
Abstract
The two primary mechanisms by which iodinated contrast media (CM) causes contrast-induced acute kidney injury (CIAKI) are the hemodynamic effect causing intrarenal vasoconstriction and the tubular toxic effect causing acute tubular necrosis. Inhibition of 15-hydroxyprostaglandin dehydrogenase (15-PGDH), which degrades prostaglandin E2 (PGE2), promotes tissue repair and regeneration in many organs. PGE2 causes intrarenal arterial vasodilation. In this study, we investigated whether a 15-PGDH inhibitor can act as a candidate for blocking these two major mechanisms of CIAKI. We established a CIAKI mouse model by injecting a 10 gram of iodine per body weight (gI/kg) dose of iodixanol into each mouse tail vein. A 15-PGDH inhibitor (SW033291), PGE1, or PGE2 were administered to compare the renal functional parameters, histologic injury, vasoconstriction, and renal blood flow changes. In addition, human renal proximal tubular epithelial cells were cultured in a CM-treated medium. SW033291, PGE1, or PGE2 were added to compare any changes in cell viability and apoptosis rate. CIAKI mice that received SW033291 had lower serum levels of creatinine, neutrophil gelatinase-associated lipocalin, and kidney injury molecule 1 (p < 0.001); lower histologic injury score and TUNEL positive rates (p < 0.001); and higher medullary arteriolar area (p < 0.05) and renal blood flow (p < 0.001) than CM + vehicle group. In cell culture experiments, Adding SW033291 increased the viability rate (p < 0.05) and decreased the apoptosis rate of the tubular epithelial cells (p < 0.001). This 15-PGDH inhibitor blocks the two primary mechanisms of CIAKI, intrarenal vasoconstriction and tubular cell toxicity, and thus has the potential to be a novel prophylaxis for CIAKI. Abbreviations: 15-PGDH: 15-hydroxyprostaglandin dehydrogenase; AMP: adenosine monophosphate; CIAKI: contrast-induced acute kidney injury; CM: contrast media; EP: prostaglandin E2 receptor; hRPTECs: human-derived renal proximal tubule epithelial cells; KIM-1: kidney injury molecule-1; MTT: 3-(4,5-Dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide; NGAL: neutrophil gelatinase-associated lipocalin; PBS: phosphate-buffered saline; PGE1: prostaglandin E1; PGE2: prostaglandin E2; RBF: renal blood flow; TUNEL: terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling; α-SMA: α-Smooth muscle actin
Introduction
Contrast-induced acute kidney injury (CIAKI) is defined as a decrease in renal function (increase in serum creatinine ≥0.3 mg/dl or ≥1.5–1.9 times baseline) that is not explained by other causes, occurring 48–72 h after administration of iodinated contrast media (CM) according to American College of Radiology and the European Society of Urogenital Radiology [Citation1–3]. Although the incidence of CIAKI is as low as 0.6–2.3% in the general population when the CM is administered intravenously, the frequency of CIAKI is reported as high as 14.5% with intraarterial CM, and up to 50% in high-risk groups, such as elderly patients with diabetes, heart failure, or underlying kidney disease [Citation1,Citation4–7]. The typical clinical course of CIAKI is that the serum level of creatinine peaks 3–5 days after CM administration and then recovers to, or near to, baseline after 1–3 weeks [Citation3]. However, CIAKI increases socioeconomic costs by inducing dialysis, prolonging hospital stays, and increasing the in-hospital mortality rate and long-term mortality [Citation8,Citation9]. Therefore, it is particularly important to devise a way to prevent CIAKI.
Multiple substances, including sodium bicarbonate, N-acetylcysteine, statins, ascorbic acid, and phosphodiesterase-5 inhibitors, have been suggested as candidates for CIAKI prevention [Citation2,Citation10–13]. Recently, prostaglandin E1 (PGE1) can act as a vasodilator and prevent CIAKI by alleviating vasoconstriction, part of the pathophysiology of that condition [Citation14].
Nonetheless, the most effective means to prevent CIAKI to date is isotonic saline hydration. The guidelines of the American College of Radiology guidelines, the European Society of Urogenital Radiology, and the European Society of Cardiology/European Association for Cardio-Thoracic Surgery recommend isotonic saline hydration as a standard preventive method for CIAKI [Citation2,Citation13,Citation15]. In spite of these guidelines, CIAKI has yet to be prevented effectively. Therefore, it is urgent to develop a new prophylactic drug for CIAKI.
The two primary mechanisms in the pathophysiology of CIAKI are the hemodynamic effect that causes medullary hypoxia through vasoconstriction and the tubular toxic effect that causes apoptosis and necrosis of tubular cells [Citation16]. If there is a substance that can block both of these, it could be a novel and outstanding candidate for preventing CIAKI. Zhang et al. reported resistance of colonic epithelial cells to dextran sulfate sodium, a harmful substance, through 15-hydroxyprostaglandin dehydrogenase (15-PGDH) inhibition, and found that it can promote tissue regeneration in various organs such as bone marrow and liver [Citation17]. 15-PGDH is an enzyme that rapidly degrades the PGE2 produced by cyclooxygenase from arachidonic acid. Therefore, PGE2 levels are increased when 15-PGDH is inhibited. PGE2 acts in the kidney to cause relaxation of the intrarenal artery [Citation18]. Thus, we investigated whether a 15-PGDH inhibitor can protect the kidney from CM by blocking intrarenal vasoconstriction and direct tubular toxicity simultaneously, being the two major pathophysiology mechanisms of CIAKI.
