
ABSTRACT
The effective spread of COVID-19 cases in several countries produces more protein sequences that are released in genomic public sources. It provides some awareness and indications for virus classification of COVID-19 and HIV-1 that are essential for drug discovery of COVID-19. This paper reveals the importance of machine learning algorithms to handle the recognition of two different viruses. Therefore, 18,476 protein sequences for both COVID-19 and HIV-1 and 9238 for each virus are applied to the proposed model based on feature extraction, data labeling, and six classifiers. Amino acid classification according to their dipoles and volumes is employed as a feature extraction tool based on the creation of eight features from twenty amino acids by using the conjoint triad (CT) method. The data labeling is employed as a coding tool by binary numbers refereeing zero for COVID-19 and one for HIV-1. The random forest (RF) model achieved the highest classification accuracy of 99.89% for eight features and 97.80% for two features. The experimental results significantly confirmed that eight features required more computational time than two features, but the accuracy rate was nearly similar in the two cases. This classification strategy of COVID-19 and HIV-1 will prompt the prediction of protein sequences of the new virus.
Introduction
Recently, coronavirus 2019 (COVID-19) has been a vigorous human pathogen that creates a significant hazard to global health (Zhou et al. Citation2020). According to virology vision, this virus originated in a subfamily of Orthocoronavirinae that is divided into four kinds: Alphacoronavirus (αCoV), Betacoronavirus (βCoV), Gammacoronavirus (γCoV) and Deltacoronavirus (δCoV) (Yang and Leibowitz Citation2015). Infection from mammals is caused by αCoV and βCoV, while infection from birds is caused by δCoV and γCoV. The World Health Organization (WHO) has substantiated that COVID-19 is officially called severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which is related to βCoV (Li, Shi, and Yu et al. Citation2005). The CoV genome consists of four structural proteins, including the spike (S) surface glycoprotein, the membrane (M) protein, the envelope (E) protein, and the nucleocapsid (N) protein (Wu et al. Citation2020). The coronavirus S protein is essential for host cells that support drug design (Li Citation2016). In January 2020, the whole genome of this virus was rapidly sequenced on the public GenBank database (NCBI virus Citation2021) for virus analysis and phylogenetic tree related to bioinformatics applications. A genomic study revealed that COVID-19 is different from coronaviruses that cause SARS and MERS (De Wit et al. Citation2016).Xiaowei et al. (Citation2020) proposed a review for the evaluation of COVID-19 by molecular immune mechanisms to provide a guide for the drug production of COVID-19 disease. This survey depends on recent advances in SARS and MERS studies. Menachery et al. (Citation2018) explained the gene expression associated with antigen presentation after MERS infection. Thus, the disruption of the immune resistance of COVID-19 is crucial in its therapy and production of different vaccines.Currently, there are many methods for COVID-19 diagnosis, including epidemiological studies, clinical symptoms, and certain tests for blood analysis, nucleic acid detection (Corman et al. Citation2020), CT screening (Xie, Zhong, and Zhao et al. Citation2020), and immune recognition technology (Woo et al. Citation2005). However, the limitations of nucleic acid detection and CT scans for COVID-19 encourage the development of immune technology for detecting COVID-19 (Woo et al. Citation2005). The viral information of COVID-19, such as its risk, period, mutation, reinfection, and recovery, is poorly understood.Generally, the virus’s invasion is developed because of no fight off infections based on a shortage of immune system performance (Gao et al. Citation1999). Xiaodi et al. (Citation2020) developed the protein-protein interaction (PPI) for predicting human-virus relationships that lead to positive implications for a treatment plan. There are some speculations for the biological relation between COVID-19 and the HIV-1 genome that are imperative to target antibodies (Pradhan, Kumar, and Akhilesh et al. Citation2020). Lately, COVID-19 protein sequences were classified according to their countries using machine learning algorithms (Afify and Zanaty Citation2021). The results indicated that the binary array labeling method and the linear support vector machine (SVM) classifier had significantly higher accuracy, sensitivity, and specificity when applied to COVID-19 protein sequences.Therefore, the objective of the proposed model is to classify known viruses, e.g., HIV-1, and recent viruses, e.g., COVID-19. The assigning of a new virus structure is based on its relationship to known viruses using genomic classification techniques. This model acts as a key to investigating the appropriate extraction features for protein sequence classification of COVID-19 and HIV-1.
