
Abstract
Renal fibrosis is the inevitable common end-point of all progressive chronic kidney diseases. The underlying mechanisms of renal fibrosis are complex, and currently there is no effective therapy against renal fibrosis. Renal microvascular rarefaction contributes to the progression of renal fibrosis; however, an imbalance between proangiogenic and antiangiogenic factors leads to the loss of renal microvasculature. Vascular endothelial growth factor (VEGF) is the most important pro-angiogenic factor. Recent studies have unraveled the involvement of VEGF in the regulation of renal microvascular rarefaction and fibrosis via various mechanisms; however, it is not clear whether it has anti-fibrotic or pro-fibrotic effect. This paper reviews the available evidence pertaining to the function of VEGF in the fibrotic process and explores the associated underlying mechanisms. Our synthesis will help identify the future research priorities for developing specialized treatments for alleviating or preventing renal fibrosis. Abbreviation: VEGF: vascular endothelial growth factor; CKD: chronic kidney disease; ESKD: end-stage kidney disease; ER: endoplasmic reticulum; VEGFR: vascular endothelial growth factor receptor; AKI: acute kidney injury; EMT: epithelial-to-mesenchymal transition; HIF: hypoxia-inducible factor; α-SMA: α smooth muscle actin; UUO: unilateral ureteral obstruction; TGF-β: transforming growth factor-β; PMT: pericyte-myofibroblast transition; NO: nitric oxide; NOS: nitric oxide synthase; nNOS: neuronal nitric oxide synthase; iNOS: inducible nitric oxide synthase; eNOS: endothelial nitric oxide synthase; sGC: soluble guanylate cyclase; PKG: soluble guanylate cyclase dependent protein kinases; UP R: unfolded protein response
1. Introduction
Chronic kidney disease (CKD) is a global public health concern. Approximately 850 million people currently suffer from different types of kidney diseases worldwide [Citation1]. A vast majority of patients with CKD progress to end-stage kidney disease (ESKD) requiring renal replacement therapy as a life-saving measure, which imposes a great health and economic burden on the affected individuals, families, and the society at large. In 2010, the estimated number of people who were on renal replacement therapy worldwide was 2.5 million, and this number is projected to nearly double by 2030, reaching almost 5.4 million [Citation2].
Renal fibrosis is the inevitable common end-point of all progressive CKDs. Renal damage is closely related to hypoxia and inflammation. Persistent insults cause an imbalance of extracellular matrix homeostasis, and cause sustained activation of fibroblast cells, leading to extensive collagen deposition and scar formation, and ultimately glomerulosclerosis and fibrosis [Citation3]. More recently, renal microvascular rarefaction, lymphangiogenesis, and endoplasmic reticulum stress (ER stress) have been implicated in the pathophysiology of renal fibrosis. Currently, there is no effective cure for renal fibrosis because the pathophysiologic mechanisms are yet to be fully elucidated. Therefore, exploration of measures that can help delay the progress of renal fibrosis is a key research imperative.
Renal microvascular rarefaction can be defined as loss of renal capillaries. Peritubular capillaries are an integral part of the renal microvasculature. Branches of glomerular efferent arterioles consist of a capillary network which supplies oxygen and nutrients to tissues along the proximal and distal tubules [Citation4]. Rarefaction of peritubular capillaries is a characteristic feature of renal fibrosis. A close link between capillary rarefaction and fibrosis has been demonstrated in animal models of diabetic nephropathy, chronic allograft nephropathy, obstructive nephropathy, and anti-glomerular basement membrane glomerulonephritis [Citation5–8]. These findings strongly suggest the involvement of peritubular capillaries rarefaction in fibrosis progression. Exposure of kidney tissue to pathogenic factors disturbs the blood flow in peritubular capillaries, resulting in decreased perfusion in the adjacent renal interstitial region, which may lead to a loss of capillaries and renal fibrosis. The consequent hypoxia promotes the infiltration of inflammatory cells and the deposition of extracellular matrix, inducing renal fibrosis. The most dominant mechanism of renal microvascular rarefaction is believed to involve an imbalance between proangiogenic and anti-angiogenic factors [Citation9]; for example, upregulation of antiangiogenic factors thrombospondin-1 and endothelin, and downregulation of angiogenic factors such as vascular endothelial growth factor (VEGF) and angiogenin. Among these, VEGF is the most important regulator of angiogenesis. The role of VEGF in kidney diseases has attracted increasing attention over the years, because in addition to regulation of angiogenesis, it also plays a role in the progression of renal fibrosis ( and ).