This study analyzed whether a 15-PGDH inhibitor in a CIAKI mouse model has a protective effect against renal deterioration and histological damage by CM, and against direct tubular cell toxicity by culturing renal tubular cells in vitro. In addition, we investigated the changes in intrarenal hemodynamic and renal blood flow (RBF) as a mechanism, and the role of receptors related to PGE2.
Materials and methods
Mice and reagents
Female C57/BL6 mice (age, 10 weeks; body weight, 20–25 g) were purchased from Orient Bio Inc. (Daejeon, Republic of Korea). Before the experiments, all mice were housed individually in standard cages and were allowed to acclimate under specific-pathogen-free conditions in the animal care facility of the College of Medicine of Inje University, Republic of Korea. The care of and experimental procedures involving the animals were approved by the Institutional Animal Care and Use Committee of Inje University (protocol no. 2018-019).
Induction of CIAKI
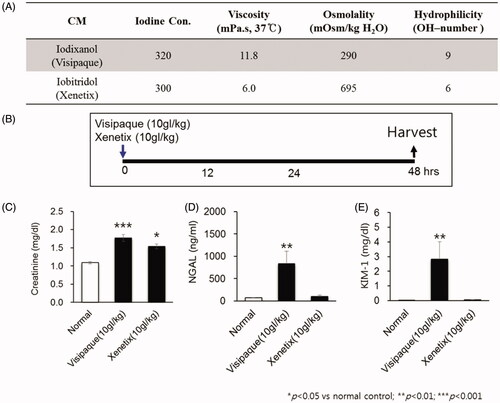
The two different radioactive iodine in terms of viscosity and osmolality were administered intravenously to 5 mice for each group. Ten grams of iodine per body weight (gI/kg) of each CM was injected by a 26-gauge syringe through the tail vein. The blood samples for the analysis of functional assessment of kidney injury such as Cr, NGAL, and KIM-1 were taken at 48 h after administration ().
Figure 1. Establishment of contrast induced acute kidney injury (CIAKI).
Study design of each group with drug administration
Female mice were injected with 10 gI/kg of Visipaque® (iodixanol) via the tail vein. SW033291 (5 mg/kg; Cayman, Ann Arbor, MI, USA), PGE1 (20 µg/kg; Cayman), or PGE2 (5 mg/kg; Sigma-Aldrich, St. Louis, MO, USA) or a vehicle (10%, ethanol; 5%, cremophor EL; and 85%, dextrose 5% in water) were intraperitoneally administered 1 h before, immediately after, and 8, 16, and 24 h after the iodixanol injection (). For inhibition of the EP4s, mice were treated with 0.2 mg/kg/day of ONO-AE3-208 by subcutaneous injection for 14 days. Serum and kidney tissue were collected 48 h after the iodixanol injection.
Figure 2. 15-Hydroxyprostaglandin dehydrogenase (15-PGDH) inhibition of contrast-induced acute kidney injury decreases the levels of renal injury biomarkers. (A) Arachidonic acid prostaglandin biosynthesis pathway and the biological activity of 15-PGDH inhibitor (SW033291). (B) Prostaglandin E2 (PGE2) levels in kidney tissue 1 h after intraperitoneal injection of 5 mg/kg SW033291 or a vehicle. (C) Experimental setup: mice received SW033291, PGE1, PGE2, or vehicle at 1 h before, immediately after, and 8, 16, and 24 h after administration of 10 gI/kg iodixanol. (D–F) Serum levels of creatinine, neutrophil gelatinase-associated lipocalin (NGAL), and kidney injury molecule-1 (KIM-1), respectively. Renal function was evaluated at 48 h after contrast medium (CM) injection. *p < 0.05; **p < 0.01; ***p < 0.001.
Measurement of PGE2 levels
Aside from the CIAKI experiment design, we first confirmed whether SW033291 administration actually increases PGE2 levels in kidney tissue in normal mice. Kidney tissues were harvested 1 h after the SW033291 injection, rinsed in ice-cold phosphate-buffered saline (PBS) containing indomethacin (10 μg/mL), and snap-frozen in liquid nitrogen. Next, 20 mg kidney tissue was homogenized in 500 µL cold PBS containing indomethacin (10 μg/mL) using a tissue homogenizer. The suspension was sonicated in an ice-water bath for 1 min using cycles of 10 s of sonication with 10 s of cooling, and then they were centrifuged for 10 min at 12,000 rpm. The supernatant was collected for PGE-2 assay. Protein concentrations were determined by bicinchoninic acid assay (Cat. #23225, Thermo Scientific). The PGE2 level in the supernatant was measured using a PGE2 enzyme-linked immunosorbent assay kit (R&D Systems, Minneapolis, MN, USA) in triplicate. PGE-2 levels were expressed as ng of PGE2/mg protein.
Assessment of renal function
Renal function was assessed by determining the serum levels of creatinine (Arbor Assays, Ann Arbor, MI, USA), NGAL (R&D Systems), and KIM-1 (R&D Systems) at 48 h after the iodixanol injection.