Materials and Methods
Text classification is one of the most challenging tasks in the feature extraction stage and machine learning algorithms (Onan et al. Citation2016). To build a strong classification schema, feature extraction is an important stage for representing the text database (Onan Citation2018).
The proposed model has constructed three phases including feature extraction, data labeling, and prediction. The feature extraction is used as data preprocessing for converting the amino acids to numbers while the data labeling is used as binary coding to refer to the zero number for COVID-19, and one number for HIV-1. Finally, the prediction phase is used for classification between two viruses databases. shows the workflow of the classification of COVID-19 and HIV-1.
Figure 1. The workflow for protein sequences classification of COVID-19 and HIV-1viruses.
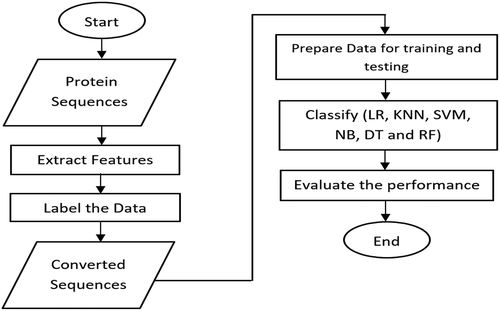
To make the predictions, we trained several classification models using the extracted features of the protein sequences. The output of this proposed model is based on the calculation of accuracy values for each classifier to obtain the best classifier for two viruses including COVID-19 and HIV-1.
Data Analysis
The dataset used was extracted from the (NCBI virus Citation2021) which is available in two formats: CSV (Comma Separated Values) and FASTA.
To date, the available dataset contains 9238 protein sequences for COVID-19 (COVID virus Citation2021) and 1,202,973 protein sequences for HIV-1 (HIV Dataset Citation2021) on the NCBI website. In this paper, the dataset is balanced between the two viruses by selecting only 9238 sequences randomly from each virus. For COVID-19, the maximum sequence length was 7098 amino acids, while the minimum sequence length was 13 amino acids. For HIV-1, the maximum sequence length was 1499 amino acids, while the minimum sequence length was 5 amino acids. It is noted that the protein sequence of HIV-1 is shorter than the protein sequence of COVID-19. Additionally, it was found that the protein sequence of COVID-19 has a high capability for mutation rather than HIV-1 because of protein sequence length. The sample size of COVID-19 virus is 10 MB while the sample size of HIV-1virus is 393 MB. Each protein sequence consists of twenty amino acids (Chen et al. Citation2010). Regarding the ranking of amino acids in (Dayhoff et al. Citation1965), the initial symbols of amino acids are alanine (A), glycine (G), leucine (L), proline (P), and threonine (T). Additionally, there are some phonetic correlations for amino acids such as phenylalanine (F), arginine (R), tryptophan (W), asparagine (N), glutamine (Q), aspartic acid (D), glutamic acid (E), lysine (K), and tyrosine (Y). In partial distributions, two additional symbols are required, such as (B) that consists of (D) or (N) and (Z) that consists of (Q) or (E). The (X) amino acid is undetermined. Then, the founding is applied to the two databases for viruses under the Python program version 3.7.0. which is an open-source programming language and released in 2018.
Feature Extraction Phase
Data preprocessing is a critical task for protein sequences to extract efficient features that are used in the classification process.
The feature extraction phase used the conjoint triad (CT) method (Shen et al. Citation2007) to convert the amino acids in each sequence to numbers according to their side chain dipoles and volumes, as shown in . The dipole and volume values are calculated using two methods: density-functional theory and molecular modeling. The twenty amino acids were divided into seven common groups: C, AGV, DE, FILP, HNQW, KR, and MSTY.