Figure 1. Pro-fibrotic effects of VEGF in chronic kidney disease. VEGF promotes the expression of VCAM-1, ICAM-1, and E-selectin through activation of the NF-κB signaling pathway. Persistent ER stress can initiate CHOP and caspase-12 apoptosis signaling pathway via three UPR branches: PERK-eIF2α-ATF4, IRE1, and ATF6 signaling pathways, eventually resulting in renal fibrosis. In turn, PERK also stimulates VEGF/VEGFR system. VEGF synergizes with PTHrP, TGF-β, and EGF in activating ERK1/2, thus promoting EMT that is related to renal fibrosis. (Abbreviations: VEGF, vascular endothelial growth factor; VCAM-1, vascular cell adhesion molecule-1; ICAM-1, intercellular adhesion molecule-1; ER, endoplasmic reticulum; UPR, unfolded protein response; VEGFR, vascular endothelial growth factor receptor; PTHrP, parathyroid hormone-related protein; TGF-β, transforming growth factor-β; EGF, epidermal growth factor; EMT, epithelial-to-mesenchymal transition; ZO-1, zonula occludens-1; α-SMA, α smooth muscle actin).
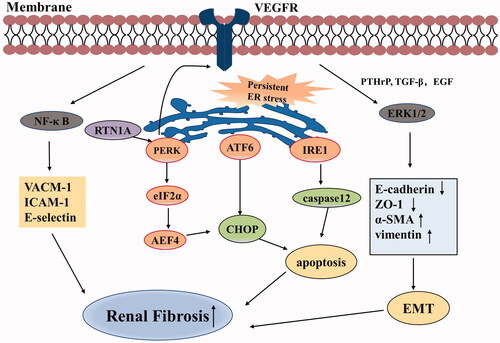
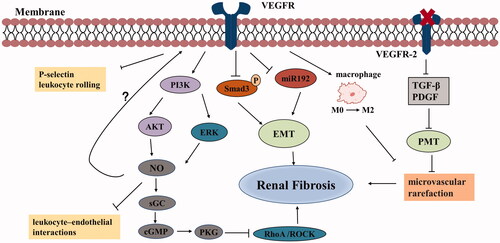
Figure 2. Anti-fibrotic effects of VEGF in chronic kidney disease. VEGF can inhibit the expression of Smad3 and miR192, thereby suppressing TGF-β-induced EMT and improving renal fibrosis. VEGF stimulates NO production by PI3K/AKT and ERK pathways. NO activates sGC to synthesize cGMP, which subsequently activates PKG. PKG inhibits RhoA/ROCK pathway, thus reducing fibrosis. VEGF induces a phenotypic shift of macrophages from M0 to M2, ameliorates fibrosis and vascular rarefaction. Inhibition of VEGFR-2 can block PMT, leading to improve microvascular rarefaction and fibrosis. VEGF can also suppress the expression of inflammatory mediators. (Abbreviations: VEGF: vascular endothelial growth factor; TGF-β: transforming growth factor-β; EMT: epithelial-to-mesenchymal transition; NO: nitric oxide; sGC: soluble guanylate cyclase; PKG: cGMP dependent protein kinases; VEGFR: vascular endothelial growth factor receptor; PMT: pericyte-myofibroblast transition; PDGF: platelet-derived growth factor).