Necrotic and apoptotic cell death assays
Kidneys were harvested at 48 h after the CM injection, fixed in 4% phosphate-buffered formalin, and embedded in paraffin. To evaluate necrosis, 5 µm thick paraffin sections were stained with hematoxylin and eosin. Tubular injury was scored semi-quantitatively according to a system by a pathologist who examined at least 20 separate fields (400× magnification) in the outer medulla, which is the zone most sensitive to ischemic injury. The scoring system was as follows: 0, no damage; 1, patchy isolated unicellular necrosis; 2, tubular necrosis < 25%; 3, tubular necrosis 25–50%; and 4, tubular necrosis > 50%. At least 20 consecutive high-power fields per section were scored by two operators blind to the details of the experiment. To analyze the frequency of apoptosis, 5 µm thick paraffin sections were subjected to terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling (TUNEL) assay (Millipore, Temecula, CA, USA) according to the manufacturer’s protocol. Four 5 µm thick paraffin sections were incubated with the TUNEL reaction mixture at 37 °C for 1 h, followed by incubation with a horseradish peroxidase-conjugated detection antibody. The signals were visualized using diaminobenzidine (Sigma-Aldrich). After counterstaining with Mayer’s hematoxylin, TUNEL-positive cells were counted in at least five separate fields (640× magnification) in the outer medulla, and the apoptosis index (%, number of apoptosis cells/total number of cells) was calculated using GENASIS software.
Human renal proximal tubular epithelial cells culture
human renal proximal tubular epithelial cells (hRPTECs) were purchased from the American Type Culture Collection (#PCS-400-010™, Manassas, VA, USA). These cells were grown in 75 cm2 flasks in Renal Epithelial Cell Basal Medium (PCS-400-030™, ATCC®) supplemented with Renal Epithelial Cell Growth Kit (PCS-400-040™, ATCC®).
Cell viability assay
3-(4,5-Dimethyl thiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay was used to assess cell viability. The hRPTECs were cultivated in 96-well plates at a density of 105 cells/mL and then incubated for 24 h. They were treated with SW033291 (1 μM), PGE1 (100 ng/mL), or PGE2 (100 nM) simultaneously with Visipaque® (iodixanol, 50 mgI/mL) for 24 h. Then, 10 μM MTT (Sigma-Aldrich) was added to each well for an additional 4 h. The blue MTT formazan precipitate was dissolved in 100 µL dimethyl sulfoxide. The absorbance at 540 nm was measured with a multi-well plate reader. Cell viability was expressed as a percentage of the no-treatment cells, as the mean value ± standard deviation of the six independent experiments.
Analyses of apoptosis by flow cytometry
After the treatment described in the section above, the hRPTECs were pelleted by centrifugation at 1800 rpm for 10 min and incubated with annexin V fluorescein isothiocyanate and propidium iodide using an Apoptosis Detection Kit I (#556547, BD Biosciences, San Jose, CA, USA) according to the manufacture’s instruction. Then quantification was conducted using a FACSC flow cytometer with Cell Quest software (BD Biosciences).
Assessment of renal vasodilation in the outer medulla
48 h after CM injection, kidney tissues were harvested, fixed in 4% phosphate-buffered formalin, and embedded in paraffin. To quantify vasodilation, the inner arteriole area of the outer medulla was determined using α-SMA-stained sections. They were incubated for 1 h with an α-SMA antibody. 3,3′-diaminobenzidine (0.7 g/tablet; Sigma-Aldrich) was added, followed by washing three times with PBS for 1 min each. After counterstaining with Mayer’s hematoxylin, the inner areas of α-SMA-positive vessels in the outer medulla (25× magnification) were measured using ImageJ software. The results were expressed as the average area of all of the renal arteries in each outer medulla section.
Renal blood flow (RBF) assessment
Total RBF was assessed by measuring renal Doppler flux using noninvasive laser Doppler flowmetry (PeriFlux System 5000, Perimed AB, Sweden). Laser Doppler probes were placed on the kidney surface to measure the renal flux [Citation19]. The flux was measured 48 h after CM administration. The relative increase represented the percentage increase in renal blood flow from baseline to peak for each test. Statistical significance was set as p < 0.05.
Measurement of adenosine monophosphate (AMP) and adenosine levels
48 h after CM administration, serum and kidney tissues were harvested. The adenosine levels in the serum and kidney tissues were measured using high-performance liquid chromatography.
Statistical analyses
Results are presented as mean ± standard error of the mean. Statistical analyses were performed with one-way analysis of variance followed by the Bonferroni post-test when three or more experimental groups were compared. Values of p < 0.05 were considered indicative of statistical significance.
Results
CIAKI is occurred in mice with intravenous injection of visipaque® (iodixanol)
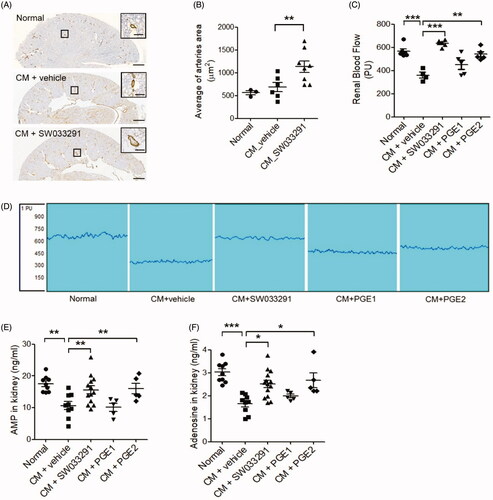
While Xenetix® and Visipaque® have the same iodine concentration, Visipaque® has a relatively lower osmolality and a higher viscosity than Xenetix® (). When compared with normal control, Xenetix® showed a significant increase in Cr only; NGAL and KIM-1 did not show significant differences. On the other hand, Visipaque® showed elevation in all three renal damage markers, Cr, NGAL and KIM-1, when compared with normal control (). Furthermore, Visipaque® showed a significantly higher necrosis and apoptosis shown by Renal injury score and TUNEL assay than normal control (). Therefore, it can be said that Visipaque®, an isosmolar and high viscosity agent, successfully induced CIAKI in mouse, and thus we chose Visipaque® as the contrast media for CIAKI mouse model.
