
Abstract
Background
Fabry disease (FD) is an X-linked lysosomal storage disorder caused by the mutation of the GLA gene, encoding the α-galactosidase, which is responsible for the catabolism of neutral glycosphingolipids. Microalbuminuria or low-grade proteinuria, and continuously progressive renal failure are common manifestations in FD males. However, sudden onset of nephrotic syndrome in FD, is rarely reported.
Case report
A 32-year-old Chinese man was admitted to our hospital because of sudden onset of generalized edema due to nephrotic syndrome. He denied hypohidrosis, nocturia, and any history of episodic hand or foot pain. A few scattered angiokeratoma can be found on the low back skin on examination. Except for the similar locating pattern of angiokeratoma, no evident abnormality was found in the laboratory work up and physical examination of his younger brother. The patient was diagnosed with FD companying with minimal change disease by renal biopsy. Genetic analysis on our patient and his sibling revealed a nonsense GLA gene variant (c.707G > A, p.Trp236*), which has been previously reported in FD. Immunotherapy alone (steroids and tacrolimus), but without enzyme replacement therapy, much improved the massive proteinuria. Follow up to date, his 24-h urine protein is stable at about 0.5 g, and renal function keeps normal.
Conclusion
Sudden onset of nephrotic syndrome, although rare, may occur in FD, even as the primary renal manifestation, but this usually suggests additional renal disease. Immunosuppressive treatment should be considered in such FD patient companying with nephrotic syndrome.
Introduction
Fabry disease (FD, OMIM: 301500) is a multisystem, X-linked inherited, lysosomal storage disorder caused by the deficient activity of the lysosomal enzyme alpha-galactosidase A (α-Gal A) [Citation1]. Deficient enzymatic activity of α-Gal A induces the progressive glycosphingolipids deposition in intra-lysosomal of the vascular endotheliocytes, cardiomyocytes, a range of renal cell types, eyes, peripheral nerves, and brain, eventually leading to diverse clinical manifestations, including acroparesthesia, pain, angiokeratoma, hypohidrosis, and progressive vascular disease of the kidney, heart, and central nervous system [Citation2,Citation3].
It has been estimated that the incidence of FD ranges between 1/40,000 and 1/117,000 in male [Citation4], however, neonatal screening has suggested a higher incidence from 1/4100 to 1/3100 [Citation5,Citation6]. FD has a wide spectrum of phenotypes and severities, and the clinical manifestations are depended on factors like genotype, the level of enzyme activity, gender, age and still as yet unknown genetic modifiers. FD patients with classic form occur mainly in male hemizygotes, presenting with pain crises, acroparesthesia, angiokeratoma, hypohidrosis, and gastrointestinal symptoms in children or adolescence, and develop cardiac, renal and cerebrovascular involvement in adulthood, but some male FD patients are characterized by late onset form that exhibit relatively milder symptoms limited to the kidney and/or heart in adulthood without any manifestations in children [Citation7,Citation8].
Whereas, heterozygous females mainly manifest late onset Fabry disease, usually suffering from a milder form of the disease, or they may be asymptomatic, but they can be affected as severely as the hemizygous males occasionally [Citation9]. According to Pinto LLC et al., the main differences in clinical expression of heterozygous females, could be explained by the mechanisms of the skewed X-inactivation and cross-correction or cross-inducing mechanisms [Citation10]. And there is a progression to renal and cardiovascular disease after adulthood, which may lead, at the fourth or fifth decade for males and the seventh for females, to premature death [Citation11].
Although microalbuminuria or low-grade proteinuria, and progression to end-stage renal failure are common renal dysfunctions in FD males, FD patients presenting with nephrotic syndrome are unusual. Herein, we report a patient initially presenting with sudden onset of nephrotic syndrome (NS), the following renal biopsy and pathologic examination, as well as GLA gene sequencing analysis confirmed his diagnosis of FD unexpectedly.
