
Abstract
Objective
This study was designed to uncover the mechanism of miR-34b-5p-mediated aquaporin-2 (AQP2) in sepsis-induced injury using human renal tubular epithelial cells (HK-2).
Methods
Serum levels of miR-34b-5p, TNF-α, IL-1β, IL-6, serum creatinine (SCr), and blood urea nitrogen (BUN) in septic patients with acute kidney injury (AKI) and healthy controls were detected. Lipopolysaccharide (LPS) was used to induce sepsis in HK-2 cells. LPS-induced HK-2 cells were transfected with miR-34b-5p inhibitor, miR-34b-5p mimic, pcDNA3.1-AQP2, si-AQP2, miR-34b-5p inhibitor + si-NC, or miR-34b-5p inhibitor + si-AQP2. The expressions of miR-34b-5p, AQP2, Bax, Bcl-2, cleaved caspase-3, TNF-α, IL-1β, and IL-6 in HK-2 cells were detected. TUNEL staining revealed the apoptosis of HK-2 cells. Dual-luciferase reporter assay verified the binding between miR-34b-5p and AQP2.
Results
The expression of miR-34b-5p and the inflammatory responses were augmented in septic AKI patients. miR-34b-5p was up-regulated and AQP2 was down-regulated in LPS-induced HK-2 cells. miR-34b-5p inhibition or AQP2 overexpression ameliorated apoptosis and inflammation in LPS-induced HK-2 cells. In contrast, overexpressing miR-34b-5p deteriorated LPS-induced injury in HK-2 cells. AQP2 was a downstream target of miR-34b-5p. AQP2 silencing abolished the suppressive effects of miR-34b-5p inhibition on LPS-induced apoptosis and inflammatory response in HK-2 cells.
Conclusion
miR-34b-5p inhibits AQP2 to promote LPS-induced injury in HK-2 cells.
Introduction
Sepsis is a life-threatening organ dysfunction induced by a disordered host response to infection [Citation1]. Sepsis is identified in up to 39% of intensive care unit (ICU) patients, resulting in an overall mortality rate of 26% ICU patients [Citation2]. A normal host response to infection involves activation of phagocytic cells and production of inflammatory mediators, and contributes to the repair of injured tissue. Uncontrolled generation of pro-inflammatory cytokines, which exceeds the boundary of local infected environment and leads to a more generalized response, will give rise to the initiation of sepsis [Citation3]. Acute kidney injury (AKI) is a common disorder which gives rise to approximately 1.7 million deaths and affects about over 13 million people annually, among which patients living in developing countries account for the majority [Citation4]. AKI is associated with considerable morbidity and high costs since patients who have survived though are likely to suffer from long-term impacts of AKI such as development of chronic kidney disease and end-stage renal disease [Citation5]. Septic AKI is a commonly diagnosed AKI syndrome which occurs in nearly half of all AKI cases and is associated with higher risk of in-hospital mortality [Citation6]. The pathogenic mechanisms of septic AKI involve the direct interaction of resident renal cells with circulating pro-inflammatory and pro-apoptotic mediators, and apoptosis is one major cellular injury induced during sepsis [Citation7]. Although there are consensus definitions of sepsis and AKI, the molecular mechanisms for the pathogenesis of septic AKI remain largely undiscovered.
MicroRNA (miRNA), a diverse group of small noncoding RNAs, has emerged as a focal research object for its critical role in post-transcriptional regulation. MiRNA-34b-5p (miR-34b-5p) has been found to function in a wide range of biological activities such as the angiogenesis of thyroid carcinoma cells [Citation8] and recurrent seizure-induced astrocyte apoptosis [Citation9]. Recently, studies have reported that miR-34b-5p is associated with sepsis-induced organ injury. For instance, down-regulation of miR-34b-5p suppressed lung inflammation and apoptosis in sepsis-induced acute lung injury [Citation10,Citation11]. However, what role miR-34b-5p plays in septic AKI is barely known.
Aquaporin-2 (AQP2) is one of the water channels expressed by renal collecting duct principal cells and plays an essential role in maintaining body water homeostasis [Citation12]. Deficiency of AQPs is observed in many renal diseases, including AKI, nephrogenic diabetes insipidus, diabetic nephropathy, polycystic kidney disease, renal cell carcinoma, and renal fibrosis [Citation13]. Urinary exosomal release of AQP2 was reduced in ischemia/reperfusion-induced AKI [Citation14]. AQP2 accumulation at the apical plasma membranes of the renal-collecting ducts was decreased in cisplatin-induced AKI [Citation15]. Renal injury can be partially alleviated by restoring the renal expression of AQP2. For instance, NF-κB inhibition ameliorated sepsis-induced acute renal failure by promoting AQP2 via vasopressin V2 receptor [Citation16]. Moreover, pretreatment with continuous erythropoietin receptor activator restored tubular function and AQP2 expression in septic AKI [Citation17].