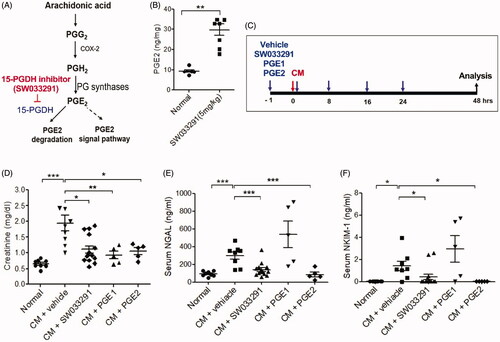
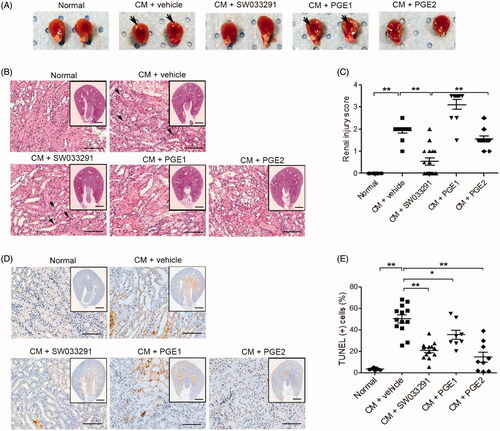
Figure 3. 15-Hydroxyprostaglandin dehydrogenase inhibition ameliorates renal tubular cell death in contrast-induced acute kidney injury mice. Before and after contrast medium (CM) administration, mice were injected intraperitoneally with a vehicle, SW033291 (15-PGDH inhibitor; 5 mg/kg), prostaglandin E1 (PGE1; 20 µg/kg), or PGE2 (5 mg/kg). Assessments were performed at 48 h after intravenous CM injection. (A) Representative gross appearances of the left and right kidneys of normal mice, and of those after injection with CM + vehicle, CM + SW033291, CM + PGE1, or CM + PGE2. Renal congestion in the outer medullary region is indicated by a black arrow. (B) Representative images of tubular injury in the outer zone of the renal medulla (hematoxylin and eosin staining). Scale bars in small panels, 500 μm and those in enlarged images, 50 μm. (C) Statistical analyses of tubular injury scores (n = 20 per group). (D) Representative images of apoptosis in the outer zone of the renal medulla (terminal deoxynucleotidyl transferase dUTP nick end labeling staining). Scale bars in small panels, 500 μm, those in enlarged images, 25 μm. (E) Statistical analyses of apoptosis (n = 20 per group). *p < 0.05; **p < 0.001.
15-PGDH inhibitor attenuates renal dysfunction in CIAKI mouse model
Renal PGE2 levels measured 1 h after administration of a 15-PGDH inhibitor (SW033291) increased by an average of three times compared to vehicle-administered, normal, control mice (9.29 ± 0.67 ng/mg in normal vs. 29.71 ± 2.79 ng/mg in SW033291, p < 0.01; ). Compared to the control group, creatinine, neutrophil gelatinase-associated lipocalin (NGAL), and kidney injury molecule-1 (KIM-1) levels were significantly increased in CM-treated mice (CM + vehicle; creatinine, 62 ± 0.02 mg/dL [normal] vs. 1.94 ± 0.24 mg/dL [iodixanol (10 gI/kg)], p < 0.001; NGAL, 63.55 ± 8.88 ng/mL [normal] vs. 299.71 ± 38.64 ng/mL [iodixanol (10 gI/kg)], p < 0.001; KIM-1, 0.03 ± 0.001 ng/mL [normal] vs. 1.41 ± 0.41 ng/mL [iodixanol (10 gI/kg)], p < 0.001; ).
SW033291 treatment in CIAKI mice led to marked reductions in the levels of creatinine, NGAL, and KIM-1 (creatinine, 1.94 ± 0.24 mg/dL [CM + vehicle] vs. 1.10 ± 0.11 mg/dL [CM + SW033291], p < 0.05; NGAL, 299.71 ± 38.64 ng/mL [CM + vehicle] vs. 140.41 ± 25.52 ng/mL [CM + SW033291], p < 0.001; KIM-1, 1.41 ± 0.41 ng/mL [CM + vehicle] vs. 0.43 ± 0.30 ng/mL [CM + SW033291], p < 0.05; ). In addition, in a group treated with PGE2, the reduction in renal injury biomarkers was similar to that of the SW033291 administration group. However, PGE1 decreased serum levels of creatinine in CIAKI mice, but not NGAL and KIM-1.
15-PGDH inhibitor ameliorates renal necrosis and apoptosis in CIAKI
In the gross findings of the mice kidneys, two groups, those treated with CM + vehicle and CM + PGE1, showed prominent vascular congestion (red blood cells sludging and medullary hyperemia) in the outer medullary region compared to the normal group, whereas two other groups, those treated with CM + SW033291 and CM + PGE2, showed less-congested outer medullary areas (). In the microscopic findings of themouse kidneys, CM + vehicle mice showed distinct tubular dilation, necrotic cells, and loss of brush border compared to normal renal tissue (). The microscopic renal injury score of the SW033291 administration group was significantly lower than that of the CM + vehicle mice (1.90 ± 0.10 (CM + vehicle) vs. 0.53 ± 0.16 (CM + SW033291), p < 0.001; ). The CM + PGE2 group also showed significantly lower renal injury scores than the CM + vehicle group, but significantly higher than the SW033291 administration group (0.53 ± 0.16 [CM + SW033291] vs. 1.55 ± 0.14 [CM + PGE2], p < 0.001). No reduction in renal injury score was observed in the PGE1 group (). The frequency of apoptosis detected by terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) staining was higher in the CIAKI mice than normal ones, and administration of SW033291 reduced the proportion of TUNEL-positive cells caused by CM (46.77 ± 4.77% [CM + vehicle] vs. 24.01 ± 2.90% [CM + SW033291], p < 0.001). The mice treated with PGE1 and PGE2 also showed significantly lower TUNEL-positive cell proportions compared to the CIAKI mice ().