Case report
The pedigree of the family is shown in . The proband (IIb) was a 32-year-old man, who was admitted to our hospital because of generalized edema for 1 month. He denied other any abnormal symptoms, such as hypohidrosis, nocturia, and any history of acroanesthesia, episodic hand or foot pain. Physical examination revealed his blood pressure of 145/96 mmHg, a few scattered angiokeratoma on his low back skin (), and edema in his both lower extremities and eyelid. shows the proband’s laboratory data. In the laboratory work-up, repeat urinalysis demonstrated 3+ protein. The urinary total protein excretion was 6.38 g/d and the serum albumin level was 28 g/L. The concentrations of serum urea and creatinine were 6.21 mmol/L and 95 μmol/L (CKD-EPI estimated GFR = 90.9 mL/min/1.73 m2), respectively. The electrocardiogram showed sinus tachycardia with high P waves in II, III, and aVF leads, and left ventricular high voltage. Ultrasonography of his both kidneys manifested elevated echogenicity of parenchyma without obvious kidney shrinkage. Examination of transthoracic echocardiography, audiometry, ophthalmology, and brain MRI were normal. Therefore, he was then diagnosed as NS. Immediately after, oral steroid methylprednisolone was initiated at a dose of 32 mg/day, and immunosuppressant tacrolimus was prescribed at a single dose of 1.5 mg, twice per day. In addition, angiotensin II receptor blocker valsartan was used to reduce proteinuria and blood pressure.
Figure 1. Pedigree of the kindred with Fabry disease. Y: years old.
Figure 2. Skin injury. (A) Skin angiokeratoma (arrow) on the back of the proband; (B) Skin angiokeratoma (arrow) on the lower abdomen of the proband’s younger brother.
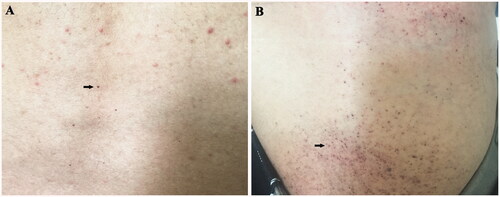
Table 1. The laboratory results of the proband.
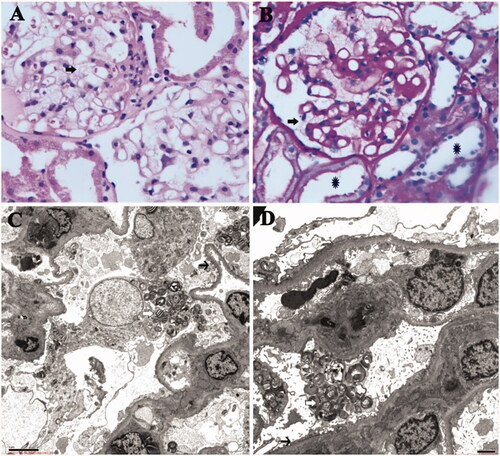
Subsequently, renal biopsy and pathologic examination were performed. Light microscopy showed that vacuolization was frequently observed in podocyte cytoplasm (), along with mild chronic tubulointerstitial lesions and mild acute lesions. Findings with immunofluorescence staining were unremarkable, although focal mesangial deposition of C3, immunoglobulin (Ig) M, Ig A in a nonspecific fashion were found. Electron microscopy demonstrated that deposits appear as typical osmiophilic inclusion bodies with a characteristic onion-skin or ‘zebra’ appearance, characterized by concentric lamellation of clear and dark layers mainly within lysosomes of podocytes (). In addition, widespread foot process fusion was also observed in the podocytes. Thus, the renal pathological findings of the proband well corresponded with renal FD concurrent with minimal change disease (MCD).
Figure 3. (A) (HE 400×) and (B) (PAS 400×): Light microscopy showed normal glomerular volume, well-opened capillary loops, and mild segmental mesangial widening with increasing mesangial matrix. Swollen podocytes with abundant, foamy, clear cytoplasm were observed (arrow); mild chronic tubulointerstitial lesions and mild acute lesions were presented, along with flattened tubular epithelial cell and brush border loss (asterisk). (C and D) By electron microscopy, glomerular capillary loops were well-opened and basement membranes were of normal thickness. Extensive foot process fusion (black arrow) with microvillus transformation was observed in the podocyte. And osmiophilic lamellar inclusions were abundantly presented in the podocyte cytoplasm, some of which showed appearance of zebra body (white arrow).
To further clarify the diagnosis of this family, genomic DNA was extracted from the peripheral blood of the proband and his family members by GenElute blood genomic DNA kit (Sigma, NA2010). By next generation sequencing, a previously reported nonsense variant c.707G > A (p.Trp236*) in GLA gene was identified in the proband. Sanger sequencing validation of all family members revealed that the proband’s mother (Ia, 56 years old), younger brother (IIc, 28 years old), and daughter (IIIa, 5 years old) carried the same heterozygous variant in GLA gene, whereas other family members did not harbor this variant. Meanwhile, a heterozygous variant c.988A > G (p.Asn329Ser) in PKD1 gene associated with polycystic kidney was only found in the proband, but not other family members. Soon afterwards, the peripheral white blood cells of this family members were collected for testing activity of α-Gal A, and its activity level was found to be 0.31 nmol/h/mg for the proband (normal range: 31.30–51.00 nmol/h/mg), 41.0 for his mother, 45.2 for his father, 0.45 for his younger brother, 32.1 for his daughter, and 40.2 for his son.