Considering the negative role of miR-34b-5p in sepsis-induced organ injury, we hypothesized that the deficiency of AQP2 might be associated with dysregulated miR-34b-5p in septic AKI. Online database further predicted that miR-34b-5p could bind AQP2. Therefore, we conducted experiments to validate the interaction of miR-34b-5p with AQP2 in septic AKI using a HK-2 cell model.
Materials and methods
Clinical samples
The study involved 30 septic AKI patients from Fujian Provincial Hospital, including 19 males and 11 females, aged from 24 to 57 years, with an average age of 39.3 ± 7.8 years. The normal group consisted of 30 healthy volunteers, including 17 males and 13 females, aged from 24 to 55 years, with an average age of 37.8 ± 8.1 years. Diagnosis of septic AKI conformed to the diagnostic standards outlined in Kidney Disease Improving Global Outcomes (KDIGO) [Citation18] and the diagnostic criteria for sepsis [Citation19]. The diagnosis of AKI was confirmed by absolute increase (26.5 μmol/L) of serum creatinine (SCr) within 48 h, increase of SCr by ≥50% within 7 days of disease onset, or urine output ≤0.5 mL/(kg·h) for consecutive 6 h. Sepsis was determined by the following two or more symptoms: hypothermia or pyrexia (<36 °C or >38.5 °C), tachycardia (>90 beats/min), tachypnea (>20 breaths/min or PaCO2 < 32 mmHg), leukocytosis (>12,000/mm3), leukopenia (<4000/mm3), and increase of immature cells (>10%). Patients with any of the following conditions were excluded: end-stage renal disease, a history of kidney transplantation, cancer, acquired immunodeficiency syndrome, and treatment of high doses of steroid. All patients received venous blood tests at the time of diagnosis. Blood samples were centrifuged at 3500 rpm for 10 min, and the serum was collected and preserved at −80 °C for later use. The sample collection was approved by the Ethics Committee of Fujian Provincial Hospital and all participants had signed the informed consents.
Cell cultivation and treatment
Human renal tubular epithelial cells (HK-2) were purchased from Shanghai Institute of Cell Research, Chinese Academy of Sciences. HK-2 cells were cultivated in DMEM (Gibco, Grand Island, NY, USA) supplemented with 10% fetal bovine serum, 1% penicillin, and 1% streptomycin at 37 °C with 5% CO2. Cell model of sepsis was induced by lipopolysaccharide (LPS). HK-2 cells were treated with 1 μg/mL LPS (Sigma Aldrich, St Louis, MO, USA) for 24 h and then seeded into a culture plate. The cells were incubated in the plate for 16 h and then subjected to transfection.
Apart from the control group, LPS-induced HK-2 cells were accordingly divided into the following groups: LPS group, LPS + inhibitor group (transfected with 30 nM miR-34b-5p inhibitor), LPS + inhibitor NC group (transfected with the negative control of miR-34b-5p inhibitor), LPS + mimic group (transfected with 30 nM miR-34b-5p mimic), LPS + mimic NC group (transfected with the negative control of miR-34b-5p mimic), LPS + pcDNA3.1 group (transfected with empty pcDNA3.1), LPS + pcDNA3.1-AQP2 group (transfected with 2 μg of pcDNA3.1-AQP2), LPS + si-AQP2 group (transfected with 2 μg of si-AQP2), LPS + si-NC group (transfected with the negative control of si-AQP2), LPS + inhibitor NC + si-NC group (co-transfected with the negative controls of miR-34b-5p inhibitor and si-AQP2), LPS + inhibitor + si-NC group (co-transfected with miR-34b-5p inhibitor and the negative control of si-AQP2), and LPS + inhibitor + si-AQP2 group (co-transfected with miR-34b-5p inhibitor and si-AQP2). All plasmids were obtained from GenePharma (Shanghai, China) and transfected into the cells via LipoFiterTM (Hanbio, Shanghai, China). The transfection experiment was performed in triplicate and the following experiments were conducted 24 h after the transfection.
Dual-luciferase reporter assay
The binding site between miR-34b-5p and AQP2 was predicted by the online software Jefferson (https://cm.jefferson.edu/rna22/Precomputed/OptionController?species=HomoSapiens&type=mRNA&version=MB21E78v2). Wild and mutated sequences of the binding site on AQP2 (mut-AQP2 and wt-AQP2) were designed and synthesized according to the prediction. The sequences were inserted into vectors (pGL3-Basic, Promega, MADISON, WI, USA) and then co-transfected with miR-34b-5p mimic or the corresponding negative control into HEK293T cells. Luciferase intensity in the cells was tested using a dual-luciferase reporter kit (Promega) for determining the binding between miR-34b-5p and AQP2. The HEK293T cells were divided into mimic + mut-AQP2 group, mimic + wt-AQP2 group, mimic NC + mut-AQP2 group, and mimic NC + wt-AQP2 group. The assay was performed in triplicate.