Table 1. Amino acid classification based on their dipole and side chains volumes
In this paper, the amino acid conversions created eight classes from 0 to 7 numbers, as shown in . The unknown amino acid was added with a feature of zero number and labeled with (X). Additionally, the unknown amino acids are (B) and (Z) amino acids because of the uncertainty of the sequences. The (B) amino acid is considered an unknown feature because (D) and (N) amino acids have different features. The (Z) amino acid is considered an unknown feature because (E) and (Q) have different classes. The (J) amino acid is considered the second feature because of (I) and (L) in the same feature, while the sixth feature consists of (D) and (E) amino acids.
Table 2. Amino acid classes
The meaning of the symbols found in is as follows:
Dipole scale: (-), Dipole<1.0/(+), 1.0< Dipole<2.0/(++), 2.0< Dipole<3.0 /(+++), Dipole>3.0/(+’+’+’), Dipole>3.0 with opposite orientation
Volume scale: (-), Volume<50;/(+), Volume> 50
Cysteine (C) is transferred from class 3 to class 7 because of its ability to form disulfide bonds.
Features extracted from the converted protein sequences by counting the number of the eight amino acid classes found in the sequences. Each class count is considered a feature. Then, the counts of each sequence were divided by the sequence length to normalize the frequency values. Therefore, each record has a feature vector consisting of eight features.
Data Labeling Phase
Binary data labeling was utilized to rate the protein sequences for COVID-19 and HIV-1. The zero number is labeled for COVID-19, and one number is labeled for HIV-1.
Classification
The classification models have been successfully supported the machine learning algorithms, whose performance in terms of accuracy values on the amount of data available to determine the best classifier (Onan et al. Citation2016). The text classification domain is applied for eliminating redundant, and noisy data from the training dataset to present an effective testing model (Onan et al. Citation2016).
All eight features extracted from protein sequences are used for classification across six different models to train the proposed model and evaluated in terms of accuracy level. The six models (Ridder de et al. Citation2013) are linear regression (LR), K-nearest neighbor (KNN) with various numbers of K parameters, support vector machine (SVM), naive Bayesian (NB), decision tree (DT), and random forest (RF).
To generate a classification model, two datasets were used: the training set 80% to train the model and the testing set 20% to exam the model. After choosing the model with the highest performance, we used the testing set to create the final results (Marsland Citation2014). The performance is measured in terms of accuracy across the eight and two features. Accuracy is calculated by summation of four values including true positive (correctly classified), true negative (correctly rejected), false positive (incorrectly classified), and false negative (incorrectly rejected).
The six machine learning models (Almeida et al. Citation2014) are summarized in the following:
The LR model (Wang et al. Citation2004) is simply managed for the prognosis of HIV-1 drug resistance.
The KNN model (Altman Citation1992) is commonly used to solve classification problems where there is no prior knowledge about the data distribution. It is based on computing the distance between such test example and each point in the training set. Then, this model keeps the closest training examples (k number of examples) and looks for the label that is most common among these examples. It is carried out to build the biological network of human protein interactions (Xu and Li Citation2006).
The SVM model (Cortes and Vapnik Citation1995) is based on the principle of a binary classification. It is used to find the largest radius around a classification boundary (margin) where no data points are placed. The closest points to this margin are called support vectors that are used to determine a decision boundary in the classification problem. It is also used for the prediction of the HIV-1 gene (Keerthikumar et al. Citation2009). In this paper, the SVM model is applied with different kernel functions, such as linear, sigmoid, polynomial, and radial basis function (RBF) (Guo et al. Citation2008).
The NB model (Rish Citation2001) is used for the recognition of human disease genes (Calvo et al. Citation2006).
The DT model (Quinlan Citation1986) is based on building a tree starting at the root (base) of the tree and processing down to the leaves to provide the classification decision. It is performed early to classify the disease proteins (Adie et al. Citation2005).
The RF model (Breiman Citation2001) subsequently exploits many decision trees to improve the learning speed for a large database. This model was applied to the HIV-1 biological phenotype for effective prediction (Xu et al. Citation2007). This model achieved the highest performance for satire detection in Turkish articles (Onan and Alp Citation2020). The implementation of this model is identified by diverse estimators such as 5, 10, 15, and 20.
