Abstract
The spectrum of acute renal failure in Falciparum malaria varies from mild urinary abnormalities to acute renal failure. Acute tubular necrosis has been reported in 1% patients, and acute cortical necrosis has rarely been reported. We present a case of acute cortical necrosis in a young patient with Falciparum malaria who had a prolonged oligo-anuric course followed by partial recovery of renal function.
INTRODUCTION
Falciparum malaria is widely prevalent in tropical areas, and infection from this species is associated with life-threatening complications.Citation[1] The spectrum of renal abnormalities extends from fluid-electrolyte abnormalities, to asymptomatic urinary abnormalities, to acute renal failure. The underlying renal pathologies reported are glomerulonephritis, tubulointerstitial nephritis, and micro-vascular involvement. Acute tubular necrosis (ATN) is reported in less than 1% of cases.Citation[2] The renal injury occurs as a consequence of the acute systemic effects of parasitic infection and immune-mediated renal injury resulting from the concomitant host-parasite interaction.Citation[3] Acute cortical necrosis in Falciparum malaria is rare, and anecdotal case reports have been described in literature. We report a case of Falciparum malaria that presented with acute renal failure (ARF), and renal histology showed features of acute cortical necrosis.
CASE REPORT
A 22-year-old healthy male, working in Eastern India, presented with fever with chills and rigors accompanied by upper abdominal pain, yellow discoloration of eyes, and diminished urine output of three days' duration. There was no history of flank pain, gross hematuria, vomiting, skin rash, polyarthralgia, or any respiratory symptoms. Physical examination revealed a febrile patient with a temperature of 101°F who was alert and conscious; there was mild icterus and pallor. The pulse rate was 110/min and regular, blood pressure was 110/70 mm Hg, and respiratory rate was 18/min. Abdominal examination revealed the liver to be palpable by 2 cms. It was tender and soft, and liver span was 15 cms. The spleen was not palpable. Examinations of neurological, cardiovascular, and respiratory systems were unremarkable.
Laboratory investigation revealed a hemoglobin of 7.4 gm/dL and a normochromic, normocytic, total leukocyte count of 6800/cumm with polymorphs of 90% and lymphocytes 8%. The platelet and reticulocyte counts were 20,000/cmm and 1.5%, respectively. The peripheral blood smear showed red blood corpuscles with ring forms of plasmodium Falciparum and a parasite index of 2%. Blood screen for glucose 6 phosphate dehydrogenase (G6PD) deficiency was negative. Blood biochemistry revealed a blood sugar (random) of 78 mg/dL, blood urea of 118 mg/dL, serum creatinine of 3.1 mg/dL, uric acid of 6.2 mg/dL, calcium of 8.4 mg/dL, phosphorus of 4.6 mg/dL, serum sodium of 135 mEq/L, and serum potassium of 4 mEq/L. Liver function tests revealed serum bilirubin of 7 mg/dL (predominantly unconjugated). The total proteins were 6.6 g/dL with serum albumin of 3.7g/dL, serum globulin of 2.9 g/dL, and serum alkaline phosphatase of 182 IU/L; aspartate aminotransferase (AST) and alanine aminotransferase (ALT) were 45 and 48 IU/L, respectively. The prothrombin time (PT) and activated partial thromboplastin time (APTT) were normal. Serum amylase was 58 U/dL, CPK was 32 U/L, and lactate dehydrogenase (LDH) was 1028 IU/L. Blood cultures were sterile. Urinalysis showed proteins 2+, 4–8 red blood cells, and 2–4 white blood cells/high power fields. There were a few epithelial cells and granular casts but no pigment casts. Urine was positive for hemoglobin. The spot urinary sodium was 69 mEq/L. Ultrasound examination of the kidneys revealed the left kidney to be 10.5 cm and the right kidney to be 11 cm in length. There was increased echogenicity of both kidneys.
The patient was diagnosed to have complicated Falciparum malaria and managed with intravenous quinine, given initially in a loading dose of 20 mg/kg body weight. Thereafter, he received a dose of 600 mg every 8h for 48 hrs and later continued in a dose of 300 mg every 12hr (adjusted for creatinine clearance) for another two days, followed by oral quinine for five days. Oral doxycycline was added for a period of one week. He received platelet transfusion for his severe thrombocytopenia.