qRT-PCR
TRIzol reagents (Invitrogen, Carlsbad, CA, USA) were used to extract total RNA from blood samples and HK-2 cells. The extraction process of the total RNA from blood samples was as follows. An RNase-free centrifuge tube was added with 0.5 mL of blood and 1.5 mL of TRIzol reagent, and maintained at a still position at room temperature after vibration. The mixture was centrifuged at 4 °C, 12,000 g/min for 15 min. Supernatant obtained after the centrifugation was transferred to another RNase-free centrifuge tube using a pipette. This tube was added with chloroform (TRIzol:chloroform = 5:1), and placed at a still position for 3 min after 15 s of vortex. After that, the mixture was centrifuged at 4 °C, 12,000 g/min for 15 min. Supernatant obtained (450 μL) and pre-chilled isopropanol (TRIzol:isopropanol = 2:1) were added into a clean 1.5-mL RNase-free centrifuge tube. The well-mixed liquid was maintained stationary at room temperature for 10 min, followed by centrifugation at 4 °C, 12,000 g/min for 15 min. Supernatant was carefully removed, and the remaining sediments were mixed with 1 mL of 75% ethanol prepared with DEPC (diethyl pyrocarbonate)-treated water, followed by centrifugation at 4 °C, 8000 g/min for 5 min. Supernatant obtained after the centrifugation was discarded, and the tube was put upside down on a clean filter paper for a while. After ethanol volatilization for 2–3 min, the tube was added with 15 μL of DEPC-treated water. The mixture was treated with a water bath at 55–60 °C for 10 min. HK-2 cells collected for RNA extraction were centrifuged at 800 r/min for 3–5 min. The culture medium was discarded, and the cells were mixed with 1 mL of TRIzol reagent. The cell suspension was transferred to a dry EP tube that had been soaked in DEPC-treated water. The tube was placed on ice for 10 min. The remaining process was the same as the extraction of total RNA from blood samples.
Concentration and purity of the RNA samples were measured after the extraction. Qualified RNA samples were adjusted to an appropriate concentration and then reverse-transcribed by reverse transcription kits (TaKaRa, Tokyo, Japan) using random primers. LightCycler 480 (Roche, Indianapolis, IN, USA) was used to detect gene expressions, and the experiments were carried out based on the instruction of SYBR Green Mix (Roche Diagnostics, Indianapolis, IN) as described below: 5 min at 95 °C followed by 40 cycles of 10 s at 95 °C, 10 s at 60 °C, and 20 s at 72 °C. Quantitative PCR of each RNA sample was performed in triplicate. U6 and GAPDH acted as the internal references of miRNA and mRNA, respectively. The 2−ΔΔCt method was used for data analysis. Sequences of the primers are exhibited in .
Table 1. Primer sequences.
Western blot
HK-2 cells were digested by trypsin and centrifuged at 1,000 rpm for 5 min. Supernatant was discarded and the cells were washed twice with pre-cooled phosphate buffered saline (PBS). The PBS was then discarded, and the petri dish was added with an appropriate volume of RIPA buffer (Beyotime, Shanghai, China) containing protease inhibitors and gently shaken to let the solution completely cover the bottom of the petri dish. After that, the dish was placed on ice for a few minutes. The cells were scraped from the bottom of the dish using a cell scraper, and the cell lysates were transferred into an EP tube using a pipette. The cell lysis and transfer of cell lysates were operated on ice. The EP tube was placed on ice for 15 min. The lysates were centrifuged at 32,000 rpm for 15 min at 4 °C. Supernatant was carefully pipetted into a new EP tube and centrifuged to acquire protein samples. GAPDH acted as the internal reference. The concentration of each protein sample was measured using a BCA kit (Beyotime) to ensure the same loading amount of each sample. The BCA working reagent was made by mixing Reagent A and Reagent B at a ratio of 50:1. The newly prepared BCA working reagent was stable at room temperature for 24 h. Concentration of the standards was set to 0, 0.05, 0.1, 0.2, 0.3, 0.4, and 0.5 mg/ml. The 96-well plate was added with an appropriate volume of protein samples, and each well was added with 200 μL of BCA working reagent. The protein samples were incubated at 37 °C for 30 min. The absorbance value was measured at 562 nm by a microplate reader. Concentration of the protein samples was calculated according to the standard curve. After the measurement of concentration, proteins of corresponding volumes were mixed with loading buffer (Beyotime) and denatured in boiling water for 3 min. The proteins were separated by 10% SDS-PAGE which was run at 80 V until bromophenol blue came into the gel and then continued at 120 V for 1–2 h. The transfer of proteins was conducted in an ice bath at 300 mA for 60 min. After that, the membrane was rinsed for 1–2 min and then soaked in blocking buffer at room temperature for 60 min or at 4 °C overnight. The proteins were incubated with primary antibodies of GAPDH (rabbit anti-human, 1:1000, Cell Signaling, Boston, USA), Bax (rabbit anti-human, 1:2000, Abcam, Cambridge, UK), Bcl-2 (rabbit anti-human, 1:800, Abcam), cleaved caspase-3 (rabbit anti-human, 1:1000, Abcam) and AQP2 (rat anti-human, 1:500, Santa Cruz, TX, USA) on a shaking bed for 1 h. The membrane was washed for 3 × 10 min and then transferred to horseradish peroxidase-marked secondary antibody solution (goat anti-rabbit IgG, 1:10,000 or goat anti-rat IgG, 1:500) (Thermo Fisher Scientific, MA, USA) to incubate the proteins at room temperature for 1 h. After that, the membrane was subjected to another round of washing, and added with color-developing solution. Protein expression was detected by a chemiluminescence imaging system (Bio-Rad, Hercules, CA, USA).