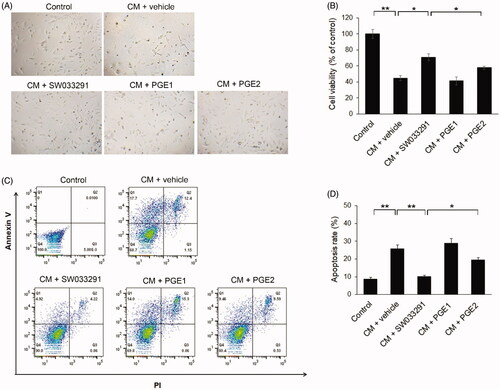
15-PGDH inhibitor protects human renal proximal tubular epithelial cells from iodixanol-induced apoptosis
We assessed the viability of hRPTECs using a MTT assay. This was significantly lower in the CM group than in the normal, control group. However, SW033291 treatment significantly increased cell viability compared to the CM + vehicle group (44.77 ± 3.01 [CM + vehicle] vs. 70.87 ± 1 [CM + SW033291], p < 0.05), but the PGE1 and PGE2 treatment groups did not show any difference in cell viability from the CM + vehicle group (). The apoptosis rate of hRPTECs also increased more than 2.5 times in the CM group compared to the normal group, but when SW033291 was added, the apoptosis rate caused by CM significantly decreased (25.78 ± 2.17% [CM + vehicle] vs. 10.06 ± 0.85% [CM + SW033291], p < 0.001). The PGE2 group tended to have a lower apoptotic rate than the CM + vehicle group (25.78 ± 2.17% [CM + vehicle] vs. 19.43 ± 1.38% [CM + PGE2], p = 0.069), but this was not statistically significant. However, comparing the SW033291 and PGE2 treatment groups, SW033291 had a significantly higher anti-apoptosis effect (10.06 ± 0.85%[CM + SW033291] vs. 19.43 ± 1.38 [CM + PGE2], p < 0.05; ). There were no differences in apoptotic rate between the PGE1 treatment and CM + vehicle groups.
Figure 4. 15-Hydroxyprostaglandin dehydrogenase inhibitor effects on iodixanol-induced apoptosis in human renal proximal tubular epithelial cells (hRPTECs). hRPTECs were treated with SW033291 (15-PGDH inhibitor), prostaglandin E1 (PGE1), or PGE2 simultaneously with Visipaque® (iodixanol 50 mgI/mL). (A) Representative pictures of hRPTECs viability 24 h after Visipaque® (iodixanol 50 mgI/mL) treatment. (B) Quantification of hRPTEC viability by MTT assay. (C) Density plot of annexin V fluorescein isothiocyanate and propidium iodide (PI) expression in hRPTECs assessed by flow cytometry. (D) Quantification of hRPTECs apoptosis by flow cytometry; *p < 0.05; **p < 0.001. CM: contrast medium.
15-PGDH inhibitor induces renal vasodilation via the PGE2 receptor 4–adenosine monophosphate–adenosine pathway in the outer medulla
SW033291 treatment in CIAKI mice significantly increased the average renal arteriole area in the outer medulla compared to untreated CIAKI mice (683.63 ± 111.11 μm2 [CM + vehicle] vs. 1132.97 ± 159.86 μm2 [CM + SW033291], p < 0.05; ). CIAKI mice exhibited a decrease in RBF, but SW033291 treatment significantly prevented the reduction of RBF by CM injection (360.0 ± 24.86 [CM + vehicle] vs. 635.2 ± 11.10 [CM + SW033291], p < 0.001; ). PGE2 treatment also significantly preserved RBF from the CM-induced reduction of RBF (360.0 ± 24.86 [CM + vehicle] vs. 541.4 ± 22.65 [CM + PGE2], p = 0.001), but PGE1 did not. Levels of AMP and adenosine in renal tissue were all significantly decreased in CIAKI mice compared to the sham, but these changes were substantially reversed by SW033291-treated mice (AMP, 10.64 ± 1.27 ng/mL [CM + vehicle] vs. 15.60 ± 1.26 ng/mL [CM + SW033291], p < 0.05; adenosine, 1.65 ± 0.14 ng/mL [CM + vehicle] vs. 2.65 ± 0.15 ng/mL [CM + SW033291], p < 0.01; ). Administering PGE2 also increased AMP and adenosine levels compared to CIAKI mice ().
Figure 5. 15-Hydroxyprostaglandin dehydrogenase inhibition induces renal vasodilation in the outer medulla via the adenosine monophosphate (AMP)–adenosine signaling pathway. (A) Representative images of arterioles in the outer zone of the renal medulla. Magnified images are enlargements of the outlined areas. (B) Statistical analyses of the inner arteriole area of the outer medulla. (C) Statistical analyses of renal blood flow following administration of a vehicle, SW033291 (inhibitor), prostaglandin E1 (PGE1), or PGE2 in contrast-induced acute kidney injury mice. (D) Representative images of renal blood flux measurements of the study groups. (E,F) Statistical analyses of AMP and adenosine levels in kidney tissue. *p < 0.05; **p < 0.01; ***p < 0.001. Scale bars, 500 μm; scale bar in the enlarged image, 50 μm. CM: contrast medium.