Performance of all classification models can be measured by computing an accuracy percentage
which refereed to the predicted and the actual classes. Accuracy represented the overall effectiveness of a classifier.
Results and Discussion
Due to the high mortality rate of COVID-19 disease, the classification paradigms are indispensable to effectively fight against COVID-19. This paper demonstrated the classification-based supervised learning techniques between the 9238 protein sequences for each virus across COVID-19 and HIV-1. The proposed model is based on the extraction of eight features by using amino acid classification. Then, binary data labeling is applied to facilitate the differentiation between the two classes of viruses. In the last phase, different models utilized 80% for the training sequences and 20% for the testing sequences. Furthermore, the proposed model is repeated for all classification steps according to two features by reducing the number of features from eight to only two selected features (class 2 and class 6). The comparison of classification results for eight features and two features are displayed in , respectively.
Table 3. Results of classification of COVID-19 and HIV-1 for eight features
Table 4. Results of classification of COVID-19 and HIV-1 for two features
For eight extracted features, the excellent KNN model for k = 5 achieved an accuracy of 99.83% rather than other parameters of k. For two extracted features, the excellent KNN model for k = 3 and K = 10 achieved an accuracy of 97.60% rather than other parameters of k.
As shown in , it is notable that the RF model with 20 estimators obtained an accuracy of 99.89%, which has the highest accuracy when compared with other models. On the other hand, the KNN model with K = 5 achieved an accuracy of 99.83%, while the SVM model with a polynomial function achieved an accuracy of 99.60%. The LR model achieved an accuracy of 91.10%, the NB model achieved an accuracy of 91.40% and the DT model achieved an accuracy of 99.70%. The weak classifier referred to SVM with the sigmoid function that achieved an accuracy of 79.10%.
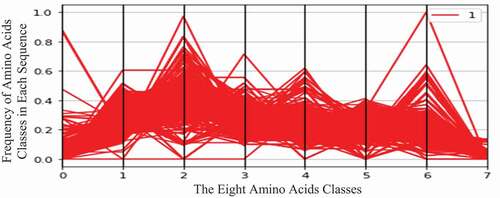
display the two virus datasets according to the eight classes of amino acids. In these figures, the horizontal axis represents eight amino acid classes (features), and the vertical axis represents the frequency (number of occurrences) of amino acid classes in each protein sequence. The green curve represents the COVID-19 virus (zero class), and the red curve represents the HIV-1 virus (one class).
Figure 2. COVID-19 and HIV-1 samples according to eight features.
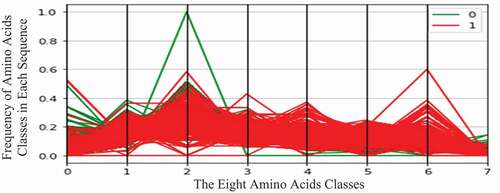
Figure 3. COVID-19 samples according to eight features.
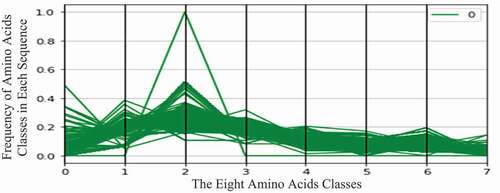
Figure 4. HIV-1 samples according to eight features.
As shown in , the COVID-19 protein sequences contain high values of the second amino acid class. This class contains the following amino acids: Isoleucine (Ile, I), Leucine (Leu, L), Phenylalanine (Phe, F), and Proline (Pro, P).
Moreover, HIV-1 protein sequences contain high values of the sixth amino acid class. This class contains the following amino acids: Glutamic Acid (Glu, E) and Aspartic Acid (Asp, D).
As shown in , it is notable that the RF model with 15 estimators obtained an accuracy of 97.80%, which has the highest accuracy when compared with other models. On the other hand, the KNN model with K = 3 and 10 achieved an accuracy of 97.60%, while the SVM model with the RBF function achieved an accuracy of 85.40%. The LR model achieved an accuracy of 76.90%, the NB model achieved an accuracy of 59.10% and the DT model achieved an accuracy of 97.60%. The weak classifier referred to SVM with the sigmoid function that achieved an accuracy of 49.70%.