TUNEL staining
HK-2 cells were fixed in 4% paraformaldehyde for 30 min and in 70% cold ethanol for 15 min. The cells were incubated in PBS containing 0.3% Triton X-100 at room temperature for 5 min and then stained in TUNEL solution (Beyotime, Shanghai, China) in dark at 37 °C for 60 min. After three PBS washes, the cells were mounted with antifluorescence quenching sealing solution and observed under a fluorescence microscope. Cell nuclei were dyed with DAPI. Apoptosis rate = (TUNEL-positive cell number/total cell number) × 100%.
ELISA
The levels of TNF-α, IL-1β, IL-6, SCr, and blood urea nitrogen (BUN) in the serum of septic AKI patients and the levels of TNF-α, IL-1β, and IL-6 in the supernatant of HK-2 cells were measured using ELISA kits (Abcam, Cambridge, UK).
Statistical analysis
GraphPad 7.0 was applied for statistical analysis with all data presented as mean ± SD. t test and one-way analysis of variance were used to compare the differences between two or more groups. Tukey's multiple comparisons test was applied for multiple comparisons post hoc. Correlation between the expression levels of miR-34b-5p and TNF-α, IL-1β, IL-6, SCr, and BUN in the septic AKI patients was measured by the Pearson correlation coefficient. p < 0.05 was considered of statistical significance.
Results
miR-34b-5p is highly expressed in septic patients with AKI
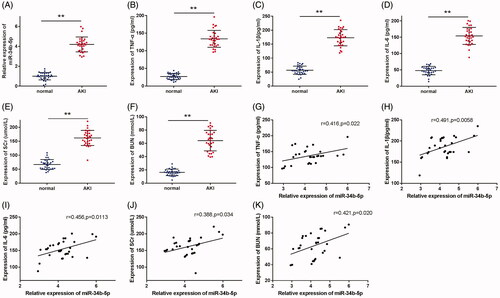
miR-34b-5p was detected to have a higher expression in the serum of septic patients with AKI (, p < 0.01, compared to the normal group). The serum levels of TNF-α, IL-1β, IL-6, SCr, and BUN were also up-regulated in patients with septic AKI in comparison with the normal group (, p < 0.01). Moreover, the Pearson correlation coefficients showed that the expression of miR-34b-5p was positively correlated with that of TNF-α, IL-1β, IL-6, SCr, and BUN in the patients with septic AKI (, p < 0.05).
Figure 1. miR-34b-5p is highly expressed in septic AKI patients. Notes: (A) qRT-PCR detected the expression of miR-34b-5p in the serum of septic AKI patients and healthy volunteers; ELISA detected the serum levels of TNF-α (B), IL-1β (C), IL-6 (D), SCr (E), and BUN (F) in septic AKI patients and healthy volunteers; Pearson correlation coefficient measured the correlation between the expression level of miR-34b-5p and the levels of TNF-α (G), IL-1β (H), IL-6 (I), SCr (J), and BUN (K) in the septic AKI patients. **P < 0.01, compared to the normal group. AKI: acute kidney injury.
miR-34b-5p promotes LPS-induced renal cell apoptosis and inflammatory response
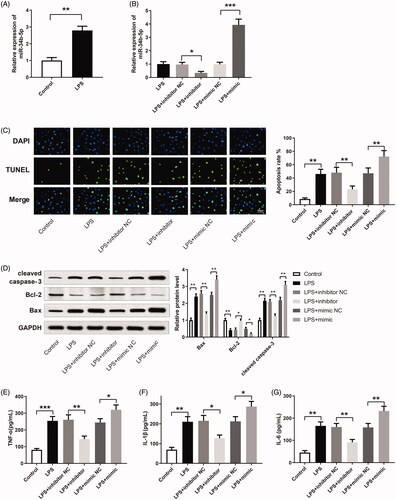
miR-34b-5p was up-regulated in the LPS group compared to the control group (, p < 0.01). After transfection of miR-34b-5p inhibitor or miR-34b-5p mimic, miR-34b-5p was successfully inhibited or overexpressed in LPS-induced HK-2 cells (, p < 0.05). TUNEL staining showed that LPS increased the apoptosis in HK-2 cells (, p < 0.01, vs the control group). The apoptosis rate was decreased in the LPS + inhibitor group while increased in the LPS + mimic group (, p < 0.01, vs the LPS + inhibitor NC group and LPS + mimic NC group respectively). The expression levels of apoptosis-related proteins (Bax, Bcl-2, and cleaved caspase-3) in HK-2 cells were detected by Western blotting. Bax and cleaved caspase-3 were increased while Bcl-2 was decreased in the LPS group (, p < 0.01, vs the control group). The expression levels of Bax and cleaved caspase-3 were down-regulated in the LPS + inhibitor group while up-regulated in the LPS + mimic group (, p < 0.01, vs the LPS + inhibitor NC group and LPS + mimic NC group, respectively). In contrast, Bcl-2 was increased in the LPS + inhibitor group while decreased in the LPS + mimic group (, p < 0.05, vs the LPS + inhibitor NC group and LPS + mimic NC group, respectively). TNF-α, IL-1β, and IL-6 were increased in the LPS group in comparison with the Control group (, p < 0.01). TNF-α, IL-1β, and IL-6 were down-regulated in the LPS + inhibitor group while up-regulated in the LPS + mimic group (, p < 0.05, vs the LPS + inhibitor NC group and LPS + mimic NC group, respectively). The above experiment results reveal that miR-34b-5p promotes LPS-induced apoptosis and inflammatory response in HK-2 cells.