On the 8th week of immunotherapy, the dose of methylprednisolone for the proband was reduced to 24 mg/day, and his 24-h proteinuria quantitation has dramatically decreased to about 1.0 g. Tacrolimus was discontinued after 3 months. Over the next 6 months, methylprednisolone was slowly tapered to 8 mg/day, and then maintained at a dose of 4 mg/day for more than 1 year. Follow up to date, the patient’s proteinuria has been stabilized at around 0.5 g/d, and his serum albumin has restored to 44 g/L, and no other organ involvement has been found. Enzyme replacement therapy (ERT) was not administered due to his financial constraints.
Besides, his younger brother (IIc), also had the presentation of angiokeratoma on his lower abdomen (). Except from this, no obvious abnormality was found in the laboratory work up, physical examination and ophthalmologic examination. And he refused further imaging examination because he lacked any discomfort. No anomalies have been found so far in other remaining family members including his mother and daughter.
Discussion
FD was first described independently by two dermatologists, Johannes Fabry and William Anderson in 1898, was also named Andeson–Fabry Disease [Citation12,Citation13]. It is the second most common lysosomal storage disorder after Gaucher disease [Citation14]. FD is caused by the deficient activity of α-Gal A, resulting from inherited or de novo pathogenic variants in the GLA gene [Citation15]. α-Gal A is a dimeric glycoprotein that catalyzes the removal of terminal nonreducing α-D-galactose residues in ceramide trihexoside [Citation3,Citation15]. Impairment of α-Gal A activity may result in obstacle of the glycolipid conversion from globotriosylceramide (GL-3) to lactosylceramide (GL-2), leading to widespread intralysosomal accumulation of glycolipid in different tissue and organs [Citation16].
Kidney is the most common organ involved in FD, and the older the patient, the more severe the kidney damage [Citation17]. Microalbuminuria and proteinuria, which is long considered the first clinical manifestation of Fabry nephropathy, usually occur at 20–30 years of age. With the progression of proteinuria, especially the level of more than 1 g per 24 h, is a powerful predictor of progression to end-stage renal disease in FD. Then, chronic kidney disease is usually found at the ages of 30–50 years in all hemizygous males and in some heterozygous females, and renal dysfunction with progression to end-stage renal disease is commonly manifested in 50% of FD males [Citation2,Citation8,Citation17]. Research by Linthorst reported that among patients who underwent renal replacement therapy, 0.33% of male patients and 0.10% of females had FD [Citation18]; and the prevalence of FD in chronic kidney disease patients who were not on dialysis was found to be 0.2% by Yeniçerioğlu (0.4% in male, 0.0% in female) [Citation19]. Although FD can coexist with several renal diseases, including IgA nephropathy, lupus nephritis, membranous GN, and crescentic nephritis, nephrotic syndrome as the first clinical clue of Fabry-related kidney damage remains rare, with only nine cases reported (shown in ), of which five were male and four were female [Citation20–27]. Zarate et al. reported a 16-year-old male patient with minimal change nephrotic syndrome and underlying FD in 2010, who was responded well to steroid and ERT treatment, eventually his proteinuria became undetectable [Citation26]. Similarly, Fujisawa et al. reported a 67-year-old male patient with late onset FD who also suffered from nephrotic syndrome probably due to FSGS or MCD, he finally achieved complete remission after immunosuppressive (steroids and cyclosporine A) therapy and ERT [Citation20]. Although it is not fully understood why FD patients are prone to secondary nephrotic syndrome, there is convincing evidence that FD may contribute in some way to cause progressive podocyte damage with foot process effacement and podocyte loss, that ultimately leads to proteinuria and even glomerulosclerosis, which may be related to the persistent accumulation of toxic glycosphingolipid in the podocytes, specific or unspecific signal pathway (e.g. ERK, JNK, and SAPK), and local inflammatory response [Citation28]. Renal pathology of our patient revealed that MCD was the major histological finding which were associated with NS, and massive proteinuria responded well to immunotherapy. Although FD itself has podocyte damage, which may significantly increase the susceptibility to MCD, we could not completely exclude the possibility that FD coexists with MCD occasionally in a patient, because either diseases are related to various etiologies. Although the proband did not achieve complete recovery from NS, the application of immunotherapy (steroids and tacrolimus), but not ERT, significantly improved his massive proteinuria, whose proteinuria stabilized at about 0.5 g/d, suggesting that the use of immunotherapy in FD patients who are complicated with NS and other kidney disease could be beneficial, due to the fact that it significantly lowered the level proteinuria which is an important factor for progression of renal disease in FD. In the future, careful monitoring of the patient is warranted and ERT therapy should be initiated if possible.
