
Abstract
In this study we investigate methods for attribute clustering and their possible applications to the task of computation of decision reducts from information systems. We focus on high-dimensional datasets, that is, microarray data. For this type of data, the traditional reduct construction techniques either can be extremely computationally intensive or can yield poor performance in terms of the size of the resulting reducts. We propose two reduct computation heuristics that combine the greedy search with a diverse selection of candidate attributes. Our experiments confirm that by proper grouping of similar—in some sense interchangeable—attributes, it is possible to significantly decrease computation time, as well as to increase a quality of the obtained reducts (i.e., to decrease their average size). We examine several criteria for attribute clustering, and we also identify so-called garbage clusters, which contain attributes that can be regarded as irrelevant.
INTRODUCTION
In many applications, available information about objects from a considered universe has to be reduced. This reduction might be required in order to limit resources that are needed by algorithms analyzing the data or to prevent crippling their performance by noisy or irrelevant attributes (Kohavi and John Citation1997; Mitchell Citation1997). Many of the popular attribute subset selection methods are derived from the theory of rough sets (Pawlak Citation1991, Świniarski and Skowron Citation2003).
In the rough set approach, the reduction of an object description is usually done by following the notion of a reduct—a minimal set of attributes that sufficiently preserves information allowing the discernment of objects with different properties, for example, belonging to different decision classes. The techniques for computation of decision reducts have been widely discussed in literature related to data analysis and knowledge discovery. Their practical significance for tasks such as attribute selection, rule induction, and data visualization is unquestionable (Janusz and Stawicki Citation2011; Widz and Ślęzak Citation2012).
The discussed approach to attribute reduction can be used for a wide spectrum of high-dimensional data types. In this article, we focus on gene expressions. There is a great deal of literature showing how to apply the rough set approach to microarray data analysis (Fang and Grzymała-Busse Citation2006; Janusz and Stawicki Citation2011; Midelfart et al. Citation2002). In Grużdź, Ihnatowicz, and Ślęzak (Citation2006), Janusz and Ślęzak (Citation2012), and Ślęzak (Citation2007), it was discussed that, given such a huge amount of attributes in microarray datasets, it is indeed better to combine the standard computation mechanisms with some elements of attribute clustering. Therefore, this work aims at experimental verification of these ideas by combining rough set algorithms for attribute reduction with rough set inspired methods for attribute clustering.
This study is a continuation of research described in Janusz and Ślęzak (Citation2012), in which we focused on identification of attribute dissimilarity measures that are appropriate for finding groups of interchangeable attributes. We extend this work by an in-depth investigation of the selected gene-clustering results. We also propose two algorithms for computation of multiple decision reducts. Those algorithms combine the greedy heuristic approach and attribute clustering results in order to obtain a set of diverse and short reducts. We evaluate the proposed methods in a series of experiments, and we discuss the impact of attribute clustering on the performance of greedy reduct computation heuristics.
This article is organized as follows: “Rough Set-Based Attribute Selection and Clustering” discusses basic notions from the rough set theory, that are related to the attribute reduction problem and recalls some popular algorithms for the computation of reducts. It also outlines our intuition behind combining attribute reduction with attribute clustering. “Framework for Experimental Validation” reports our experimental framework for utilizing gene-clustering methods for computation of reducts, and “Experiments with Dissimilarity Functions” presents the evaluation of the proposed modification to the permutation-based reduct computation algorithm. “Analysis of Selected Gene-Clustering Results” investigates selected gene-clustering results. “Randomized Greedy Computation Decision Reducts” shows how those observations can be used to overcome major issues related to the greedy computation of reducts for high-dimensional datasets. “Concluding Remarks” concludes the paper.
ROUGH SET-BASED ATTRIBUTE SELECTION AND CLUSTERING
In the rough set theory, the available information about the considered universe is usually represented in a decision system understood as a tuple , where U is a set of objects, A is a set of attributes, and d is a distinguished attribute called a decision. By a decision reduct of
, we usually mean a compact yet informative subset of available attributes. The most basic type of decision reduct is a subset of attributes
satisfying the following conditions:
For any pair
of objects belonging to different decision classes (i.e.,
There is no proper subset
A decision reduct is a set of attributes that are sufficient to discern objects from different decision classes. At the same time, this set has to be minimal, in a sense that no further attributes can be removed from DR without losing the discernibility property. For example, and
are decision reducts of the decision system
from . The first listed condition for a decision reduct is often replaced by some other requirements for preserving information about the decision while reducing attributes. In this article, for simplicity, we restrict ourselves to the aforementioned discernibility-based criterion, which is well documented in the rough set literature (Bazan et al. Citation2000; Nguyen Citation2006).