Figure 2. miR-34b-5p promotes LPS-induced apoptosis and inflammatory response in HK-2 cells. Notes: (A) qRT-PCR detected the expression of miR-34b-5p in LPS-induced HK-2 cells; **P < 0.01, compared to the control group. After transfection of miR-34b-5p inhibitor or miR-34b-5p mimic in LPS-induced HK-2 cells, (B) qRT-PCR detected the expression of miR-34b-5p in HK-2 cells; (C) TUNEL staining measured the apoptosis rate of HK-2 cells; (D) Western blotting detected the expressions of Bax, Bcl-2 and cleaved caspase-3 in HK-2 cells; ELISA measured the levels of TNF-α (E), IL-1β (F), and IL-6 (G) in the supernatant of HK-2 cells; scale bar = 50 μm; *P < 0.05, **P < 0.01, ***P < 0.001, compared to the control group, LPS + inhibitor NC group or LPS + mimic NC group. LPS: lipopolysaccharide.
AQP2 suppresses LPS-induced renal cell apoptosis and inflammatory response
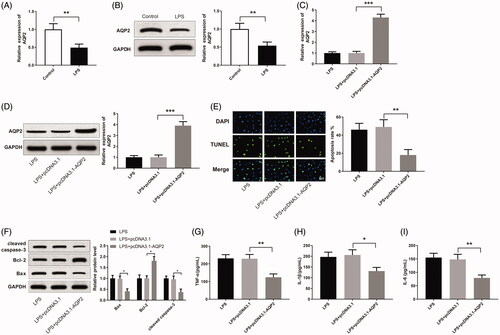
AQP2 was detected to be down-regulated in LPS-induced HK-2 cells in comparison with the cells in the control group (, p < 0.01). AQP2 was up-regulated in LPS-induced HK-2 cells after the transfection of pcDNA3.1-AQP2 (, p < 0.001). The LPS + pcDNA3.1-AQP2 group obtained less apoptotic cells than the LPS + pcDNA3.1 group (, p < 0.01). Furthermore, Bax and cleaved caspase-3 were reduced, and Bcl-2 was increased in the LPS + pcDNA3.1-AQP2 group compared to the LPS + pcDNA3.1 group (, p < 0.05). The expression levels of TNF-α, IL-1β, and IL-6 also declined in the LPS + pcDNA3.1-AQP2 group compared to the LPS + pcDNA3.1 group (, p < 0.05). From the above, AQP2 can inhibit LPS-induced apoptosis and inflammatory response in HK-2 cells.
Figure 3. Overexpressing AQP2 suppresses LPS-induced apoptosis and inflammatory response in HK-2 cells. Notes: qRT-PCR (A) and Western blotting (B) detected the expression of AQP2 in LPS-induced HK-2 cells; **P < 0.01, compared to the control group. After transfection of pCDNA3.1-AQP2 in LPS-induced HK-2 cells, qRT-PCR (C) and Western blotting (D) detected the expression of AQP2 in the cells; (E) TUNEL staining measured the apoptosis rate of HK-2 cells; (F) Western blotting detected the expressions of Bax, Bcl-2 and cleaved caspase-3 in HK-2 cells; ELISA measured the levels of TNF-α (G), IL-1β (H). and IL-6 (I) in the cell supernatant; scale bar = 50 μm; *P < 0.05, **P < 0.01, ***P < 0.001, compared to the LPS + pcDNA3.1 group. LPS: lipopolysaccharide.
miR-34b-5p targets AQP2
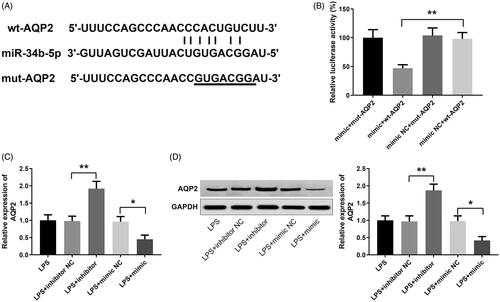
Plasmids inserted with wild and mutated three prime untranslated region (3′-UTR) of AQP2 were constructed for dual-luciferase reporter assay of the binding between miR-34b-5p and AQP2 (). Luciferase activities of the mimic + mut-AQP2 group, mimic NC + mut-AQP2 group, and mimic NC + wt-AQP2 group had no significant differences. The mimic + wt-AQP2 group obtained a significantly reduced luciferase activity (, p < 0.01, vs the mimic NC + wt-AQP2 group). qRT-PCR and Western blotting were applied to detect the expression of AQP2 in LPS-induced HK-2 cells after transfection with miR-34b-5p inhibitor or miR-34b-5p mimic. AQP2 was up-regulated in the LPS + inhibitor group while down-regulated in the LPS + mimic group (, p < 0.05), indicating that miR-34b-5p negatively regulated AQP2 in HK-2 cells.