2. VEGF function
VEGF is a family of secreted polypeptides, which includes VEGF-A, -B, -C, -D and placental growth factor in humans. The biological role of VEGF-B is yet to be defined. VEGF-C and VEGF-D mainly regulate lymphangiogenesis, while placental growth factor is believed to induce peripheral arteriogenesis. VEGF-A, also named VEGF, participates in embryonal angiogenesis and is a key mediator that promotes vasculogenesis, and angiogenic remodeling [Citation10]. VEGF is expressed as multiple isoforms (VEGF121, VEGF165, VEGF189, and VEGF206) showing different biological activities [Citation11]. This paper focuses on the function of VEGF (VEGF-A) in renal microvascular rarefaction and fibrosis. In the normal kidney, VEGF is constitutively localized to glomerular podocytes and thick ascending limbs of Henle’s loop with lower levels of expression in the proximal and distal tubules [Citation12,Citation13]. It exerts its specific effects on angiogenesis, vasodilation, and vascular permeability via the activation of the tyrosine kinase receptors: VEGF receptor 1 (VEGFR-1) and VEGF receptor 2 (VEGFR-2). Firstly, it is well known that VEGF plays a pivotal role in renal microcirculation. Basile’s team reported that endothelial mesenchymal transition may cause interstitial deposition and vascular dropout after acute kidney injury (AKI). VEGF-121 treatment was found to contribute to the maintenance of the vascular integrity but it did not enhance the number of endothelial cells; the likely reason was that endothelial mesenchymal transition blockade by VEGF protected vascular rarefaction following AKI [Citation14]. Chade and colleagues demonstrated the vasculoprotective effect of VEGF in chronic renal ischemia. In renovascular disease, microvascular loss of the stenotic kidney was driven by VEGF reduction. Intra-renal administration of VEGF stimulated vascular proliferation and maturation, promoted endothelial cell migration and preserved microvascular density [Citation15]. Secondly, VEGF signaling was mainly mediated through VEGFR-2 located on glomerular endothelial cells. Reports have also suggested that VEGF may exert its effects via autocrine mechanism [Citation16], but Quaggin et al. discovered that podocytes did not secrete VEGFR. A whole-body knockout of VEGFR-2 caused severe vascular and glomerular injury, while podocyte-specific loss of VEGFR-2 did not impair glomerular development and functioning. This suggested that VEGF produced by podocytes predominantly bound to VEGFR-2 via the paracrine pathway, thus ensuring the normal function of the glomerular filtration barrier [Citation17,Citation18]. VEGF expression in renal tubular cells was found to be essential for maintaining peritubular capillaries. Mice with selective deletion of VEGF-A in renal tubules exhibited a small but histologically normal kidney, followed by an obvious decrease in peritubular capillaries density [Citation13]. As reported, VEGF produced by podocytes may collaborate with platelet-derived growth factor to recruit mesangial cells [Citation19]. VEGF secretion from podocytes was decreased, which resulted in the absence of the endothelial cells and mesangial cells and the destruction of glomerular structure soon after birth; subsequently, these mice died of severe renal failure at the age of 3 weeks. This research indicated that VEGF also has a protective effect on mesangial survival [Citation20]. In addition, there are several studies of VEGF in the context of diabetic nephropathy. VEGF exhibited different expression levels in animal models and humans. Reduced VEGF levels were observed in kidney biopsy specimens from diabetic patients, which were shown to be associated with the loss of podocytes [Citation21]. However, VEGF expression was increased in early diabetic nephropathy in animal models [Citation22]. This discrepancy may be attributable to different stages of the disease. Renal pathological changes were often observed in the early stage of diabetic nephropathy animal model, but most patients who underwent renal biopsy were in an advanced stage due to the insidious nature of the disease. A genetics-based study revealed that reduced VEGF expression in podocytes aggravated high-glucose-mediated kidney impairment through accelerated endothelial cell apoptosis, thereby resulting in massive albuminuria and glomerulosclerosis [Citation23]. The balance of VEGF is important, and both down- or over-expression can lead to different types of renal diseases [Citation17].
VEGF expression showed a strong correlation with renal fibrosis and the loss of glomerular and peritubular capillaries during progression of CKD. In particular, reduced VEGF in the outer medulla and medullary rays of the aging kidney was found to be linked to rarefaction of peritubular capillaries, but VEGF was increased in focal areas of the cortex, which was considered as a compensatory response to cortical hypoperfusion [Citation24]. In a study, supplementation of VEGF in the remnant kidney model alleviated vascular injury and fibrosis [Citation25]. In another study, Gremlin (a family of bone morphogenetic protein antagonists) was found to promote renal tubulointerstitial fibrosis by upregulating epithelial-to-mesenchymal transition (EMT), and VEGFR-2 was implicated in this process [Citation26]. Hence, it is not clear whether the presence of VEGF is beneficial in the context of renal fibrosis. In this review article, we summarize the role of VEGF in kidney diseases, with the objective to offer new insights into the potential therapeutic strategies for CKD patients.
3. Role of VEGF in renal fibrosis
3.1. VEGF and hypoxia
Hypoxia is one of the main mechanisms leading to renal fibrosis, usually secondary to renal microvascular rarefaction. Hypoxia-inducible factor (HIF) is the master transcription factor for cellular adaptation to hypoxia, consisting of two isoforms HIF-1α and HIF-2α [Citation27]. Exposure of renal tissue to hypoxia induces activation of HIF for self-repair. Angiogenesis is a key step in the repair process. Studies have shown that hypoxia activates HIF-VEGF signaling and thereby mediates renal angiogenesis [Citation28]. Hypoxia promotes the expression of VEGF through HIF-1α [Citation29], while hyperoxia inhibits it [Citation30].