TABLE 1 An Exemplary Decision Table with a Binary Decision
Many algorithms for attribute reduction are described, utilizing various greedy or randomized search approaches (Janusz and Stawicki Citation2011). Most of them refer to the search for an optimal (shortest, generating minimum number of rules, etc.) decision reduct or some larger ensembles of decision reducts that constitute efficient classification models (Janusz Citation2012). We can also consider their approximate versions, which are especially useful for noisy datasets (Ślęzak Citation2000). For instance, we can require that only a percentage of object pairs satisfies the first condition for a decision reduct. Moreover, we may extend the discernibility notion toward the criteria of a sufficient dissimilarity or a discernibility in a degree, which are useful in the case of numeric data (Jensen and Shen Citation2009).
A commonly used technique for the computation of decision reducts is the greedy approach explained by Algorithm 1. In this algorithm, corresponds to an attribute quality measure that is monotonic, in a sense that it decreases with an increasing size of the set from its second argument. This function also needs a property that it equals 0 if the second argument is a superreduct, that is, a set of attributes that discern all objects from different decision classes. A number of such functions were adopted for the purposes of reduct computation (Janusz and Stawicki Citation2011; Ślęzak Citation2000).
The utilization of the greedy heuristic usually leads to a short reduct (by means of its cardinality). However, two major disadvantages of this approach are its high computational complexity with regard to the total number of attributes in the data and the fact that it can be used to construct only a single reduct. A viable alternative that can overcome those issues is the permutation-based method, called ordered reducts (Janusz and Ślęzak Citation2012; Wróblewski Citation2001), explained by Algorithm 2.
Appropriately extended classical notions of the rough set theory can be successfully applied as an attribute selection framework for the analysis of large and complex data sets, such as microarray data. However, there is yet another possibility for scaling the original rough set notions with regard to the number of attributes. The basic idea is to utilize additional information about groups of attributes that can potentially replace each other while constructing reducts from the data.
For a moment, let us imagine that some miraculous oracle could identify such groups of attributes in the data. In Janusz and Ślęzak (Citation2012), we proposed a method that allows us to incorporate this additional knowledge into the reduct computation process by influencing the generation of permutations in the ordered reducts algorithm. This process is explained by Algorithm 3. We refer to a fusion of Algorithm 3 into the permutation-based method as ordered reducts with diverse attribute drawing (OR-DAD).
The knowledge regarding groups of interchangeable attributes can often be acquired from domain experts or external knowledge bases such as domain ontologies (e.g., the Gene Ontology). It can also be obtained automatically by utilizing attribute clustering methods (Jain, Murty, and Flynn Citation1999). From a perspective of the microarray data analysis, such an idea refers to a task of gene clustering (Baldi and Hatfield Citation2002; McKinney et al. Citation2006). In Grużdź, Ihnatowicz, and Ślęzak (Citation2006), we reported that the gene-clustering outcomes may meet expert expectations to more extent when they are based on information-theoretic measures, rather than on standard numeric and rank-based correlations. In other words, interpreting genes as attributes with some approximate dependencies between them may bring better results than treating them as numeric vectors. In Ślęzak (Citation2007), we suggested that attribute clustering can be conducted also by means of dissimilarity functions based on discernibility between objects, utilized as a form of measuring degrees of functional dependencies between attributes. We also proposed a mechanism wherein reducts could be searched in a data table consisting only of the previously computed cluster representatives, with their occurrence in reducts used as feedback for the clustering refinements.
It is reasonable to use analogous criteria for preserving information about a decision while reducing attributes and measuring distances between them. As an example, let us compare the attributes and
in . In the case of most of pairs of objects,
discerns them, iff
does. This may indicate either that there are relatively many pairs of reducts of the form
and
,
, or that the attributes
and
do not occur in reducts at all. Reducts
and
are an illustration of this kind of replaceability. The attributes that are likely to be interchangeable can be easily noticed by studying a dendrogram generated by a hierarchical clustering algorithm. An example of such a tree generated for the decision system from is presented in . As expected, the attributes
and
are merged into a single cluster as the second pair.
FIGURE 1 An attribute-clustering tree for the decision table from , obtained by applying the agglomerative nesting algorithm in combination with the direct discernibility dissimilarity function.