Table 2. Laboratory and clinical data of nine cases with NS in FD patients.
In this study, the variant c.707G > A identified in four family members, usually is presumed to cause the translation process to terminate prematurely (p.Trp236*), which may generate a truncated nonfunctional protein. In addition, this variant might also cause the disease by the nonsense mediated mRNA delay or abnormal splicing. This variant was first reported by Germain DP in a 32-year-old British FD patient who presented with classic phenotype in 2002 [Citation29]. Intriguingly, another missense variant c.707G > T (p.Trp236Leu) involving the same nucleotide and resulting in Tryptophan substituted by Leucine in position 236 was reported in a classic FD patient by Topaloglu in 1999 [Citation30]. However, it is a pity that the detailed clinical phenotypes of these patients were not mentioned in the article. Therefore, the description of the patients in this family will provide a certain reference for the study of the relationship between genotypes and phenotypes of the disease.
Various studies have shown that correlation of genotype–phenotype is poor in FD patients carrying different genotypes of variants [Citation2]; even the same variant in one family can bring about different phenotypes, including the age of onset, rate of progression, organ manifestations, and disease severity. This presumably depends on genetic (e.g., modifier genes) or non-genetic factors (e.g., environment and diet) in addition to the nature of the variant [Citation31]. In the present family, the manifestation of the proband initiated as NS, but his younger brother, despite harboring the same variant, lacked a renal phenotype. Such discrepancies might indicate a phenotype variability between these two siblings. However, considering NS as a phenotype of FD is farfetched in this proband, as we mentioned above. If regardless of NS in this proband, it is difficult to distinguish the phenotype difference between the proband and his younger brother only considering their lower level of α-Gal A activity. Although renal biopsy of his younger brother is not available, close follow-up should be taken because his renal abnormalities especially his albuminuria may occur with the progression of disease, and renal biopsy should be performed if necessary. Besides, the proband’s heterozygous mother and daughter have been presented asymptomatic to date, consistent with previous reports in which heterozygote females are generally asymptomatic or present with intermittent and mild symptoms of the disease. However, as clinical symptoms in females could develop later in life, they should be regularly monitored as well.
Conclusion
Sudden onset of NS may occur in FD and even be the first clinical manifestation, but this usually indicates a presence of a second renal disease. Appropriate diagnostic approach, often including renal biopsy is warranted in those case. Use of immunotherapy is not indicated for FD, but for secondary renal disease, and could be beneficial also for FD due to impact on lowering proteinuria. Regular monitoring of a patient and his affected relatives is important and introduction of disease specific therapy in all clinically affected as soon as possible in order to avoid additional kidney injury.
Author contributions
L.S. conceived and designed the study. R.Z and Z.C drafted the manuscript. Y.L, Q.Y and Y.S contributed to modification of the manuscript. S.S and Y.C undertook the analysis and interpretation of the data. S.W, X.S, Z.L, W.G and Y.H collected the clinical and follow-up data. All authors have approved the final version of the manuscript.
Acknowledgments
We are grateful to all subjects for their participation.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Yalın SF, Eren N, Sinangil A, et al. Fabry disease prevalence in renal replacement therapy in Turkey. Nephron. 2019;142(1):26–33.
- Sessa A, Meroni M, Battini G, et al. Chronic renal failure, dialysis, and renal transplantation in Anderson-Fabry disease. Semin Nephrol. 2004;24(5):532–536.
- Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5(1):30.
- Zarate YA, Hopkin RJ. Fabry’s disease. Lancet. 2008;372(9647):1427–1435.
- Mechtler TP, Stary S, Metz TF, et al. Neonatal screening for lysosomal storage disorders: feasibility and incidence from a nationwide study in Austria. Lancet. 2012;379(9813):335–341.
- Spada M, Pagliardini S, Yasuda M, et al. High incidence of later-onset Fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79(1):31–40.
- Mehta A, Ricci R, Widmer U, et al. Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry outcome survey. Eur J Clin Invest. 2004;34(3):236–242.
- Arends M, Wanner C, Hughes D, et al. Characterization of classical and nonclassical Fabry disease: a multicenter study. JASN. 2017;28(5):1631–1641.
- Whybra C, Kampmann C, Willers I, et al. Anderson-Fabry disease: clinical manifestations of disease in female heterozygotes. J Inherit Metab Dis. 2001;24(7):715–724.