HIF was shown to upregulate VEGF expression and consequently inhibit hypoxia-mediated myocardial injury [Citation31]. Of note, although hypoxia response and VEGF signaling pathways were activated in chronic renal fibrosis, the expression of VEGF was attenuated [Citation5,Citation32]. Hypoxia-induced VEGF production appears to require multiple regulatory factors; possibly, the positive regulators of VEGF (insulin-like growth factor and epidermal growth factor) were significantly downregulated in stable and progressive renal disease, causing reduction of VEGF [Citation32]. In the study by Lindenmeyer et al., renal tubular epithelium atrophy and capillary repair defect induced lower VEGF expression in diabetic nephropathy [Citation5]. In particular, some studies suggest that VEGF exerts pro-inflammatory effect, in addition to its pro-angiogenic effect; thus the levels of VEGF reduced presumably at the expense of hypoxia in the setting of severe inflammation hit on the kidney [Citation33].
3.2. VEGF and EMT
EMT, whereby renal tubular epithelial cells attain mesenchymal phenotype via a specific transforming program, is a key process in renal fibrosis and it is the essential source of myofibroblasts in kidneys. In this process, some epithelial markers are down-regulated such as E-cadherin and zonula occludens-1, and the mesenchymal markers, such as N-cadherin, α smooth muscle actin (α-SMA) and vimentin, are increased. Lately, many studies have identified the role of VEGF and EMT in renal fibrosis, but they showed different perspectives, and often contradictory results. In a study, administration of VEGF to unilateral ureteral obstruction (UUO) model mice led to a significant decrease in connective tissue growth factor and transforming growth factor-β (TGF-β) levels and significant increase in E-cadherin level on days 3 and 7. Thus VEGF was found to alleviate UUO-induced fibrosis by suppressing EMT in early renal disease [Citation34]. The inhibitory effect of VEGF on EMT may be explained by the inhibitory effect of VEGF on the expression and phosphorylation of Smad3, which suppresses TGF-β-induced EMT and improves renal fibrosis. Moreover, VEGF can also inhibit EMT via miR192 [Citation35]. However, other studies suggest that VEGF may contribute to fibrosis via promoting the EMT process. PI3K/AKT pathway had been reported to regulate EMT of tubular epithelial cells and fibrosis [Citation36]. A study in models of allergic airway disease demonstrated that inhibition of VEGF reduced TGF-β levels by suppressing activation of PI3K/AKT, thus preventing airway epithelium fibrosis [Citation37]. Treatment of diabetic retinopathy mice with calcium dobesilate (an effective drug for diabetic microangiopathy) led to attenuation of glomerular hypertrophy and tubulointerstitial fibrosis. The protective effect of calcium dobesilate in diabetic nephropathy was shown to be mediated through downregulating the expression of VEGF and VEGFR-2 and suppressing downstream signaling PIK3/AKT/mTOR [Citation38]. Therefore, VEGF may induce EMT via the PI3K/AKT pathway. Bone morphogenetic protein-7 belongs to the TGF-β superfamily and it can reverse EMT and exhibit a renal anti-fibrotic effect [Citation39]. Gremlin is a bone morphogenetic protein antagonist. In a study by Marquez-Exposito et al., binding of gremlin to VEGFR-2 was found to decrease E-cadherin expression and increase α-SMA and vimentin. In addition, treatment with VEGFR-2 inhibitor prominently reduced collagen deposition and ameliorated renal fibrosis in UUO [Citation26]. In fetal RPE cells, gremlin-1 was shown to promote SNAI1 (a key transcription factor for initiating EMT) and TGF-β expression, increase Smad2 phosphorylation, and induce the EMT process [Citation40]; however, the mechanisms in tubular cells is yet to be elucidated. In a study on nasopharyngeal carcinoma, VEGF was found to induce EMT via VEGFR-2 and ERK1/2 activation, which contributed to invasion and metastasis of cancer cells [Citation41]. VEGF induced the loss of zonula occludens-1 and upregulation of α-SMA in tubuloepithelial cells, indicating that it could facilitate EMT. It was perhaps relevant that VEGF synergized with parathyroid hormone-related protein, TGF-β, and epidermal growth factor in activating ERK1/2 [Citation42]. Due to these conflicting findings, further studies are required to elucidate the mechanism by which VEGF regulates EMT during renal fibrosis.