Figure 4. miR-34b-5p targets AQP2. Notes: (A) 3′UTR of AQP2 where miR-34b-5p binds to; (B) dual-luciferase reporter assay for verifying the targeting relationship between miR-34b-5p and AQP2; **P < 0.01, compared to the mimic NC + wt-AQP2 group. qRT-PCR (C) and Western blotting (D) detected the expression of AQP2 in LPS-induced HK-2 cells after transfection of miR-34b-5p inhibitor or miR-34b-5p mimic; *P < 0.05, **P < 0.01, compared to the LPS + inhibitor NC group or LPS + mimic NC group.
miR-34b-5p mediates LPS-induced renal cell apoptosis and inflammatory response by targeting AQP2
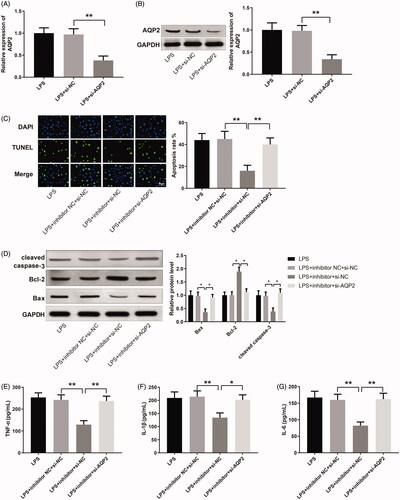
AQP2 was successfully down-regulated in LPS-induced HK-2 cells after the transfection of si-AQP2 (, p < 0.01), which guaranteed the effectiveness of si-AQP2 in later cell transfection for investigation of the miR-34b-5p/AQP2 axis. The apoptosis rate was reduced in the LPS + inhibitor + si-NC group while increased in the LPS + inhibitor + si-AQP2 group (, p < 0.01, vs the LPS + inhibitor NC + si-NC and LPS + inhibitor + si-NC group, respectively). Bax and cleaved caspase-3 were decreased while Bcl-2 was increased in the LPS + inhibitor + si-NC group compared to the LPS + inhibitor NC + si-NC group; inverse expression patterns of these apoptosis-related proteins were detected in the LPS + inhibitor + si-AQP2 group in comparison with the LPS + inhibitor + si-NC group (, p < 0.05). The expression levels of TNF-α, IL-1β and IL-6 were down-regulated in the LPS + inhibitor + si-NC group while up-regulated in the LPS + inhibitor + si-AQP2 group (, p < 0.05, vs the LPS + inhibitor NC + si-NC and LPS + inhibitor + si-NC group, respectively). Taken together, miR-34b-5p inhibits AQP2 to promote LPS-induced apoptosis and inflammatory response in HK-2 cells.
Figure 5. miR-34b-5p mediates LPS-induced apoptosis and inflammatory response in HK-2 cells by targeting AQP2. Notes: qRT-PCR (A) and Western blotting (B) detected the expression of AQP2 in LPS-induced HK-2 cells transfected with si-AQP2. After transfection of miR-34b-5p inhibitor + si-NC or miR-34b-5p inhibitor + si-AQP2 in LPS-induced HK-2 cells, (C) TUNEL staining assessed the cell apoptosis; (D) Western blotting measured the expressions of Bax, Bcl-2, and cleaved caspase-3 in HK-2 cells; ELISA measured the levels of TNF-α (E), IL-1β (F), and IL-6 (G) in the cell supernatant; scale bar = 50 μm; *p < 0.05, **p < 0.01, compared to LPS + inhibitor NC + si-NC group or LPS + inhibitor + si-NC group. LPS: lipopolysaccharide.
Discussion
Sepsis is a clinical syndrome characterized with detrimental host response to infection and may lead to life-threatening AKI when it occurs in the kidney. Diagnosis of septic AKI based on isolated urine output or SCr confers differential risk of the same staged AKI and both the two diagnosis standards have their limitations [Citation20]. Given that, emerging risk scoring systems or serum biomarkers are suggested. Early detection of septic AKI with the complementary assistance of biomarkers is of great value for disease management and recovery [Citation21]. miR-34b-5p, identified as a novel biomarker for septic AKI in this study, can regulate sepsis-induced renal injury by targeting AQP2.
