
Abstract
Hyperuricemia is a common symptom in adult population. It usually accompanies the chronic kidney disease. Less frequently, it is a primary phenomenon causing later serious clinical consequences. Familial juvenile hyperuricemic nephropathy (FJHN) is one of the hereditary conditions associated with high levels of serum uric acid and leading to dialysis in young adult age. It results from mutation in the UMOD gene, encoding the uromodulin protein, that is, Tamm–Horsfall protein. The aim of this paper was to present two families (7 affected members) with FJHN, in whom standard nephrological diagnostics did not provide clear cause of dialysis-dependent chronic kidney disease, until genetic testing was performed.
Introduction
Hyperuricemia is often detected in chronic kidney disease (CKD) patients. The excess of uric acid (UA) in the body may result from both primary (enzymatic defects) and secondary causes (e.g., decreased renal excretion).Citation1
The excessive amount of UA leads to the transformation of renal tubule cells into fibroblasts (renal fibrosis), stimulates the inflammatory processes, activates the renin–angiotensin–aldosterone system, damages and causes the dysfunction of endothelial cells (↓NO), which in turn contributes to eGFR reduction.Citation2 The risk of CKD progression is higher in subjects with elevated UA.Citation3,Citation4
The familial juvenile hyperuricemic nephropathy (FJHN) is a rare genetic condition with autosomal dominant transmission. It is one of the disorders defined under a common term of familial urate-associated nephropathies (FUAN).Citation5 The mutation in the UMOD gene, encoding the uromodulin protein, that is, Tamm–Horsfall protein, represents the pathogenetical background of FJHN.Citation6 The resulting dysfunction of renal tubules leads to hyperuricemia with decreased UA renal excretion and accumulation of its crystals in the interstitial tissue. The symptoms of progressive renal function impairment become apparent in the adolescence, while the end-stage renal disease (ESRD) develops at the age of 40–70 years old.Citation7
The aim of this paper is to present two families with FJHN (11 p.), in whom initial nephrological diagnostic did not provide clear evidence to the cause of CKD until genetic testing was performed.
Case reports
The family history () revealed that the already deceased man (SJ) of the first generation suffered from ESRD and was hemodialyzed. Four of his children from two marriages also suffer from CKD.
Figure 1. The family pedigrees of both families’ male members are indicated with rectangles. CKD = blue. V = confirmed UMOD mutation. ? = genetic testing was not performed. Crossed = deceased. Plain rectangle or ellipse = genetic testing performed, no mutation found.
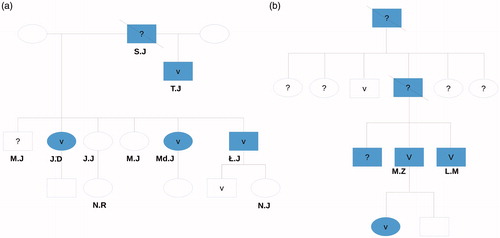
His son (TJ) was first diagnosed with abnormal renal parameters at the age of 24 years. Not later than a few months after this discovery, he was hemodialyzed due to the ESRD in the course of chronic pyelonephritis and gout.
Three out of his six offspring (two women JD and MdJ, as well as one man ŁJ) from his second marriage suffer from chronic kidney disease, as well. JD was diagnosed with CKD at the age of 27 years. She had not been treated for any chronic disease before. On admission, she reported with joint pain. The laboratory findings revealed creatinine and urea level of 7,36 mg/dL and 172 mg/dL, respectively. The blood concentration of UA was elevated as well. The urinalysis showed quite few epithelial cells and neither glucosuria nor proteinuria. The abdominal ultrasound revealed smaller than normal kidneys. The G4 of CKD was diagnosed (eGFR 15 mL/min), and after 9 months she was placed on chronic hemodialysis due to the ESRD resulting probably from glomerulonephritis.
Her sister (MdJ) was diagnosed with G2 of CKD (eGFR 60 mL/min/1.73 m2) at the age of 16 years. Until the diagnosis was established, she was perfectly healthy. The laboratory findings revealed increased blood concentration of UA, with its decreased fractional excretion (UAFE), while no abnormalities in the urinalysis were found. The abdominal ultrasound showed smaller than normal kidneys. The kidney biopsy indicated neither glomerular process nor active interstitial changes.