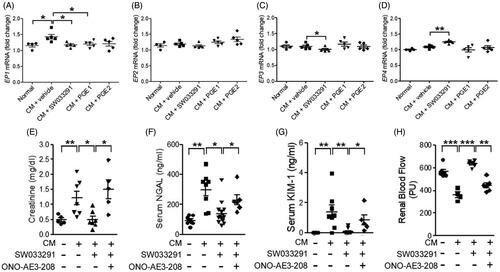
Next, we analyzed PGE2 receptor (EP) expression. EP1 increased in the CM + vehicle group, but not in the SW033291, PGE1, or PGE2 groups (). There were no differences in the expression level of EP2 between the groups (). However, the SW033291 group showed significantly lower EP3 expression than the CM group (1.10 ± 0.03-fold [CM + vehicle] vs. 1.00 ± 0.02-fold [CM + SW033291], p < 0.05; ). The EP4 expression level was significantly increased only in the SW033291 group (1.09 ± 0.02-fold [CM + vehicle] vs. 1.25 ± 0.02-fold [CM + SW033291], p < 0.01; ). An EP4 antagonist, ONO-AE3-208, attenuated the renoprotective effects of SW033291, as indicated by increases in creatinine, NGAL, and KIM-1 (creatinine, 0.50 ± 0.10 mg/dL [CM + SW033291] vs. 1.51 ± 0.31 mg/dL [CM + ONO-AE3-208 + SW033291], p < 0.05; NGAL, 140.41 ± 25.52 ng/mL [CM + SW033291] vs. 229.85 ± 27.12 ng/mL [CM + ONO-AE3-208 + SW033291], p < 0.05; KIM-1, 0.09 ± 0.05 ng/mL [CM + SW033291] vs. 0.85 ± 0.26 ng/mL [CM + ONO-AE3-208 + SW033291], p < 0.05; ). Moreover, we assessed RBF, and ONO-AE3-208 blocked the increase in RBF by SW033291 treatment in CIAKI mice ().
Figure 6. 15-Hydroxyprostaglandin dehydrogenase inhibition changes the level of prostaglandin E2 (PGE2) receptor (EP) expression in kidney tissue. The protective effects of the15-PGDH inhibitor, SW033291, can be blocked by an EP4 antagonist, ONO-AE3-208. (A–D) Statistical analyses of the EP expression level. (E–G) Serum levels of creatinine, neutrophil gelatinase-associated lipocalin (NGAL), and kidney injury molecule-1 (KIM-1) due to the absence or presence of ONO-AE3-208 in the contrast-induced acute kidney injury model. (H) Statistical analyses of renal blood flow. *p < 0.05; **p < 0.01; ***p < 0.001; CM: contrast medium.
Discussion
We confirmed that iodixanol causes functional and histological acute kidney injury in mice.
To establish a CIAKI animal model, we considered the conventional rat models using water depletion and furosemide to induce a severe dehydration, and then administering CM to achieve CIAKI; however, this method is rather complicated and time-consuming, and most importantly, far from clinical settings [Citation20,Citation21]. We specifically chose mouse over rat to establish a CIAKI animal model, and using a number of different CM with varying osmolality and viscosity, we aimed to find the appropriate CM that induces CIAKI. Visipaque® (iodixanol), a CM with high viscosity and iso-osmolality, showed a significant functional and histopathological renal damage. The main difference between our animal model and the conventional dehydration-induced rat model is that the authors identified viscosity as the starting point for CIAKI. This study has shown that CM with a high viscosity can induce CIAKI in mouse on its own, without dehydration or using other drugs. Seeliger et al. [Citation22] mentioned that high CM viscosity is a key element in the pathophysiology of CIAKI, and when CM is administrated in a dehydrated state, fluid viscosity increases exponentially and flow through medullary tubules and vessels decreases, which increases the duration of contact of CM to tubular cells, thereby increasing renal tubular toxicity.
We analyzed whether functional and histological protection against CIAKI is possible by administering a 15-PGDH inhibitor, PGE1, or PGE2. Biomarker analyses were used to detect the functional changes of the kidneys; the 15-PGDH inhibitor, PGE1, and PGE2 all showed a similar effect by reducing serum creatinine levels in the CIAKI model (). On the other hand, NGAL and KIM-1 showed varying results; with 15-PGDH inhibitor and PGE2, the concentration of the two markers decreased, whereas PGE1 did not decrease them (). Creatinine is a biomarker that reflects the kidney’s ability to excrete waste, while NGAL and KIM-1 suggest that the kidney has undergone a tubular injury [Citation23,Citation24]. Therefore, we can assume that PGE1, PGE2, and the 15-PGDH inhibitor all have a protective effect in the excretory function against CM, but only PGE2 and the 15-PGDH inhibitor have a protective effect against tubular toxicity.
To assess the degree of histological renal injury, we used the renal injury score to evaluate necrosis, and TUNEL staining for apoptosis. The 15-PGDH inhibitor effectively inhibited both necrosis and apoptosis of renal tissues by CM, whereas PGE2 inhibited apoptosis but failed to successfully reduce necrosis. PGE1 decreased apoptosis but increased necrosis (). Interestingly, the 15-PGDH inhibitor showed a greater effect in reducing renal necrosis than PGE2. The estimated half-life of PGE2 is less than 15 s because PGE2 is rapidly degraded by 15-PGDH [Citation19]. Therefore, increasing the level and extending the half-life of intrinsic PGE2 induced by blocking 15-PGDH () would be more effective for reducing the renal injury score than directly administering PGE2, which has a short half-life.