- Pinto LLC, Vieira TA, Giugliani R, et al. Expression of the disease on female carriers of X-linked lysosomal disorders: a brief review. Orphanet J Rare Dis. 2010;5:14–14.
- Hsu T-R, Chang F-P, Chu T-H, et al. Correlations between endomyocardial biopsies and cardiac manifestations in Taiwanese patients with the Chinese hotspot IVS4 + 919G > A mutation: data from the Fabry outcome survey. IJMS. 2017;18(1):119.
- Fabry J. Ein Beitrag zur Kenntniss der Purpura haemorrhagica nodularis (Purpura papulosa haemorrhagica Hebrae. Arch f Dermat. 1898;43(1):187–200.
- Garzuly F, Maródi L, Erdös M, et al. Megadolichobasilar anomaly with thrombosis in a family with Fabry’s disease and a novel mutation in the alpha-galactosidase A gene. Brain. 2005;128(Pt 9):2078–2083.
- Tuttolomondo A, Pecoraro R, Simonetta I, et al. Anderson-Fabry disease: a multiorgan disease. Curr Pharm Des. 2013;19(33):5974–5996.
- Ishii S, Chang H-H, Kawasaki K, et al. Mutant alpha-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin. Biochem J. 2007;406(2):285–295.
- Oliveira JP, Ferreira S. Multiple phenotypic domains of Fabry disease and their relevance for establishing genotype-phenotype correlations. Appl Clin Genet. 2019;12:35–50.
- Branton MH, Schiffmann R, Sabnis SG, et al. Natural history of Fabry renal disease: influence of alpha-galactosidase A activity and genetic mutations on clinical course. Medicine (Baltimore). 2002;81(2):122–138.
- Linthorst GE, Bouwman MG, Wijburg FA, et al. Screening for Fabry disease in high-risk populations: a systematic review. J Med Genet. 2010;47(4):217–222.
- Yeniçerioğlu Y, Akdam H, Dursun B, et al. Screening Fabry’s disease in chronic kidney disease patients not on dialysis: a multicenter study. Ren Fail. 2017;39(1):104–111.
- Fujisawa H, Nakayama Y, Nakao S, et al. Effectiveness of immunosuppressive therapy for nephrotic syndrome in a patient with late-onset Fabry disease: a case report and literature review. BMC Nephrol. 2019;20(1):469–469.
- Reyes Marín FA, Gómez Navarro B, Tamayo y Orozco J, et al. Nephropathy in a case of Fabry’s disease. Revista de investigacion clinica. Organo Del Hospital de Enfermedades de la Nutricion. 1991;43(4):373–376.
- Majima K, Ishizaki T, Inoue T, et al. A case of Fabry’s disease associated with lupus nephritis. Nihon Jinzo Gakkai Shi. 1992;34(11):1189–1194.
- Thamboo TP, Sivaraman P, Chan NH-L. Pathologic quiz case: an unsuspected cause of nephrotic syndrome. Heterozygous Fabry disease. Arch Pathol Lab Med. 2004;128(5):593–594.
- Inagaki S-i, Migita M, Hayakawa M, et al. An asymptomatic heterozygous female with Fabry disease: implications for enzyme replacement therapy. J Nippon Med Sch. 2005;72(6):387–390.
- Fischer EG, Moore MJ, Lager DJ. Fabry disease: a morphologic study of 11 cases. Mod Pathol. 2006;19(10):1295–1301.
- Zarate YA, Patterson L, Yin H, et al. A case of minimal change disease in a Fabry patient. Pediatr Nephrol. 2010;25(3):553–556.
- Zhou W, Ni Z, Zhang M. Hemizygous Fabry disease associated with membranous nephropathy: a rare case report. Clin Nephrol. 2018;90(3):227–231.
- Shankland SJ. The podocyte’s response to injury: role in proteinuria and glomerulosclerosis. Kidney Int. 2006;69(12):2131–2147.
- Germain DP, Shabbeer J, Cotigny S, et al. Fabry disease: twenty novel α-galactosidase A mutations and genotype-phenotype correlations in classical and variant phenotypes. Mol Med. 2002;8(6):306–312.
- Topaloglu AK, Ashley GA, Tong B, et al. Twenty novel mutations in the α-galactosidase A gene causing Fabry disease. Mol Med. 1999;5(12):806–811.
- Wang C, Wang Y, Zhu F, et al. A missense mutation of the α-galactosidase A gene in a Chinese family of Fabry Disease with renal failure. Kidney Blood Press Res. 2013;37(4–5):221–228.