This table confirmed that the two features, including the second class and sixth class of amino acids, can differentiate between COVID-19 and HIV-1 viruses. However, the RF model is a nonlinear classifier that outperforms other classification schemes with variations in the number of features. It yielded a higher classification accuracy using the eight features rather than that for two features. In this proposed model, the two features (frequency of classes) can only be applied to detect the virus more quickly than eight features. Additionally, this model indicated that the protein sequence of HIV-1 is more acidic than that of COVID-19.
Conclusion
Although COVID-19 disease is considered a global epidemic, the concerns from the mutation of this disease created the motivation to understand its protein sequences and classify between COVID-19 and HIV-1 viruses. The visibility of this proposed model confirmed that COVID-19 has lower acidity than HIV-1. Combining viral sequences with machine learning algorithms, the virus classification accuracy is often altered by selected features and classifier categories. The overall outcomes proved that the RF model based on accuracy measure has a substantial impact on the differentiation between protein sequences of COVID-19 and HIV-1 viruses. The next challenge is to gain knowledge about the selection of suitable features for COVID-19 virus diagnosis. Finally, bioinformatics applications will be advanced to cover COVID-19 genome analysis associated with favorable classification methodology.
Disclosure Statement
No potential conflict of interest was reported by the author(s).
References
- Afify H., and Zanaty M. 2021. Computational predictions for protein sequences of COVID-19 virus via machine learning algorithms, Med Biol Eng Comput. 22, 1–12. doi:https://doi.org/10.1007/s11517-021-02412-z.
- Almeida, H., M.-J. Meurs, L. Kosseim, G. Butler, A. Tsang, and A. R. Dalby. 2014. Machine learning for biomedical literature triage. PLoS One 9 (12):e115892. doi:https://doi.org/10.1371/journal.pone.0115892.
- Altman, N. S. 1992. An introduction to kernel and nearest-neighbor nonparametric regression. The American Statistician 46:175–85.
- Breiman, L. 2001. Random forests. Machine Learning 45 (1):5–32. doi:https://doi.org/10.1023/A:1010933404324.
- Calvo, S., M. Jain, X. Xie, S. A. Sheth, B. Chang, O. A. Goldberger, A. Spinazzola, M. Zeviani, S. A. Carr, V. K. Mootha, et al. 2006. Systematic identification of human mitochondrial disease genes through integrative genomics. Nature Genetics 38(5):576–82. doi: https://doi.org/10.1038/ng1776.
- Chen, C., P. B. McGarvey, H. Huang, and C. H. Wu. 2010. Protein bioinformatics infrastructure for the integration and analysis of multiple high-throughput omics data. Advances in Bioinformatics 2010:1–19. Article ID 423589. doi: https://doi.org/10.1155/2010/423589.
- Corman, V. M., O. Landt, M. Kaiser, R. Molenkamp, A. Meijer, D. K. Chu, T. Bleicker, S. Brünink, J. Schneider, M. L. Schmidt, et al. 2020. Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 25(3). doi: https://doi.org/10.2807/1560-7917.ES.2020.25.3.2000045.
- Cortes, C., and V. Vapnik. 1995. Support-vector networks. Machine Learning 20 (3):273–97. doi:https://doi.org/10.1007/BF00994018.
- COVID virus: online, https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/virus?SeqType_s=Nucleotide&VirusLineage_ss=Severe%20acute%20respiratory%20syndrome%20coronavirus%202%20(SARS-CoV2),%20taxid:2697049, (accessed on 1 March 2021).
- Dayhoff, M. O., R. V. Eck, M. A. Chang, and M. R. Sochard. 1965. Atlas of protein sequence and structure. Silver Spring, Maryland: National Biomedical Research Foundation.
- De Wit, E., N. Van Doremalen, D. Falzarano, and V. J. Munster. 2016. SARS and MERS: Recent insights into emerging coronaviruses. Nature Reviews Microbiology 14 (8):523–34. doi:https://doi.org/10.1038/nrmicro.2016.81.