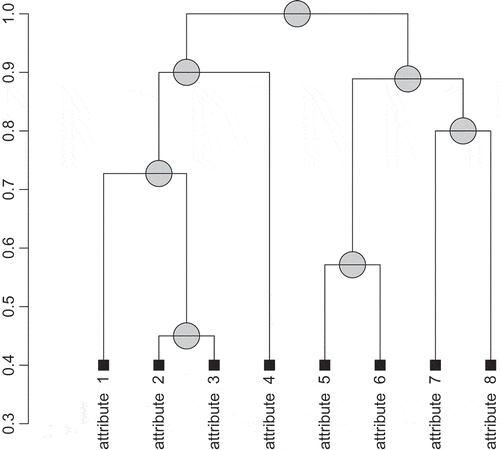
The methods of an attribute reduction and grouping can be put together in many different ways. As an example, in Abeel and colleagues. (Citation2010), it is noted that so-called signatures (irreducible subsets of genes providing enough information about probabilities of specific types of cancer—the reader may notice an interesting correspondence of this notion to a probabilistic version of a decision reduct in Ślęzak Citation2000) can contain genes that are interchangeable with the others because of data correlations or multiple explanations of some biomedical phenomena. Moreover, such an interchangeability can be observed not only for single elements but also for whole sets of attributes.
FRAMEWORK FOR EXPERIMENTAL VALIDATION
We conducted a series of experiments to verify usefulness of the attribute clustering for scalable computation of decision reducts. We wanted to find answers to two main questions. The first was whether the attribute grouping can speed up searching for reducts. The second question was related to a quality of reducts generated using different clustering methods—we wanted to check if such reducts are more concise. The minimal number of attributes is not the only possible optimization criterion for decision reducts (Ślęzak Citation2000; Wrόblewski 2001). However, it is indeed the most straightforward idea to rely on minimal reducts in order to clearly visualize the data dependencies.
In the experiments, we use a microarray dataset from the Rough Sets and Current Trends in Computing (RSCTC) 2010 conference competition aimed at constructing classifiers with the highest possible accuracy (Wojnarski et al. Citation2010). We focus on this specific dataset because—although, currently, we do not evaluate the obtained reducts by means of accuracy of classifiers that they are yielding—this will be the next step of our investigation, leading toward the ability to compare reduct-based classification models with the competition winners.
Microarrays are usually described by many thousands of attributes whose values correspond to expression levels of genes. The considered dataset is related to the investigation of a chronic hepatitis C virus role in the pathogenesis of HCV-associated hepatocellular carcinoma. It contains data on 124 tissue samples described by 22,277 numeric attributes (genes). It was obtained from the ArrayExpress repository (Parkinson et al. Citation2009; dataset accession number: E-GEOD-14323). The gene expression levels in this dataset were obtained using Affymetrix GeneChip Human Genome U133A 2.0 microarrays.
We preprocessed the data by discretizing attributes using an unsupervised method. Every expression-level value of a given gene was replaced by one of the three labels: over_expressed, normal, or under_expressed. A label for an attribute a and a sample u is decided as follows:
We operate on relatively simple rough set-motivated dissimilarity functions that refer to the comparison of attributes’ abilities to discern important pairs of objects. The first considered function, called a direct discernibility function, is a ratio between a number of pairs of objects from different decision classes that are discerned by exactly one attribute to a number of such objects discerned by at least one of the compared attributes. It can be written down in a way that emphasizes its analogy to some standard measures used in data clustering (Jain, Murty, and Flynn Citation1999; Kaufman and Rousseeuw Citation1990).
We also verified the usefulness of two other discernibility-based dissimilarity functions. The relative discernibility, described in more detail in Janusz and Ślęzak (Citation2012); takes into account the fact that some pairs of objects belonging to different decision classes are more difficult to discern than the others. It assigns higher weights to pairs of objects from different decision classes, which are discerned by a lower number of attributes. The full discernibility function does not take into account decision classes. Instead, it measures a ratio between the total number of objects discerned by exactly one attribute to the total number of objects discerned by at least one of the attributes. These definitions should be regarded as just a few of many possible mathematical formulations of the basic intuition that an attribute dissimilarity measure should help to identify groups of attributes that are interchangeable within the same reducts.
In order to assess an impact of different attribute-clustering methods on computation of reducts, in experiments we clustered the genes using several techniques. We combined the discernibility-based functions with the agglomerative nesting (agnes), which is a hierarchical grouping algorithm (Jain, Murty, and Flynn Citation1999; Kaufman and Rousseeuw Citation1990). We compared it with k-means and agnes algorithms, working on dissimilarities computed using Euclidean distance on nondiscretized data. We also checked clusterings based on correlations between values of attributes, coupled with the agnes algorithm. As a reference, we took results obtained for a random clustering, which is actually equivalent to no clustering at all. We additionally checked the worst-case scenario in which the attributes are grouped so that the most dissimilar genes (according to the direct discernibility function) are in the same clusters.
In each repetition of the experiment, we generated 100 reducts for all the compared clustering methods. For the reduct computation we used Algorithm 2. The permutations for each run of the algorithm were generated based on the clusterings corresponding to the tested grouping methods. Algorithm 3 explains the permutation construction process. In practice, there is no need to pregenerate a permutation for the reduct computation, because it might be an integral part of the algorithm. However, in experiments, we explicitly generated the permutations for the sake of reproducibility of the results.