We performed cell culture experiments using hRPTECs to determine whether CM has a direct cellular toxicity to renal tubular cells and whether the 15-PGDH inhibitor, PGE1, and PGE2 have protective effects. CM showed a decrease in cellular viability and increase in apoptosis on the hRPTECs, as reported by other previous studies [Citation25–27]. However, such harmful effects of CM were significantly reversed by the administration of the 15-PGDH inhibitor (); PGE1 had no protective effect against cellular toxicity, and PGE2 showed only a minimal effect, much less than that of the 15-PGDH inhibitor. Therefore, the protective effect on the renal tubular cells against CM could be better achieved by inhibiting the 15-PGDH to block the catabolism of the endogenous PGE2, rather than administering exogenous PGE2.
CM is also known as a toxic agent that decreases renal blood flow and increases vasoconstriction in many studies [Citation16,Citation28]. Our renal hemodynamic study also showed that CM has obviously decreased renal blood flow (). However, this deteriorating effect of CM on renal hemodynamics was definitely reversed by the administration of PGE2 or 15-PGDH inhibitor, but not by PGE1. Moreover, 15-PGDH inhibitor has increased renal vasodilation more than that of CM group, confirmed by measuring the size of arterioles. Furthermore, the analysis of vasoactive substances to increase the renal vasodilation showed that both adenosine, involved in renal blood vessel expansion, and its precursor, AMP, were increased in the 15-PGDH inhibitor or PGE2 administration group than CM group. Thus, we can suggest that the protective effects of PGE2 and 15-PGDH inhibitor from renal vasoconstriction of CM are induced by increasing adenosine and AMP, which cause renal vasodilation.
To investigate the mechanism behind the renal protective effect of 15-PGDH inhibitor, we examined the change in expression of PGE2 receptors. According to many studies regarding the contribution of prostaglandin EP receptors to renal microvascular reactivity, EP1 and EP3 mediate renal vasoconstrictor responses, whereas EP2 and EP4 mediate vasodilation [Citation29–31]. Our results are consistent with these observations that vasodilatory effect of 15-PGDH inhibitor is mediated by the decreased EP 3 expression and increased EP4 expression. Moreover, ONO-AE3-208, an EP4 antagonist, clearly offsets the renal protective effect of the 15-PGDH inhibitor. We can assume that the 15-PGDH inhibitor maximizes the protective action against renal damage by CM through the EP4.
There are certain limitations of this study. We have identified that SW003291 has a protective effect against cellular toxicity by CM, but we have not discovered the exact mechanism of apoptosis and pathways involved in it. Moreover, SW033291 was administered systemically; although SW033291 induces both renal PGE2 and renal EP4 receptors, we cannot completely rule out the possibility that some of the effects observed in the kidney may be secondary to systemic effects of increased PGE2. Further studies are mandatory to elucidate these questions.
In summary, administering the 15-PGDH inhibitor, SW033291, before and after CM in the CIAKI mouse model had a functional and histological protection against CIAKI. This was achieved by providing protection against both hemodynamic and tubular toxic effects, the two main mechanisms of CIAKI. The intrarenal vasodilation and increased renal blood flow induced by SW033291 were via the EP4s, which were strongly associated with the increase in AMP and adenosine levels. The protective effect of SW033291 on the tubular cell toxicity of CM was achieved by inhibiting tubular cell apoptosis. Therefore, we suggest that 15-PGDH inhibitor may be a novel prophylactic agent for CIAKI, and further studies are needed for clinical implications.
Disclosure statement
Sanford Markowitz is a founder, shareholder, consultant, recipient of a sponsored research grant award from, and a member of the Board of Directors of, Rodeo Therapeutics, and has a royalty interest in technology on use of 15-PGDH inhibitors for renal protection from technology optioned to Rodeo Therapeutics. These conflicts are overseen and managed by Case Western Reserve University in accord with institutional polices. Ki Beom Bae has a royalty interest in technology on use of 15-PGDH inhibitors for renal protection from technology optioned to Rodeo Therapeutics. These conflicts are overseen and managed by Inje University in accord with institutional polices.
Additional information
Funding
References
- van der Molen AJ, Reimer P, Dekkers IA, et al. Post-contrast acute kidney injury – part 1: definition, clinical features, incidence, role of contrast medium and risk factors: recommendations for updated ESUR Contrast Medium Safety Committee guidelines. Eur Radiol. 2018;28:2845–2855.
- ACR Committee on Drugs and Contrast Media. ACR manual on contrast media, version 10.3. 2020.
- Mehran R, Nikolsky E. Contrast-induced nephropathy: definition, epidemiology, and patients at risk. Kidney Int Suppl. 2006; 69:S11–S15.
- Goldenberg I, Matetzky S. Nephropathy induced by contrast media: pathogenesis, risk factors and preventive strategies. CMAJ. 2005;172:1461–1471.
- Neumann FJ, Sousa-Uva M, Ahlsson A, et al., ESC Scientific Document Group. 2018 ESC/EACTS Guidelines on myocardial revascularization. Eur Heart J. 2019;40:87–165.
- Jeong BY, Lee HY, Park CG, Kang J, et al. Oxidative stress caused by activation of NADPH oxidase 4 promotes contrast-induced acute kidney injury. PLoS One. 2018;13:e0191034.
- Shen J, Wang L, Jiang N, et al. NLRP3 inflammasome mediates contrast media-induced acute kidney injury by regulating cell apoptosis. Sci Rep. 2016;6:34682.
