immunological research, and the use of immortalized cell lines in research dates back as early as the 1900s (Mirabelli et al., Citation2019). Although there are many benefits to the use of cell lines in research (cost, cultivability, and published body of literature) limitations of cell lines include altered phenotype with increasing passage, differential native responses and functions, and genotypic drift (Castell et al., Citation2006; Parson et al., Citation2005). As a result, many researchers began to also utilize primary cells in their research. Here, we described an easy to follow and comprehensive protocol for the collection, isolation, cryopreservation, and resuscitation of primary murine bone marrow derived macrophages (BMDMs). We showed that the cryopreservation of these BMDMs did not result in marked alterations to their cellularity or morphology compared to “fresh” BMDMs. Additionally, cryopreservation of primary BMDMs did notimpact their viability following resuscitation and culture protocol. We did find, however, that resuscitated “frozen” BMDMs exhibited reduced capacity for clearance ofS. aureus compared to “fresh” BMDMs. DMSO in cryopreservation media is known to affect the epigenetic landscape of mammalian cells (Verheijen et al., Citation2019; Wei et al., Citation2020). Additionally, residual DMSO in “frozen” BMDMs could have provided a survival advantage to S. aureus. Supporting this, one study showed that DMSO inhibited killing of S. aureus by polymorphonuclear leukocytes (PMNLs) (Repine et al., Citation1981). Several studies also demonstrated protective effects of DMSO on bacterial survival (Han et al., Citation2023; Mi et al., Citation2016). Taken together, these factors could have contributed to the observed reduction of S. aureus clearance in “frozen” vs “fresh” BMDMs. Finally, while “fresh” and “frozen” BMDMs in our study were exposed to the same culture conditions, after 6 days in culture, “frozen” BMDMs but not “fresh” BMDMs were additionally co-cultured with fixed BMDMs for 1–12 hours. While fixed BMDMs are not viable, this additional variable could have affected S. aureus clearance by “frozen” BMDMs. Additional experimentation is needed to further investigate our findings. Taken together, however, we demonstrated that “frozen” BMDMs were both viable and functional following our protocol.
Although there is a myriad of different protocols for the isolation of primary cells either from blood or bone marrow (Barcelo et al., Citation2018; Haag & Murthy, Citation2021; Mendoza et al., Citation2022; Nasser et al., Citation2020; Trouplin et al., Citation2013; Weischenfeldt & Porse, Citation2008), we have found that these protocols can be difficult to follow for individuals newly entering immunology who have limited to no experience in mouse or wet lab work. Additionally, to the best of our knowledge, an up-to-date comprehensive protocol that encompasses the entire process of cell isolation, differentiation, cryopreservation, and resuscitation, especially as it pertains to primary murine bone marrow derived macrophages has not been published. Furthermore, we are not aware of a protocol that includes all of these components and also addresses the viability and functionality of cryopreserved BMDMs for subsequent assay use. Here, we present a comprehensive and easy to follow protocol for the collection, isolation, cryopreservation, and resuscitation of primary bone marrow derived macrophages as well as data showing the viability and functionality of primary BMDMs that have undergone liquid nitrogen cryopreservation.
Authorship
Conceptualization: AML, ARA
Methodology: AML, KMS
Data Curation: AML
Project Administration/Oversight: AML, ARA
Writing Original Draft: AML
Writing Revisions and Editing: AML, KMS, ARA
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
All data generated or analyzed during this study are included in this article and its supplementary material files. Further enquiries can be directed to the corresponding author.
Additional information
Funding
References
- Barcelo, H., Faul, J., Crimmins, E., & Thyagarajan, B. (2018, April). A practical cryopreservation and staining protocol for immunophenotyping in population studies. Current Protocols in Cytometry, 84(1). https://doi.org/10.1002/cpcy.35
- Bisgaard, L. S., Mogensen, C. K., Rosendahl, A., Cucak, H., Nielsen, L. B., Rasmussen, S. E., & Pedersen, T. X. (2016, October). Bone marrow-derived and peritoneal macrophages have different inflammatory response to oxLDL and M1/M2 marker expression – implications for atherosclerosis research. Scientific Reports, 6(1), 35234. https://doi.org/10.1038/srep35234
- Boltz-Nitulescu, G., Wiltschke, C., Holzinger, C., Fellinger, A., Scheiner, O., Gessl, A., & Förster, O. (1987, January). Differentiation of rat bone marrow cells into macrophages under the influence of mouse L929 cell supernatant. Journal of Leukocyte Biology, 41(1), 83–91. https://doi.org/10.1002/jlb.41.1.83
- Castell, J. V., Jover, R., Martnez-Jimnez, C. P., & Gmez-Lechn, M. J. (2006, April). Hepatocyte cell lines: Their use, scope and limitations in drug metabolism studies. Expert Opinion on Drug Metabolism and Toxicology, 2(2), 183–212. https://doi.org/10.1517/17425255.2.2.183