EXPERIMENTS WITH DISSIMILARITY FUNCTIONS
Table 2 summarizes measurements of computation times. For each clustering method, the mean and standard deviations of 20 independent repetitions of the experiment are given. The results clearly show the advantage of using the direct discernibility function in combination with a hierarchical clustering algorithm to speed up the generation of decision reducts. Times obtained by this method are significantly lower than those of all other approaches. The significance was measured using a t-test (Demšar Citation2006), and the p-values obtained at 0.95 confidence level were all lower than . For instance, the times obtained by this method when grouping was made into 1000 clusters are, on average, lower by
than the corresponding times for the random method. Moreover, robustness of the previously discussed tendency is confirmed in by the stability with regard to a number of considered clusters.
TABLE 2 Average Computation Times of 100 Reducts for Permutations Produced Using Different Clusterings
The results obtained for the relative discernibility function may be regarded as disappointing. The tested weighting schema seems to degrade the performance of the reduct computation algorithm, especially when a low number of gene clusters is considered. The explanation of this behavior will be within the scope of our future research. The experiments show that distinguishing between the cases that are easier or more difficult to discern might not be necessary; however, a better-adjusted mathematical formula for such distinguishing may lead to more promising outcomes.
The results from obtained for the two Euclidean distance-based clusterings also show a clear advantage of using hierarchical methods for grouping genes in microarray data. Actually, the times for the k-means clustering with the Euclidean settings cannot be regarded as statistically different from the results of random clusterings at the level of 1000 generated clusters.
For each clustering method, we also measured an average size of the generated reducts. This statistic reflects a quality of reducts, both by means of data-based knowledge representation and ability to construct efficient classification models. These results are displayed in . The standard deviations given in this table are not computed directly from the sizes of the reducts but from the average sizes of 100 reducts in each of the 20 experiment runs. This explains such low values of this statistic.
TABLE 3 Average Sizes of 100 Reducts Computed for Different Clusterings
The direct discernibility method significantly outperformed other approaches also in terms of the reduct size. As before, the significance was checked using a t-test. On average, decision reducts generated by using the hierarchical clustering based on the direct discernibility function are shorter than those computed from the random clusterings by nearly 1.5 genes. They were also shorter than the reducts computed for the agnes algorithm and for the Euclidean distances by over 0.5 gene. It confirms that a proper attribute clustering increases efficiency of the reduct computation methods.
Because reducts are often computed in order to create a concise representation of data (e.g., for a convenient visualization, see Widz and Ślęzak Citation2012) we also measure sizes of the shortest reducts computed in each of the 20 repetitions of the experiment. These results are shown in . They additionally confirm the importance of considering a specific decision problem as a context when forming groups of genes. The attribute dissimilarity functions that do not refer to a given decision task perform worse than those taking the decision attribute into account. The best illustration for this fact are the results obtained for the full discernibility function, which are significantly worse than random. The full discernibility is a measure that is similar to the direct discernibility measure but it, neglects the decision attribute. This leads to a radical change in the obtained results—from the best to worse-than-random.
TABLE 4 Average Minimal Sizes among 100 Reducts Computed for Different Clusterings
ANALYSIS OF THE SELECTED GENE-CLUSTERING RESULTS
We manually investigated results of different clusterings in order to gain some insights on factors that influence the reduct computation efficiency. We noticed that the most successful clusterings, which are based on the direct discernibility method combined with the agnes algorithm, are significantly imbalanced with regard to the number of attributes in each group. For instance, the distribution of attributes for the clustering into 10 groups is shown in . Analogical distributions obtained for the relative discernibility-, full discernibility-, correlation- and Euclidean-based clusterings are given for a reference.
TABLE 5 Distribution of Attributes in Clusterings into 10 Groups Using the Agnes Algorithm
The first group in the direct discernibility clustering is highly over represented, whereas the distribution of genes for the correlation measure is quite uniform. The distributions for the relative and Euclidean measures can be placed between the distributions of the direct discernibility and correlation groupings. Finally, the distribution of the full discernibility clustering is the most imbalanced—nearly of genes are placed in a single group. This result confirms that the full discernibility function is unable to capture different roles played by particular genes in the decision problem.
When we compared the clustering trees obtained for those measures, we found that the direct discernibility measure leads to a skewed outcome, whereas the trees for the other functions (apart from the full discernibility measure) are wellbalanced (see ). However, the dissimilarities between the clusters—corresponding to relative differences in height of the tree nodes—are usually larger for the direct discernibility measure.
FIGURE 2 A visualization of the clustering trees for five different gene dissimilarity measures, which were cut at a height corresponding to the division into 10 groups.