Increased creatinine (2,5 mg/dL) and UA (decreased UAFE) levels were first observed in their brother (ŁJ) at the age of 21 years (no chronic diseases in his medical record). The kidneys observed in the abdominal ultrasound were smaller than average. The microscopic examination of renal bioptates suggested the presence of chronic, interstitial, and low-active changes. The entire clinical presentation leads to the diagnosis of chronic tubulointerstitial nephritis while UA nephropathy was suspected (G3 of CKD).
The blood concentration of UA was normal in another two sisters, who do not suffer from CKD, yet the UAFE was reduced. Similar results were observed in each child of the mentioned siblings, excluding 3-year-old KJ. His UA concentration was raised (9.6 mg/dL, reference value of 5.0 mg/dL), which distinguishes him from other healthy members of this family.
Eleven members of the first family were genetically tested for uromodulin gene mutation (March 2015). The ADTKD-UMOD OMIM 162000 mutation was discovered in 5 of them (sequence variant c (0).2297 > C in exon 4, leading to Cys77Arg exchange). The complete data set is presented in .
Table 1. Data set for the first family (J).
The clinical course in the second affected family was similar (). The 16-year-old female patient (KM) was admitted to the clinic due to the high levels of creatinine and UA. She complained of knee and hip pain, which have lasted for a year preceding admission. Due to the positive family history, she had her renal parameters and UA level controlled every year, which remained normal till the above-mentioned hospital admission. The UA-associated nephropathy was diagnosed (G3a of CKD). The kidney biopsy was not performed as genetic testing was available.
The father of the patient (MZ) suffered from gout. As ESRD developed, he was switched to hemodialysis therapy at the age of 37 years. The siblings of the patient’s father suffer from CKD, as well. The UMOD gene mutation was confirmed in one of the father’s brothers. In his case, the renal disease was diagnosed at the age of 26 years (currently G4 stadium). The same disease was diagnosed in his second brother, yet neither he nor their sister was genetically tested. Detailed medical history revealed that KM’s grandfather was hemodialyzed due to the ESRD and died at the age of 45 years. His two sisters were diagnosed with gout and underwent kidney transplantation due to the CKD.
The presence of ADTKD-UMOD OMIM 162000 (c.916T > A, p.Cys306Ser) mutation was confirmed in 3 affected members of this family (the other patient did not give consent for testing).
Discussion
The genetic testing allowed diagnosing FJHN in 7 members of both families and raising a suspicion of such disorder in one, in whom clinical symptoms could not have developed due to young age. The clinical course of FJHN in both families corresponds with descriptions available in the literature, which emphasize the importance of acquiring family history.
Demet et al. presented the case of FJHN in 14-year-old patient (eGFR 30 mL/min/1.73 m2). The family history was of significant importance as well. The CKD was diagnosed in the girl’s grandmother (at the age of 34 years), her mother (29 years) and her uncle (24 years). All of them were hemodialyzed. The girl’s UAFE was reduced and the blood concentration of UA was increased (characteristic laboratory finding in this group of patients). The kidney biopsy revealed the chronic interstitial nephritis, as in the patient described in our cohort.Citation7
Dong Hun presented the first Korean patient with genetically confirmed FJHN. The 16-year-old patient was diagnosed with gout and renal function impairment, whereas urinalysis remained normal, and fractional excretion of UA was reduced. The kidney biopsy was not performed in this patient. The brother, father, grandfather, and the uncle of the patient had also suffered from CKD and gout. The genetic testing was performed in patient and his father revealing the presence of UMOD gene mutation in both of them.Citation8
Moskowitz et al. analyzed 31 papers published since October 2011 concerning the UMOD gene mutation. They presented 202 patients, members of 74 families from America, Asia, and Europe, with 59 different UMOD mutations. They calculated the mean age of hyperuricemia, gout, and ESRD to be 24, 40, and 56 years old, respectively. They also proved that the type of mutation influences the age at which the disease reaches the end-stage stadium and demonstrated the association between the gender and the first symptoms of gout and ESRD (earlier in men).Citation9 Our cohort of patients entered the CKD G5 earlier than those described by Moskowitz.