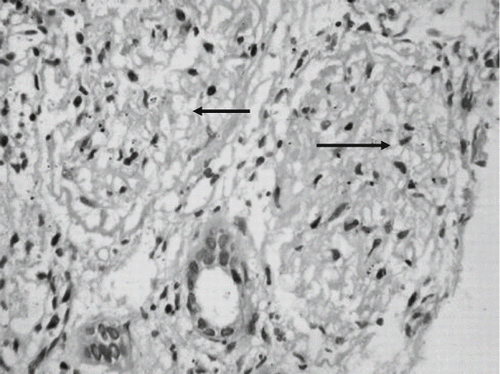
The patient responded well to antimalarial therapy in that he became afebrile within 24hrs and no parasitemia was detected in the peripheral smear. However, he remained oliguric and required hemodialysis three times per week for rising serum creatinine and blood urea nitrogen (maximum of 12 and 220 mg/dL). Six weeks into the illness, the urine output gradually increased to about 1.5 liters a day, but the serum creatinine remained persistently elevated around 3–3.5mg/dL. Kidney biopsy done four weeks after the onset of illness revealed patchy necrosis of glomeruli and proximal convoluted tubules predominantly in the cortex, with a clear zone of demarcation with medullary areas, focal interstitial inflammatory infiltrate, and interstitial edema, thereby suggesting acute cortical necrosis (see and ). A 99m Technitium DTPA scan done after 12 weeks of illness revealed a corrected total GFR of 35.2 mL/min, and a repeat ultrasonography of the kidneys revealed a distinct decrease in the length of both kidneys (right kidney 8.2 cm, left kidney 8.0 cm), with increased echogenicity.
Figure 1. Renal cortical necrosis. Kidney biopsy shows glomerular necrosis with a margin between viable and ischaemic renal tissue (arrow) (H&E, ×150).
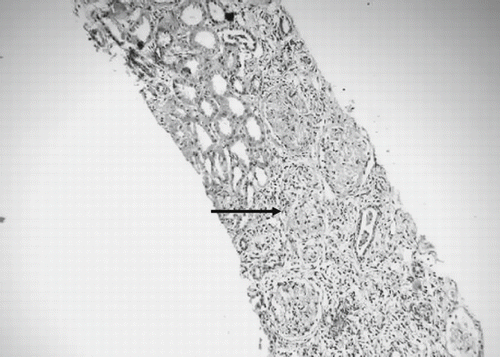
Figure 2. Renal cortical necrosis. Renal biopsy shows necrosis of two glomeruli (H&E, ×300).
DISCUSSION
The spectrum of renal involvement in Falciparum malaria varies from mild proteinuria accompanied by urinary sediment changes to acute renal failure. The incidence of ARF in patients with Falciparum malaria ranges from 1% to 4%.Citation[4] The incidence rises to as high as 60% in patients having heavy parasitemia (i.e., more than 10% P. Falciparum infested RBCs).Citation[5]
In malarial ARF, acute tubular necrosis (ATN) is the most consistent finding, though occasionally a mixture of ATN, interstitial nephritis, and glomerulonephritis has been described.Citation[6] Renal cortical necrosis is rarely reported.Citation[7],Citation[8] Over a 28-year period, 113 out of 2,986 patients (3.8%) dialyzed for acute renal failure at a referral center in North India was diagnosed to have acute renal cortical necrosis (ACN). Obstetric causes were responsible for ACN in 56.6% patients and non-obstetric causes in 43.4%. Within the obstetric group, ACN developed in association with complications of late pregnancy in 37.1% and following septic abortion in 19.5%. The various non-obstetric causes included viperine snake bite in 14.2%, hemolytic uremic syndrome in 11.5%, renal allograft rejection in 5.3%, acute gastroenteritis in 4.4%, acute pancreatitis in 3.5%, septicemia in 2.7%, and trauma- and drug-induced intravascular hemolysis in 0.9% patients.Citation[9] In a retrospective analysis of 1,822 patients presenting with acute renal failure in Eastern India over a period of twenty-two years, acute cortical necrosis was diagnosed in 57 (3.12%) of all cases; obstetric 32 (56.2%), and non-obstetric 25 (43.8%). The latter included extensive burns, sepsis, pancreatitis, snakebite, hemolytic-uremic syndrome, dehydration in children, and organophosphorus poisoning.Citation[10] Falciparum malaria as a cause of acute cortical necrosis was not reported.