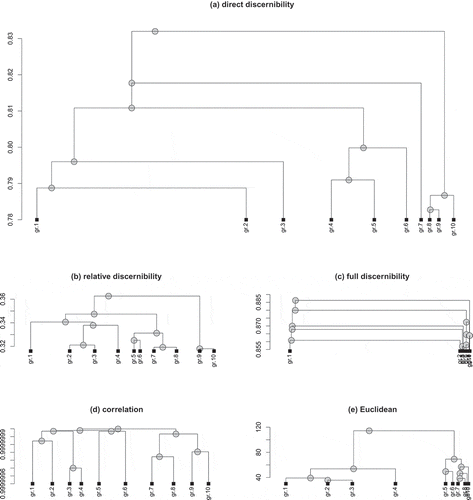
The presence of a majority cluster among groups of genes may have a very intuitive explanation. It is common that only a small portion of genes in data is truly related to a problem indicated by a decision attribute. A majority of genes do not bring any important information, hence, intuitively, a good gene-clustering algorithm should place them in a separate cluster. We decided to perform an additional series of tests in order to verify whether this hypothesis is true for the direct discernibility clustering.
TABLE 6 Performance of the Ordered Reducts (OR) and Ordered Reducts with Diverse Attribute Drawing (OR-DAD) Algorithms with and without Attributes from the Group 1 of the Direct Discernibility Clustering into 10 Groups (Results of 20 Independent Repetitions of the Experiment)
We checked the performance of the permutation-based reduct computation heuristic (the OR-DAD algorithm) in a case when we drop the attributes from the majority cluster of the direct discernibility clustering into 10 groups (using agnes). We modified the permutation generation process so that it does not include attributes from the majority cluster. The results of the comparison are shown in . By removing attributes from the majority cluster, we decreased average computation time of 100 reducts by and their average size by 0.142. Although such an improvement is not very large, its statistical significance was confirmed by the t-test at 0.95 confidence level. It is worth mentioning that the time complexity of the permutation-based heuristic is constant with regard to the number of attributes in the data, so this difference in performance was solely a result of the omission of unnecessary (uninformative) attributes from the majority cluster. We additionally computed reducts from random permutations of genes from the majority cluster and from random permutations of the remaining attributes. The reducts constructed from attributes placed in the majority cluster were, on average, longer by nearly
, and their construction was over
more timeconsuming. In fact, their statistics were comparable or even worse than those obtained for the worst-case clustering scenario (see , , and ). We may use this observation to improve the existing reduct computation heuristics.
RANDOMIZED-GREEDY COMPUTATION OF DECISION REDUCTS
Our experiments described in previous sections showed that a proper attribute-clustering method can significantly improve the permutation-based reduct algorithm by indicating groups of attributes that are potentially interchangeable in many reducts. In our research, we were interested in whether this observation holds also for the greedy reduct computation methods. The greedy heuristic often allows us to find a much shorter reduct than those obtained from the randomized algorithms. For instance, Algorithm 1 combined with the gini gain measure for evaluation of attribute quality, applied to the hepatitis C data, generates a reduct consisting of only six genes. When we compare this result to the minimal sizes of the reducts constructed using the OR-DAD algorithm (see ), it is clearly visible that the greedy reduct is, on average, smaller by two to four attributes. The gini gain measure could be used also to reformulate constraints in the decision reduct definition, as proposed in Ślęzak (Citation2000). However, in this study we keep our focus on standard decision reducts and we treat gini gain just as an example of a greedy evaluation function.
Two major disadvantages of the greedy heuristic are its computational inefficiency for datasets with a significantly large number of attributes and the fact that it can be used to generate only a single reduct. For example, in the described experiment, the computation time needed to construct the greedy reduct was 544 seconds, which is over times slower than in the case of the permutation-based algorithm.
The above observation motivated us to measure an impact of attribute grouping on a computation time of the greedy reduct. We introduced constraints to the greedy algorithm that allow selection of only a single attribute from each cluster. The selection itself was still done in the greedy fashion. This modification resulted in a significant decrease of time needed for computation of a single greedy reduct—it took 392 seconds when grouped into 10 clusters with the direct discernibility measure (about less). The size of a reduct obtained in this way was six, which is equal to the classical case. However, those two greedy reducts differed on two out of six attributes. In particular, this shows that searching for a single-decision reduct provides highly incomplete knowledge about dependencies in the data, especially for such large numbers of attributes. Hence, the approaches aimed at extraction of larger families of reducts should be preferred (Widz and Ślęzak Citation2012; Wrόblewski 2001).
Following the previous study, we wanted to check if a more significant improvement could be obtained. We decided to investigate the possibility of introducing some intelligent attribute search strategies into the greedy algorithm to accelerate its execution. We also wanted to check whether our previous observation regarding the majority cluster can bring some benefit for the greedy computation of decision reducts.