- Dick de Ridder, Jeroen de Ridder, and Marcel J T Reinders. 2013. Pattern recognition in bioinformatics. Briefings in Bioinformatics 14(5):633–47. doi: https://doi.org/10.1093/bib/bbt020.
- Euan A Adie, Richard R Adams, Kathryn L Evans, David J Porteous, and Ben S Pickard. 2005. Speeding disease gene discovery by sequence based candidate prioritization. BMC Bioinformatics 6(1):55. doi: https://doi.org/10.1186/1471-2105-6-55.
- Gao, F., E. Bailes, D. L. Robertson, Y. Chen, C. M. Rodenburg, S. F. Michael, L. B. Cummins, L. O. Arthur, M. Peeters, G. M. Shaw, et al. 1999. Origin of HIV-1 in the chimpanzee pan troglodytes troglodytes. Nature 397(6718):436–41. doi: https://doi.org/10.1038/17130.
- Guo, Y., L. Yu, Z. Wen, and M. Li. 2008. Using support vector machine combined with auto covariance to predict protein-protein interactions from protein sequences. Nucleic Acids Research 36 (9):3025–30. doi:https://doi.org/10.1093/nar/gkn159.
- HIV Dataset: online, https://www.ncbi.nlm.nih.gov/labs/virus/vssi/#/virus?SeqType_s=Nucleotide&VirusLineage_ss=Human%20immunodeficiency%20virus%201%20(HIV-1),%20taxid:11676, (accessed on 1 March 2021).
- Keerthikumar, S., S. Bhadra, K. Kandasamy, R. Raju, Y. L. Ramachandra, C. Bhattacharyya, K. Imai, O. Ohara, S. Mohan, A. Pandey, et al. 2009. Prediction of candidate primary immunodeficiency disease genes using a support vector machine learning approach. DNA Research 16(6):345–51. doi: https://doi.org/10.1093/dnares/dsp019.
- Li, F. 2016. Structure, function, and evolution of coronavirus spike proteins. Annual Review of Virology 3 (1):237–61. doi:https://doi.org/10.1146/annurev-virology-110615-042301.
- Marsland, S. 2014. Machine learning: An algorithmic perspective. 2nd ed. Chapman and Hall/CRC.
- Menachery, V. D., A. Schafer, K. E. Burnum-Johnson, H. D. Mitchell, A. J. Eisfeld, K. B. Walters, C. D. Nicora, S. O. Purvine, C. P. Casey, M. E. Monroe, et al. 2018. MERS-CoV and H5N1 influenza virus antagonize antigen presentation by altering the epigenetic landscape. Proceedings of the National Academy of Sciences of the United States of America 115(5):E1012e–E1021. doi: https://doi.org/10.1073/pnas.1706928115.
- NCBI virus: online, https://www.ncbi.nlm.nih.gov, (accessed on 1 March 2021).
- Onan, A. 2018. An ensemble scheme based on language function analysis and feature engineering for text genre classification. Journal of Information Science 44 (1):28–47. doi:https://doi.org/10.1177/0165551516677911.
- Onan, A., and T. M. Alp. 2020. Satire identification in Turkish news articles based on ensemble of classifiers. Turkish Journal of Electrical Engineering & Computer Sciences 28 (2):1086–106. doi:https://doi.org/10.3906/elk-1907-11.
- Onan A., Korukoglu S., and Bulut H.(2016). Ensemble of keyword extraction methods and classifiers in text classification. Expert Systems With Applications 57: 232–247.
- Patrick C. Y., Woo, Susanna K. P., Lau, Beatrice H. L., Wong, Hoi-Wah Tsoi, Ami M. Y., Fung, Richard Y. T., Kao, Kwok-Hung Chan, J. S., Malik Peiris, and Kwok-Yung Yuen. 2005. Differential sensitivities of severe acute respiratory syndrome (SARS) coronavirus spike polypeptide enzyme-linked immunosorbent assay (ELISA) and SARS coronavirus nucleocapsid protein ELISA for serodiagnosis of SARS coronavirus pneumonia. Journal of Clinical Microbiology 43(7):3054e3058. doi: https://doi.org/10.1128/JCM.43.7.3054-3058.2005.