- From AM, Bartholmai BJ, Williams AW, et al. Mortality associated with nephropathy after radiographic contrast exposure. Mayo Clin Proc. 2008;83:1095–1100.
- Goldenberg I, Chonchol M, Guetta V. Reversible acute kidney injury following contrast exposure and the risk of long-term mortality. Am J Nephrol. 2009;29:136–144.
- Mamoulakis C, Tsarouhas K, Fragkiadoulaki I, et al. Contrast-induced nephropathy: Basic concepts, pathophysiological implications and prevention strategies. Pharmacol Ther. 2017;180:99–112.
- Weisbord SD, Gallagher M, Jneid H, et al., PRESERVE Trial Group. Outcomes after angiography with sodium bicarbonate and acetylcysteine. N Engl J Med. 2018;378:603–614.
- Cho A, Lee YK, Sohn SY. Beneficial effect of statin on preventing contrast-induced acute kidney injury in patients with renal insufficiency: a meta-analysis. Medicine (Baltimore). 2020;99:e19473.
- Kolh P, Windecker S, Alfonso F, et al., European Society of Cardiology Committee for Practice Guidelines, Task Force on Myocardial Revascularization of the European Society of Cardiology and the European Association for Cardio-Thoracic Surgery, European Association of Percutaneous Cardiovascular Interventions. 2014 ESC/EACTS Guidelines on myocardial revascularization: the Task Force on Myocardial Revascularization of the European Society of Cardiology (ESC) and the European Association for Cardio-Thoracic Surgery (EACTS). Developed with the special contribution of the European Association of Percutaneous Cardiovascular Interventions (EAPCI). Eur J Cardiothorac Surg. 2014;46:517–592.
- Geng N, Zou D, Chen Y, et al. Prostaglandin E1 administration for prevention of contrast-induced acute kidney injury: a systematic review and meta-analysis of randomized controlled trials. Medicine (Baltimore). 2018;97:e11416.
- Stacul F, van der Molen AJ, Reimer P, et al., Contrast Media Safety Committee of European Society of Urogenital Radiology (ESUR). Contrast induced nephropathy: updated ESUR Contrast Media Safety Committee guidelines. Eur Radiol. 2011;21:2527–2541.
- Caiazza A, Russo L, Sabbatini M, et al. Hemodynamic and tubular changes induced by contrast media. Biomed Res Int. 2014;2014:578974.
- Zhang Y, Desai A, Yang SY, et al. Tissue Regeneration. Inhibition of the prostaglandin-degrading enzyme 15-PGDH potentiates tissue regeneration. Science. 2015;348:aaa2340.
- Eskildsen MP, Hansen PB, Stubbe J, et al. Prostaglandin I2 and prostaglandin E2 modulate human intrarenal artery contractility through prostaglandin E2-EP4, prostacyclin-IP, and thromboxane A2-TP receptors. Hypertension. 2014;64:551–556.
- Persson P, Fasching A, Teerlink T, et al. Cellular transport of L-arginine determines renal medullary blood flow in control rats, but not in diabetic rats despite enhanced cellular uptake capacity. Am J Physiol Renal Physiol. 2017;312:F278–F283.
- Cheng W, Zhao F, Tang CY, et al. Comparison of iohexol and iodixanol induced nephrotoxicity, mitochondrial damage and mitophagy in a new contrast-induced acute kidney injury rat model. Arch Toxicol. 2018;92:2245–2257.
- Sun S, Zhang T, Nie P, et al. A novel rat model of contrast-induced acute kidney injury. Int J Cardiol. 2014;172:e48–e50.
- Seeliger E, Lenhard DC, Persson PB. Contrast media viscosity versus osmolality in kidney injury: lessons from animal studies. Biomed Res Int. 2014;2014:358136.
- Martensson J, Bellomo R. The rise and fall of NGAL in acute kidney injury. Blood Purif. 2014;37:304–310.
- Bonventre JV. Kidney Injury Molecule-1 (KIM-1): a specific and sensitive biomarker of kidney injury. Scand J Clin Lab Invest Suppl. 2008;241:78–83.
- Kolyada AY, Liangos O, Madias NE, et al. Protective effect of erythropoietin against radiocontrast-induced renal tubular epithelial cell injury. Am J Nephrol. 2008;28:203–209.
- Romano G, Briguori C, Quintavalle C, et al. Contrast agents and renal cell apoptosis. Eur Heart J. 2008;29:2569–2576.
- Oh HJ, Oh H, Nam BY, et al. The protective effect of klotho against contrast-associated acute kidney injury via the antioxidative effect. Am J Physiol Renal Physiol. 2019;317:F881–F889.
- Vlachopanos G, Schizas D, Hasemaki N, et al. Pathophysiology of contrast-induced acute kidney injury (CIAKI). Curr Pharm Des. 2019;25:4642–4647.
- van Rodijnen WF, Korstjens IJ, Legerstee N, et al. Direct vasoconstrictor effect of prostaglandin E2 on renal interlobular arteries: role of the EP3 receptor. Am J Physiol Renal Physiol. 2007;292:F1094–F1101.
- Tang L, Loutzenhiser K, Loutzenhiser R. Biphasic actions of prostaglandin E(2) on the renal afferent arteriole: role of EP(3) and EP(4) receptors. Circ Res. 2000;86:663–670.
- Imig JD, Breyer MD, Breyer RM. Contribution of prostaglandin EP(2) receptors to renal microvascular reactivity in mice. Am J Physiol Renal Physiol. 2002;283:F415–F422.