Acute renal failure in Falciparum malaria is generally observed in patients with heavy parasitemia. The patho-physiologic alterations in Falciparum malaria contributing to acute renal failure are hypovolemia, intravascular hemolysis, catecholamine release, decreased deformability of RBCs, and disseminated intra-vascular coagulation. The asexual forms of P. Falciparum invade an erythrocyte, and this leads to alterations in the membrane of infected red blood cells with decreased deformability. Electron-dense protrusions or knobs are formed on the RBC surface that extrude an adhesive protein (PfEMP1), which mediates the attachment to receptors on venular and capillary endothelium.
At the same time, these infected red blood cells may adhere to other infected and non-infected red blood cells. This process of cytoadherence, rosette formation, and aggregation result in the sequestration of the red cells containing the parasites in vital organs, where they interfere with metabolism and microcirculation. This interference with microcirculation leads to ischemic in peritubular vessels in the kidney and acute tubular necrosis.Citation[11] Increased release of catecholamines, increased vascular response to catecholamines, and increased production of oxygen free radicals complement the activation, and hyperpyrexia also plays a role in renal injury. Hemolysis in P. Falciparum infection is often mild and usually extra vascular, except in patients with G6PD deficiency, when hemolysis can be severe.Citation[12] Other factors that contribute to intravascular hemolysis are changes in RBC membrane, immunologic injury, and interference with RBC ATP and ATPase.Citation[5] Hypovolemia is a common feature due to excessive insensible fluid loss, peripheral vasodilatation, and increased capillary permeability, leading to fluid leakage from the intravascular space.
Thus, prolonged renal hypoperfusion due to the interplay of red cell parasitization and monocyte activation, with the release of cytokines and other contributing factors outlined above, could be a crucial factor in the genesis of acute cortical necrosis in Falciparum malarial infection. Renal cortical necrosis may be focal and localized or it can be extensive. In most cases, the medulla, juxta medullary cortex, and a thin rim of subcapsular cortex are spared. The complete recovery of renal function following prolonged oliguria is rare.
In conclusion, Falciparum malaria is an uncommon cause of acute cortical necrosis. The pathogenesis is multifactorial. It should be suspected in patients with acute renal failure who have a prolonged phase of oligo anuria.
REFERENCES
- White NJ. Malaria. Manson's Tropical Disease20th, GC Cook. Saunders, London 1999; 1087–1164
- Boonpucknavig V, Sitprija V. Renal disease in acute plasmodium Falciparum infection in man. Kidney Int. 1979; 16: 44–52
- Sitprija V. Nephropathy in Falciparum malaria. Kidney Int. 1988; 34: 867–877
- Suvanapha R, Sitprija V, Van Y, Persele de Strihov C. Acute renal failure in the tropics and Hanta virus disease. Oxford Textbook of Clinical Nephrology, S Cameron, AM Davison, JP Grunfeld, D Kerr, E Rite. Oxford University Press, OxfordUK 1992; 1124–1146
- Rajapurkar MM. Renal involvement in malaria. J Postgrad Med. 1994; 40: 132–134
- Barsoum R. Malarial nephropathies. Nephrol Dial Transplant. 1998; 13: 1588–1597
- Chugh KS, Singhal PC, Kher V, et al. Spectrum of acute cortical necrosis in Indian patients. Am J Med. 1983; 286: 18–20
- Singhal MK, Arora P, Kher V, Pandey R, Gulati S, Gupta A. Acute cortical necrosis in Falciparum malaria. An unusual cause of end stage renal disease. Ren Fail. 1997; 19: 491–494
- Chugh KS, Jha V, Sakhuja V, Joshi K. Acute renal cortical necrosis—a study of 113 patients. Ren Fail. 1994; 16: 37–47
- Prakash J, Vohra R, Wani IA, et al. Decreasing incidence of renal cortical necrosis in patients with acute renal failure in developing countries: A single-centre experience of 22 years from Eastern India. Nephrol Dial Transplant. 2007; 22: 1213–1217
- White NJ, Breman JG. Malaria and babesiosis. Diseases caused by red blood cell parasites. Harrison's Principles of Internal Medicine16th, DL Kasper, E Braunwald, AS Fauci, LS Hauser, DL Longo, JL Jameson. McGraw Hill, New York 2005; 1218–1232
- Choudhary VP, Madan N, Sood SK. Intravascular hemolysis and renal insufficiency in children with glucose-6-phosphate dehydrogenase deficiency following anti-malarial therapy. Ind J Med Res. 1980; 71: 561–563