First, we repeated the execution of the greedy algorithm combined with the clustering into 10 groups, based on the direct discernibility clustering but without consideration of genes from the majority cluster (i.e., gr.1). The reduct was generated in which is over 3.5 times faster than in the case of the standard greedy algorithm. The obtained reduct had the same size as the original one (i.e., it consisted of six attributes). It differed, however, on three out of six genes. Interestingly, it also differed on three genes from the reduct obtained by application of the clustering but with the majority cluster included.
In the second experiment, we verified efficiency of two reduct generation heuristics that combine the greedy approach with some randomization techniques and the utilization of the attribute clustering results. They were motivated by the random forest algorithm (Breiman Citation2001) which constructs an ensemble of decision trees generated from randomized subsets of attributes. Analogically, at each step of the reduct computation algorithm, only a small subset of randomly chosen attributes can be considered. This approach is sometimes called the random reducts algorithm (Algorithm 4) and it was already used in a slightly modified version in, for example, Janusz (Citation2012) and Janusz and Stawicki (Citation2011).
FIGURE 3 Average computation times, minimal and average sizes, and average maximal overlap of reducts computed using the RR-DAS and OR-DAS algorithms based on direct discernibility. Plots correspond to different settings of the attribute sample size used in every iteration of the algorithms.
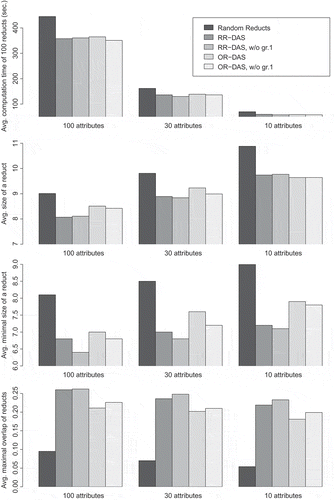

By the utilization of attribute-clustering results, we may try to bias the attribute-sampling process and improve the efficiency of the reduct construction. For this purpose we propose two heuristics. In the first, which we call random reducts with diverse attribute sampling (RR-DAS), attributes are uniformly sampled from all the clusters. At each step of the algorithm, the set of attributes to be evaluated contains approximately the same number of elements from every group. This guarantees maximal diversity of attributes considered at every step of the algorithm. The search for the best attribute in every iteration is performed using the greedy approach. This heuristic is outlined in Algorithm 5.




The second of the proposed heuristics aims to diversify sets of attributes considered during different steps of the reduct computation. In this approach, called ordered reducts with diverse attribute search (OR-DAS), groups of attributes are permuted and during each iteration the best attribute is searched within an attribute sample drawn from a single cluster (see Algorithm 6).

Performances of the aforen mentioned heuristics were compared in a series of tests on the hepatitis C data. The plots shown in present average results for computation of 100 reducts using the compared algorithms. Average computation times, average reduct sizes, and average minimal reduct sizes are displayed. The last statistic—average maximal overlap—reflects a homogeneity of a set of reducts. For each reduct DR from a set RS, it computes its maximal percentage of common attributes with other reducts in the set and takes the mean of those values:
In the plots, the approach that does not use attribute clusterings (the random reducts algorithm) is compared with both of the proposed heuristics combined with the direct discernibility clustering (see “Framework for Experimental Validation”) into 10 groups (labels RR-DAS and OR-DAS). The impact of removing the majority cluster prior to computation of reducts (the bars with the label w/o gr.1) is also assessed.
The advantage of using the attribute clustering results for computing the randomized greedy reducts is clearly visible. The usage of the direct discernibility clustering not only does speed up the computations, on average, by approximately , but it also decreases the average and minimal size of the obtained reducts (on average, by approximately
). The reducts computed with the use of the gene-clustering results were often as small as the reduct generated using the classical greedy heuristic. The combination of nondeterminism and clustering, however, allowed us to obtain several different short reducts in a much shorter time. The removal of the majority cluster brought a further improvement of those results, but in most of the cases the difference was statistically insignificant.
In all cases, the employment of the clustering results decreased the diversity of the obtained sets of reducts. This can be an issue if the reducts are to be utilized for constructing an ensemble of classification models. This problem is less conspicuous for the second of the proposed heuristics (OR-DAS), hence, it might be preferable for constructing diverse ensembles based on short decision reducts.
One should remember that the sets of reducts can be searched also for other reasons. As an example, let us consider a task of a robust attribute selection (Abeel et al. Citation2010; Świniarski and Skowron Citation2003). In such a case, one is interested in a single subset of attributes selected from the union of reducts, which would be stable over multiple runs of a given algorithm. In practice, it is often accomplished by choosing the attributes that most frequently occur in the obtained attribute subsets. Already, several attribute filtering techniques that derive from the rough set theory have investigated attributes from a union of multiple reducts (Błaszczyński, Słowi¨ski, and Susmaga, 2011; Janusz and Stawicki Citation2011). In order to better reflect stability of such an attribute subset, the aforementioned way of evaluating sets of reducts may need a revision.