First of all, we measured the level of miR-34b-5p in patients with septic AKI and healthy people. Pro-inflammatory factors (TNF-α, IL-1β, and IL-6), SCr, and BUN were increased in the collected patient serum, confirming sepsis and AKI in these patients. Meanwhile, septic AKI patients showed a higher expression of miR-34b-5p. We then established a LPS-induced cell model of septic AKI to investigate the effect of miR-34b-5p in vitro. LPS treatment increased the expressions of miR-34b-5p, pro-apoptotic proteins, and pro-inflammatory cytokines in HK-2 cells. miR-34b-5p overexpression further aggravated LPS-induced renal cell apoptosis and inflammation. Serval studies have reported the involvement of miR-34b-5p in inflammation and apoptosis. For instance, miR-34b-5p was induced by p53 to block NF-κB-inducing kinase and therefore inhibit noncanonical NF-κB signaling [Citation22]. Overexpression of miR-34b-5p promoted tumor cell apoptosis in serval cancers, including thyroid carcinoma [Citation23], colorectal cancer [Citation24], and oral cancer [Citation25]. Also, miR-34b-5p mediated recurrent seizures-induced apoptosis of hippocampal astrocytes [Citation9]. Nonetheless, the effect of miR-34b-5p in septic AKI remains obscure to date. In contrast to miR-34b-5p, the 3p strand of miR-34b (miR-34b-3p) impeded NF-κB signaling as well as the release of pro-inflammatory cytokines in sepsis-induced AKI [Citation26]. Moreover, miR-34b was detected to alleviate inflammatory response and apoptosis in high glucose-induced HK-2 cells [Citation27]. Considering the above evidence, miR-34b may exert differential effects in septic AKI by different strands and further investigation is needed to confirm the promotive effect of miR-34b-5p in septic AKI.
AQP2 is a functional protein expressed in kidney's collecting duct principal cells and is of great importance for improving renal dysfunction. Firstly, AQP2 is down-regulated in rat models of acute endotoxemia within a short time treatment with LPS [Citation28,Citation29]. Propofol pretreatment prevented AQP2 down-regulation and ameliorated LPS-induced renal injury in a rat model of AKI by inhibiting ICAM-1, TNF-α, and apoptotic proteins [Citation30]. The expression of AQP2 and renal tubular function could also be preserved by α-Lipoic acid in septic AKI, which was achieved by suppression of apoptosis and inflammation [Citation31]. Consistent with the previous findings, AQP2 suppressed LPS-induced sepsis in HK-2 cell models and AQP2 overexpression could attenuate renal injury by inhibiting inflammatory response and apoptosis. However, whether there is an interaction between AQP2 and other regulatory molecules in septic AKI is largely unknown.
The online bioinformatic database predicted the binding site between miR-34b-5p and AQP2, and vectors inserted with wild and mutated 3′UTR of AQP2 were constructed for dual-luciferase reporter assay. The experimental data indicated that miR-34b-5p could bind to the 3′UTR of AQP2 and therefore mediate the expression of AQP2. In cells with over-expressed or under-expressed miR-34b-5p, the expression of AQP2 was subsequently down-regulated or up-regulated. We then conducted experiments to verify the interaction of miR-34b-5p with AQP2 in HK-2 cells. AQP2 silencing reversed the suppression of apoptosis and inflammation by miR-34b-5p inhibitor in LPS-induced HK-2 cells, which was manifested by increased levels of pro-apoptotic and pro-inflammatory molecules.
In summary, miR-34b-5p promotes LPS-induced renal cell injury by inhibiting AQP2. As high expression of miR-34b-5p is detected in septic AKI patients, the serum level of miR-34b-5p may have diagnostic value for sepsis-induced AKI. The findings may also inspire the development of molecule-targeted therapy for septic AKI. Nonetheless, this study lacks animal experiments to confirm the regulation of septic AKI by miR-34b-5p in vivo. More studies are expected to further probe into the potential of miR-34b-5p for septic AKI diagnosis and treatment.
Acknowledgments
Thanks for all the participants and contributors.
Disclosure statement
The authors declare there is no conflict of interest regarding the publication of this paper.
Additional information
Funding
References
- Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus definitions for sepsis and septic shock (sepsis-3). JAMA. 2016;315(8):801–810.
- Sakr Y, Jaschinski U, Wittebole X, et al. Sepsis in intensive care unit patients: worldwide data from the intensive care over nations audit. Open Forum Infect Dis. 2018;5(12):ofy313.
- Taeb AM, Hooper MH, Marik PE. Sepsis: current definition, pathophysiology, diagnosis, and management. Nutr Clin Pract. 2017;32(3):296–308.
- Mehta RL, Cerda J, Burdmann EA, et al. International Society of Nephrology's 0by25 initiative for acute kidney injury (zero preventable deaths by 2025): a human rights case for nephrology. Lancet. 2015;385(9987):2616–2643.
- Zuk A, Bonventre JV. Acute kidney injury. Annu Rev Med. 2016;67:293–307.
- Bellomo R, Kellum JA, Ronco C, et al. Acute kidney injury in sepsis. Intensive Care Med. 2017;43(6):816–828.