The Austrian researchers (5 families with UMOD gene mutation) calculated that such diseases concern 1.7 persons per million inhabitants, yet in dialyzed cohort this rate is lower than 1 per 1000.Citation10
Although congenital tubulopathies in adults are very rare, the above-mentioned cases clearly demonstrate that their impact on families of hemodialyzed patients should not be underestimated. The presented patients were diagnosed with different clinical entities (i.e., chronic pyelonephritis, glomerulonephritis, uremic nephropathy, or idiopathic) and the ultimate diagnosis was established after many years. Therefore, the disorder of FUAN group should be considered in young CKD patients, with strongly positive family history for CKD (especially ESRD), with gout or increased UA concentration.
Currently, the FJHN is a not curable disease, yet attempts can be made to slow down the progression of the disease (by reducing the blood level of UA usingCitation11–13) and to select patients at risk of CKD in the future as a part of appropriate genetic counseling.
Disclosure statement
The authors report no conflicts of interest. The authors alone are responsible for the content and writing of this article.
References
- Zimmermann-Górska I. [Choroby związane z obecnością kryształów]. In: Gajewski P, et al. eds. [Interna Szczeklika], 2014, 1st ed. Warsaw, Poland: Medycyna Praktyczna; 2014:1929–1933.
- Ryu ES, Kim MJ, Shin HS, et al. Uric acid-induced phenotypic transition of renal tubular cells as a novel mechanism of chronic kidney disease. Am J Physiol Renal Physiol. 2013;304:F471–F480.
- Iseki K, Ikemiya Y, Inoue T, Iseki C, Kinjo K, Takishita S. Significance of hyperuricemia as a risk factor for developing ESRD in a screened cohort. Am J Kidney Dis. 2004;44:642–650.
- Kanbay M, Yilmaz MI, Sonmez A, et al. Serum uric acid independently predicts cardiovascular events in advanced nephropathy. Am J Nephrol. 2012;36:324–331.
- Internet site: http://rarerenal.org/clinician-information/adtkd-clinician-information/
- Raciborski F, ed. [Występowanie i leczenie dny moczanowej w Polsce. Analiza, wskazania, rekomendacje]. Raport Instytutu Ochrony Zdrowia. Instytut Ochrony Zdrowia, 2015.
- Alaygut D, Torun-Bayram M, Soylu A, Kasap B, Türkmen M, Kavukçu S. Chronic kidney disease in an adolescent with hyperuricemia: Familial juvenile hyperuricemic nephropathy. Turk J Pediatr. 2013;55:637–640.
- Lee DH, Kim JK, Oh SE, Noh JW, Lee YK. A case of familial juvenile hyperuricemic nephropathy with novel uromodulin gene mutation, a novel heterozygous missense mutation in Korea. J Korean Med Sci. 2010;25:1680–1682.
- Moskowitz JL, Piret SE, Lhotta K, et al. Association between genotype and phenotype in uromodulin-associated kidney disease. Clin J Am Soc Nephrol. 2013;8:1349–1357.
- Lhotta K, Piret SE, Kramar R, Thakker RV, Sunder-Plassmann G, Kotanko P. Epidemiology of uromodulin-associated kidney disease – results from a nation-wide survey. Nephron Extra. 2012;2:147–158.
- Levy GD, Rashid N, Niu F, Cheetham TC. Effect of urate-lowering therapies on renal disease progression in patients with hyperuricemia. J Rheumatol. 2014;41:955–962.
- Fairbanks LD, Cameron JS, Venkat-Raman G, et al. Early treatment with allopurinol in familial juvenile hyerpuricaemic nephropathy (FJHN) ameliorates the long-term progression of renal disease. QJM. 2002;95:597–607.
- Cameron JS, Moro F, Simmonds HA. Gout, uric acid and purine metabolism in paediatric nephrology. Pediatr Nephrol. 1993;7:105–118.