- Prashant Pradhan, Ashutosh Kumar Pandey, Akhilesh Mishra, Parul Gupta, Praveen Kumar Tripathi, Manoj Balakrishnan Menon, James Gomes, Perumal Vivekanandan, and Bishwajit Kundu. 2020. Uncanny similarity of unique inserts in the 2019-nCoV spike protein to HIV-1 gp120 and Gag. bioRxiv.
- Quinlan, J. R. 1986. Induction of decision trees. Machine Learning 1 (1):81–106. doi:https://doi.org/10.1007/BF00116251.
- Rish, I.: (2001). An empirical study of the naive Bayes classifier. In: IJCAI 2001 Workshop on Empirical Methods in Artificial Intelligence, vol. 3, pp. 41–46, IBM New York.
- Shen, J., J. Zhang, X. Luo, W. Zhu, K. Yu, K. Chen, Y. Li, and H. Jiang, (2007) Predicting protein-protein interactions based only on sequences information. Proceedings of the National Academy of Sciences of the USA (PNAS), USA, pp. 4337–41.
- Wang, K., E. Jenwitheesuk, R. Samudrala, and J. E. Mittler. 2004. Simple linear model provides highly accurate genotypic predictions of HIV-1 drug resistance. Antiviral Therapy 9 (3):343–52.
- Wendong Li, Zhengli Shi, Meng Yu, Wuze Ren, Craig Smith, Jonathan H Epstein, Hanzhong Wang, Gary Crameri, Zhihong Hu, Huajun Zhang, Jianhong Zhang, Jennifer McEachern, Hume Field, Peter Daszak, Bryan T Eaton, Shuyi Zhang, and Lin-Fa Wang. 2005. Bats are natural reservoirs of SARS-like coronaviruses. Science (New York, NY) 310(5748):676–79. doi: https://doi.org/10.1126/science.1118391.
- Wu, A., Y. Peng, B. Huang, X. Ding, X. Wang, P. Niu, J. Meng, Z. Zhu, Z. Zhang, and J. Wang. 2020. Genome composition and divergence of the novel coronavirus (2019-nCoV) originating in China. Cell Host & Microbe 27 (3):325–28. doi:https://doi.org/10.1016/j.chom.2020.02.001.
- Xiaodi, Y., S. Yang, Q. Li, S. Wuchty, and Z. Zhang. 2020. Prediction of human-virus protein-protein interactions through a sequence embedding-based machine learning method. Computational and Structural Biotechnology Journal 18:153–61. doi:https://doi.org/10.1016/j.csbj.2019.12.005.
- Xiaowei, L., M. Geng, Y. Peng, L. Meng, and S. Lu. 2020. Molecular immune pathogenesis and diagnosis of COVID-19. Journal of Pharmaceutical Analysis 10 (2):102–08. doi:https://doi.org/10.1016/j.jpha.2020.03.001.
- Xingzhi Xie, Zheng Zhong, Wei Zhao, Chao Zheng, Fei Wang, and Jun Liu. 2020. Chest CT for typical 2019-nCoV pneumonia: Relationship to negative RT-PCR testing. Radiology 296 (2):E41-E45.
- Xu, J., and Y. Li. 2006. Discovering disease-genes by topological features in human protein-protein interaction network. Bioinformatics 22 (22):2800–05. doi:https://doi.org/10.1093/bioinformatics/btl467.
- Xu, S., X. Huang, H. Xu, and C. Zhang. 2007. Improved prediction of coreceptor usage and phenotype of HIV-1 based on combined features of V3 loop sequence using random forest. Journal of Microbiology 45:441–46.
- Yang, D., and J. L. Leibowitz. 2015. The structure and functions of coronavirus genomic 3′ and 5′ ends. Virus Research 206:120–33. doi:https://doi.org/10.1016/j.virusres.2015.02.025.
- Zhou, P., X.-L. Yang, X.-G. Wang, B. Hu, L. Zhang, W. Zhang, H.-R. Si, Y. Zhu, B. Li, C.-L. Huang, et al. 2020. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 579(7798):270–73. doi: https://doi.org/10.1038/s41586-020-2012-7.