- Dellepiane S, Marengo M, Cantaluppi V. Detrimental cross-talk between sepsis and acute kidney injury: new pathogenic mechanisms, early biomarkers and targeted therapies. Crit Care. 2016;20:61.
- Maroof H, Islam F, Ariana A, et al. The roles of microRNA-34b-5p in angiogenesis of thyroid carcinoma. Endocrine. 2017;58(1):153–166.
- Liu L, Liu L, Shi J, et al. MicroRNA-34b mediates hippocampal astrocyte apoptosis in a rat model of recurrent seizures. BMC Neurosci. 2016;17(1):56.
- Qiu N, Xu X, He Y. LncRNA TUG1 alleviates sepsis-induced acute lung injury by targeting miR-34b-5p/GAB1. BMC Pulm Med. 2020;20(1):49.
- Xie W, Lu Q, Wang K, et al. miR-34b-5p inhibition attenuates lung inflammation and apoptosis in an LPS-induced acute lung injury mouse model by targeting progranulin. J Cell Physiol. 2018;233(9):6615–6631.
- Nedvetsky PI, Tamma G, Beulshausen S, et al. Regulation of aquaporin-2 trafficking. Handb Exp Pharmacol. 2009;(190):133–57.
- He J, Yang B. Aquaporins in renal diseases. Int J Mol Sci. 2019;20(2):366.
- Asvapromtada S, Sonoda H, Kinouchi M, et al. Characterization of urinary exosomal release of aquaporin-1 and -2 after renal ischemia-reperfusion in rats. Am J Physiol Renal Physiol. 2018;314(4):F584–F601.
- Gao J, Gu Z, Li M, et al. L-carnitine ameliorates the decrease of aquaporin 2 levels in rats with cisplatin-induced kidney injury. Nephron. 2017;135(4):315–325.
- Hocherl K, Schmidt C, Kurt B, et al. Inhibition of NF-kappaB ameliorates sepsis-induced downregulation of aquaporin-2/V2 receptor expression and acute renal failure in vivo. Am J Physiol Renal Physiol. 2010;298(1):F196–204.
- Rodrigues CE, Sanches TR, Volpini RA, et al. Effects of continuous erythropoietin receptor activator in sepsis-induced acute kidney injury and multi-organ dysfunction. PLoS One. 2012;7(1):e29893.
- Palevsky PM, Liu KD, Brophy PD, et al. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for acute kidney injury. Am J Kidney Dis. 2013;61(5):649–672.
- Levy MM, Fink MP, Marshall JC, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS international sepsis definitions conference. Crit Care Med. 2003;31(4):1250–1256.
- Poston JT, Koyner JL. Sepsis associated acute kidney injury. BMJ. 2019;364:k4891.
- Peerapornratana S, Manrique-Caballero CL, Gomez H, et al. Acute kidney injury from sepsis: current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019;96(5):1083–1099.
- Jang H, Park S, Kim J, et al. The tumor suppressor, p53, negatively regulates non-canonical NF-κB signaling through miRNA induced silencing of NF-κB-inducing kinase . Mol Cells. 2020;43(1):23–33.
- Maroof H, Islam F, Dong L, et al. Liposomal delivery of miR-34b-5p induced cancer cell death in thyroid carcinoma. Cells. 2018;7(12):265.
- Tian J, Cui P, Li Y, et al. LINC02418 promotes colon cancer progression by suppressing apoptosis via interaction with miR-34b-5p/BCL2 axis. Cancer Cell Int. 2020;20:460.
- Hsieh MJ, Lin CW, Su SC, et al. Effects of miR-34b/miR-892a upregulation and inhibition of ABCB1/ABCB4 on melatonin-induced apoptosis in VCR-resistant oral cancer cells. Mol Ther Nucleic Acids. 2020;19:877–889.
- He SY, Wang G, Pei YH, et al. miR-34b-3p protects against acute kidney injury in sepsis mice via targeting ubiquitin-like protein 4A. Kaohsiung J Med Sci. 2020;36(10):871–842.
- Lv N, Li C, Liu X, et al. miR-34b alleviates high glucose-induced inflammation and apoptosis in human HK-2 cells via IL-6R/JAK2/STAT3 signaling pathway. Med Sci Monit. 2019;25:8142–8151.
- Grinevich V, Knepper MA, Verbalis J, et al. Acute endotoxemia in rats induces down-regulation of V2 vasopressin receptors and aquaporin-2 content in the kidney medulla. Kidney Int. 2004;65(1):54–62.
- Olesen ET, de Seigneux S, Wang G, et al. Rapid and segmental specific dysregulation of AQP2, S256-pAQP2 and renal sodium transporters in rats with LPS-induced endotoxaemia. Nephrol Dial Transplant. 2009;24(8):2338–2349.
- Cui WY, Tian AY, Bai T. Protective effects of propofol on endotoxemia-induced acute kidney injury in rats. Clin Exp Pharmacol Physiol. 2011;38(11):747–754.
- Suh SH, Lee KE, Kim IJ, et al. Alpha-lipoic acid attenuates lipopolysaccharide-induced kidney injury. Clin Exp Nephrol. 2015;19(1):82–91.