Following, we propose several methods for measuring stability of attribute subsets retrieved from families of reducts. We decided to compare reducts obtained in 20 repetitions of previous experiments. We measured the average maximum overlap between the unions of reducts from each of the runs (aveMaxOverlap) and we counted how many attributes were present in the intersection of all the unions (common attrs). For each execution of this experiment we additionally checked how many attributes were present in at least 5 out of 100 reducts (frequent attrs) and we measured the average maximum overlap of those attribute sets (freqMaxOverlap). All of those statistics are presented in .
TABLE 7 Stability of an Attribute Selection Using Different Reduct Computation Algorithms
Attribute sets returned by the proposed algorithms turned out to be much more stable than those obtained without the use of clustering. Interestingly, the permutation-based method (OR-DAS) achieved slightly better results than the RR-DAS algorithm. Moreover, as it was expected, the average maximal overlap of unions of reducts increased when the attributes from the majority group were removed. However, we need to remember that the parameters considered are designed to investigate stability of results with respect to specific goals of an attribute selection. Some other measures should be introduced in order to study the stability of results in a form of sets of attribute sets, optimized for the purposes of the representation of data dependencies or the construction of classifier ensembles.
CONCLUDING REMARKS
In this article, we presented an investigation of a possibility of combining the greedy and permutation-based heuristics to facilitate fast computation of representative ensembles of short decision reducts. The choice of parameters responsible for generation of permutations and the greedy heuristic measures may have a significant influence on the results. However, in all such scenarios it is expected that attribute clustering can improve the computations and the interpretation of reducts.
We proposed a new approach to attribute clustering and its application to a task of computation of short decision reducts from datasets with a large number of attributes. We showed that by utilization of clustering results, it is possible to significantly speed up the search for decision reducts and that the obtained reducts tend to be smaller than those reached without the clustering. We also proposed a discernibility-based attribute dissimilarity measure that is particularly useful for identifying groups of attributes that are likely to be interchangeable in many reducts.
We intend to combine our methods with other knowledge-discovery approaches that involve attribute grouping and selection (Abeel et al. Citation2010; Grużdź, Ihnatowicz, and Ślęzak Citation2006). One may also consider an idea of full integration of the algorithms for attribute clustering and selection, so they can provide feedback to each other within the same learning process. Such a new process may be performed separately for particular microarray datasets or over their larger unions (Janusz and Ślęzak Citation2012).
The integration of the attribute clustering and selection procedures may bring not only significant performance improvements but may also provide a new meaning with regard to the attribute selection outcomes. Instead of subsets of individuals chosen from thousands of attributes, it may be better to deal with subsets of representatives selected from much more robust clusters of interchangeable attributes. Moreover, the outcomes of attribute clustering may help to identify truly irrelevant attributes.
FUNDING
This research was partly supported by the Polish National Science Centre (NCN) grants DEC-2011/01/B/ST6/03867 and DEC-2012/05/B/ST6/03215.
Notes
All the algorithms and experiments were implemented and conducted in R System (http://www.r-project.org/).
REFERENCES
- Abeel, T., T. Helleputte, Y. V. de Peer, P. Dupont, and Y. Saeys. 2010. Robust biomarker identification for cancer diagnosis with ensemble feature selection methods. Bioinformatics 26(3):392–398.
- Baldi, P., and G. W. Hatfield. 2002. DNA microarrays and gene expression: From experiments to data analysis and modeling. Cambridge, UK: Cambridge University Press.
- Bazan, J. G., H. S. Nguyen, S. H. Nguyen, P. Synak, and J. Wróblewski. 2000. Rough set algorithms in classification problem. In Rough set methods and applications: New developments in knowledge discovery in information systems, Studies in Fuzziness and Soft Computing 56:49–88, L. Polkowski, S. Tsumoto, and T. Y. Lin. ed. Heidelberg: Physica-Verlag.
- Błaszczyński, J., R. Słowi¨ski, and R. Susmaga. 2011. Rule-based estimation of attribute relevance. In Rough sets and knowledge technology, Lecture Notes in Computer Science 6954:36–44. Berlin, Heidelberg: Springer.
- Breiman, L. 2001. Random forests. Machine Learning 45(1):5–32.
- Demšar, J. 2006. Statistical comparisons of classifiers over multiple data sets. Journal of Machine Learning Research 7:1–30.
- Fang, J., and J. W. Grzymała-Busse. 2006. Leukemia prediction from gene expression data – A rough set approach. In International conference on artificial intelligence and soft computing, Lecture Notes in Computer Science 4029:899–908. Berlin, Heidelberg: Springer.