- Cutolo, M., Capellino, S., Montagna, P., Ghiorzo, P., Sulli, A., & Villaggio, B. (2005). Sex hormone modulation of cell growth and apoptosis of the human monocytic/macrophage cell line. Arthritis Research & Therapy, 7(5), R1124. https://doi.org/10.1186/ar1791
- Haag, S., & Murthy, A. (2021). Murine monocyte and macrophage culture. Bio-protocol, 11(6). https://doi.org/10.21769/BioProtoc.3928
- Han, H., Kang, J.-K., Ahn, K. J., & Hyun, C.-G. (2023, June). DMSO alleviates LPS-Induced inflammatory responses in RAW264.7 macrophages by inhibiting NF-κB and MAPK activation. Biochemistry (Moscow), 3(2), 91–101. https://doi.org/10.3390/biochem3020007
- Hirayama, D., Iida, T., & Nakase, H. (2017, December). The phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. International Journal of Molecular Sciences, 19(1), 92. https://doi.org/10.3390/ijms19010092
- Khadaroo, R. G., Kapus, A., Powers, K. A., Cybulsky, M. I., Marshall, J. C., & Rotstein, O. D. (2003, November). Oxidative stress reprograms lipopolysaccharide signaling via SRC kinase-dependent pathway in RAW 264.7 macrophage cell line. Journal of Biological Chemistry, 278(48), 47834–47841. https://doi.org/10.1074/jbc.M302660200
- Lendeckel, U., Venz, S., & Wolke, C. (2022, April). Macrophages: Shapes and functions. ChemTexts, 8(2), 12. https://doi.org/10.1007/s40828-022-00163-4
- Mendoza, R., Banerjee, I., Manna, D., Reghupaty, S. C., Yetirajam, R., & Sarkar, D. (2022). Mouse bone marrow cell isolation and macrophage differentiation. Methods of Molecular Biology, 2455, 85–91. https://doi.org/10.1007/978-1-0716-2128-8_8
- Mi, H., Wang, D., Xue, Y., Zhang, Z., Niu, J., Hong, Y., Drlica, K., & Zhao, X. (2016, August). Dimethyl sulfoxide protects Escherichia coli from rapid antimicrobial-mediated killing. Antimicrobial Agents & Chemotherapy, 60(8), 5054–5058. https://doi.org/10.1128/AAC.03003-15
- Mirabelli, P., Coppola, L., & Salvatore, M. (2019, August). Cancer cell lines are useful model systems for medical research. Cancers (Basel), 11(8), 1098. https://doi.org/10.3390/cancers11081098
- Moore, S. C., Anderson, S. J., & Walker, W. S. (1992, July). Mouse macrophages contain a truncated CD4 transcript. Journal of Leukocyte Biology, 52(1), 128–129. https://doi.org/10.1002/jlb.52.1.128
- Nasser, H., Adhikary, P., Abdel-Daim, A., Noyori, O., Panaampon, J., Kariya, R., Okada, S., Ma, W., Baba, M., Takizawa, H., Yamane, M., Niwa, H., & Suzu, S. (2020, July). Establishment of bone marrow-derived M-CSF receptor-dependent self-renewing macrophages. Cell Death Discovery, 6(1), 63. https://doi.org/10.1038/s41420-020-00300-3
- National Academies Press. (2011). Guide for the care and use of laboratory animals. https://doi.org/10.17226/12910
- Parson, W., Kirchebner, R., Mühlmann, R., Renner, K., Kofler, A., Schmidt, S., & Kofler, R. (2005, March). Cancer cell line identification by short tandem repeat profiling: Power and limitations. FASEB Journal, 19(3), 1–18. https://doi.org/10.1096/fj.04-3062fje
- Pearlman, E., Jiwa, A. H., Engleberg, N. C., & Eisenstein, B. I. (1988, August). Growth of legionella pneumophila in a human macrophage-like (U937) cell line. Microbial Pathogenesis, 5(2), 87–95. https://doi.org/10.1016/0882-4010(88)90011-3
- Philippeos, C., Hughes, R. D., Dhawan, A., & Mitry, R. R. (2012). Introduction to cell culture. Methods of Molecular Biology, 806, 1–13. https://doi.org/10.1007/978-1-61779-367-7_1
- Repine, J. E., Fox, R. B., & Berger, E. M. (1981, January). Dimethyl sulfoxide inhibits killing of Staphylococcus aureus by polymorphonuclear leukocytes. Infection and Immunity, 31(1), 510–513. https://doi.org/10.1128/iai.31.1.510-513.1981
- Trouplin, V., Boucherit, N., Gorvel, L., Conti, F., Mottola, G., & Ghigo, E. (2013, November). Bone marrow-derived macrophage production. Journal of Visualized Experiments, (81). https://doi.org/10.3791/50966-v
- Verheijen, M., Lienhard, M., Schrooders, Y., Clayton, O., Nudischer, R., Boerno, S., Timmermann, B., Selevsek, N., Schlapbach, R., Gmuender, H., Gotta, S., Geraedts, J., Herwig, R., Kleinjans, J., & Caiment, F. (2019, March). DMSO induces drastic changes in human cellular processes and epigenetic landscape in vitro. Scientific Reports, 9(1), 4641. https://doi.org/10.1038/s41598-019-40660-0
- Wei, F., Zhao, L., & Jing, Y. (2020, November). Mechanisms underlying dimethyl sulfoxide-induced cellular migration in human normal hepatic cells. Environmental Toxicology and Pharmacology, 80, 103489. https://doi.org/10.1016/j.etap.2020.103489
- Weischenfeldt, J., & Porse, B. (2008, December). Bone marrow-derived macrophages (BMM): Isolation and applications. Cold Spring Harbor Protocols, 2008(12), db.prot5080. https://doi.org/10.1101/pdb.prot5080
- Zajd, C. M., Ziemba, A. M., Miralles, G. M., Nguyen, T., Feustel, P. J., Dunn, S. M., Gilbert, R. J., & Lennartz, M. R. (2020, February). Bone marrow-derived and elicited peritoneal macrophages are not created equal: The questions asked dictate the cell type used. Frontiers in Immunology, 11(11). https://doi.org/10.3389/fimmu.2020.00269
- Zhang, X., Goncalves, R., & Mosser, D. M. (2008, November). The isolation and characterization of murine macrophages. Current Protocols in Immunology, 83(1). https://doi.org/10.1002/0471142735.im1401s83