- Grużdź, A., A. Ihnatowicz, and D. Ślęzak. 2006. Interactive gene clustering – A case study of breast cancer microarray data. Information Systems Frontiers 8(1):21–27.
- Jain, A. K., M. N. Murty, and P. J. Flynn. 1999. Data clustering: A review. ACM Computing Surveys 31(3):264–323.
- Janusz, A. 2012. Dynamic rule-based similarity model for DNA microarray data. In Transactions on rough sets XV, Lecture Notes in Computer Science 7255:1–25. Berlin, Heidelberg: Springer.
- Janusz, A., and D. Ślęzak. 2012. Utilization of attribute clustering methods for scalable computation of reducts from high-dimensional data. In Federated conference on computer science and information systems, 295–302. Washington, D.C.: IEEE.
- Janusz, A., and S. Stawicki. 2011. Applications of approximate reducts to the feature selection problem. In Rough sets and knowledge technology, Lecture Notes in Computer Science 6954:45–50. Berlin, Heidelberg: Springer.
- Jensen, R., and Q. Shen. 2009. New approaches to fuzzy-rough feature selection. IEEE Transactions on Fuzzy Systems 17(4):824–838.
- Kaufman, L., and P. Rousseeuw. 1990. Finding groups in data: An introduction to cluster analysis. New York, NY: Wiley Interscience.
- Kohavi, R., and G. H. John. 1997, December. Wrappers for feature subset selection. Artificial Intelligence 97:273–324.
- McKinney, B. A., D. M. Reif, M. D. Ritchie, and J. H. Moore. 2006. Machine learning for detecting gene-gene interactions: A review. Applied Bioinformatics 5(2):77–88.
- Midelfart, H., H. J. Komorowski, K. Nørsett, F. Yadetie, A. K. Sandvik, and A. Lægreid. 2002. Learning rough set classifiers from gene expressions and clinical data. Fundamenta Informaticae 53(2):155–183.
- Mitchell, T. M. 1997. Machine learning. New York, NY: McGraw-Hill.
- Nguyen, H. S. 2006. Approximate Boolean reasoning: Foundations and applications in data mining. In Transactions on rough sets V, Lecture Notes in Computer Science 4100:334–506. Berlin, Heidelberg: Springer.
- Parkinson, H. E., M. Kapushesky, N. Kolesnikov, G. Rustici, M. Shojatalab, N. Abeygunawardena, H. Berube, M. Dylag, I. Emam, A. Farne, E. Holloway, M. Lukk, J. Malone, R. Mani, E. Pilicheva, T. F. Rayner, F. I. Rezwan, A. Sharma, E. Williams, X. Z. Bradley, T. Adamusiak, M. Brandizi, T. Burdett, R. Coulson, M. Krestyaninova, P. Kurnosov, E. Maguire, S. G. Neogi, P. Rocca-Serra, S.-A. Sansone, N. Sklyar, M. Zhao, U. Sarkans, and A. Brazma. 2009. Array express update – From an archive of functional genomics experiments to the atlas of gene expression. Nucleic Acids Research 37(Database-Issue):868–872.
- Pawlak, Z. 1991. Rough sets – Theoretical aspects of reasoning about data. Boston, MA: Kluwer Academic Publishers.
- Ślęzak, D. 2000. Normalized decision functions and measures for inconsistent decision tables analysis. Fundamenta Informaticae 44(3):291–319.
- Ślęzak, D. 2007. Rough sets and few-objects-many-attributes problem: The case study of analysis of gene expression data sets. In Frontiers in the convergence of bioscience and information technologies, 437–442. Washington, D.C.: IEEE.
- Świniarski, R. W., and A. Skowron. 2003. Rough set methods in feature selection and recognition. Pattern Recognition Letters 24(6):833–849.
- Widz, S., and D. Ślęzak. 2012. Rough set based decision support – Models easy to interpret. In Selected methods and applications of rough sets in management and engineering, Advanced Information and Knowledge Processing, 95–112, G. Peters, P. Lingras, D. Ślęzak, and Y. Yao. ed. Berlin: Springer.
- Wojnarski, M., A. Janusz, H. S. Nguyen, J. G. Bazan, C. Luo, Z. Chen, F. Hu, G. Wang, L. Guan, and H. Luo. 2010. RSCTC 2010 discovery challenge: Mining DNA microarray data for medical diagnosis and treatment. In Rough sets and current trends in computing, Lecture Notes in Computer Science 6086:4–19. Berlin, Heidelberg: Springer.
- Wróblewski, J. 2001. Ensembles of classifiers based on approximate reducts. Fundamenta Informaticae 47(3–4):351–360.