Myofibroblasts are the major secretors of collagen, driving progressive fibrosis. Besides EMT, other sources of myofibroblasts include fibroblasts, pericytes, and bone marrow-derived cells [Citation43]. Especially pericytes, a field of intense research, are identified as precursors for myofibroblasts. Pericytes, which are inserted into capillary endothelium, can help maintain vascular integrity. In pathological conditions, pericytes may detach from the endothelial cells and differentiate into myofibroblasts, resulting in microvascular rarefaction and kidney damage, a process known as pericyte-myofibroblast transition (PMT) [Citation44]. Thus, PMT are also one of the major sources of myofibroblasts. In a study by Lin et al., in UUO rats, blocking VEGFR-2 signaling by circulating soluble receptor ectodomains not only attenuated PMT, but also induced marked peritubular capillaries rarefaction and fibrosis [Citation45]. Surprisingly, the authors found that the secretion of VEGF164-mediated angiogenesis was reduced in UUO, but that of VEGF120/188 (which induces abnormal angiogenesis) was increased. It can be assumed that VEGF participates in PMT through transformation into different subtypes.
3.3. VEGF and NO
Nitric oxide (NO) is a soluble gas produced by endothelial cells, which has widespread effects in human body. Within endothelial cells, NO is synthesized by nitric oxide synthase (NOS) using L-arginine, oxygen, and NADPH. There are three common isoforms of NOS: neuronal nitric oxide synthase (nNOS), inducible nitric oxide synthase (iNOS), and endothelial nitric oxide synthase (eNOS) [Citation46]. NO causes vasodilation, regulates vascular blood flow and pressure, and plays a significant role in angiogenesis. In addition, NO also contributes to the inhibition of platelet aggregation and adhesion, leukocyte activation and infiltration of inflammatory mediators, thereby playing an important role in immune defense [Citation47]. As outlined previously, VEGF is also a potent pro-angiogenic factor. NO and VEGF are closely related. In one study, NO blockade was found to reduce VEGF expression in the tubules; this suggested that VEGF-induced angiogenesis may require complete NO system [Citation48]. About the cross-regulation between the two, NO was suggested as a downstream effector of VEGF; for example, Shashar et al. reported that VEGF activated VEGFR in human umbilical vein endothelial cells, which promoted the production of NO [Citation49]. However, according to Bussolati et al., VEGF binds to VEGFR-1 and facilitates NO synthesis; moreover, NO may negatively modulate VEGFR-2-induced endothelial proliferation [Citation50]. NO was also found to regulate VEGF via autocrine or paracrine mechanisms in vascular smooth muscle cells, macrophages, keratinocytes, and tumor cells. In some studies, NO was found to promote VEGF generation via positive feedback mechanism [Citation51]. Overall, the mechanism by which NO modulates VEGF is not clear. In the kidney, VEGF was shown to stimulate eNOS phosphorylation by IRS-1/PI3K/AKT and ERK pathways, thereby inducing NO production [Citation52].
Recent studies have demonstrated the potential involvement of NO in the process of renal interstitial fibrosis. At the molecular level, NO activates soluble guanylate cyclase (sGC) to synthesize cGMP, which subsequently activates cGMP dependent protein kinases (PKG). PKG inhibits RhoA/ROCK pathway which is associated with renal fibrosis [Citation53]. NO was shown to prevent endothelin activation and to alleviate collagen deposition and renal fibrosis [Citation54]. In a diabetic nephropathic rat model, treatment with NO down-regulated the expressions of TGF-β and fibronectin, and restored Wnt5a expression and β-catenins; these findings suggest that NO may delay interstitial fibrosis in patients with diabetic nephropathy [Citation55]. Similarly, in aristolochic acid nephropathy [Citation56] and UUO rat [Citation57] models, L-arg (an NO donor) supplementation increased VEGF concentration and alleviated renal inflammatory response and fibrosis. Conversely, administration of NOS inhibitor (asymmetric dimethylarginine, N-nitro-l-arginine methyl ester) caused capillary damage and accelerated renal interstitial fibrosis [Citation57,Citation58]. These results appear to suggest that VEGF works in concert with NO to prevent renal fibrosis. However, opposite results were reported in a rat model of chronic cyclosporine nephrotoxicity, where VEGF promoted cyclosporin-mediated renal fibrosis [Citation59]. VEGF and VEGF receptors have been shown to be upregulated in chronic cyclosporine nephrotoxicity rats [Citation60], but NO was found to retard the progression of renal fibrosis via blocking TGF-β and degrading extracellular matrix [Citation61]. Interestingly, VEGF expression was downregulated in the cyclosporine nephrotoxicity model after administration of L-arg, which indicated that VEGF may also have an independent role in cyclosporine-induced fibrosis [Citation62]. Reduced NO bioavailability by high glucose was found to reduce VEGF overexpression in diabetic nephropathy. Increased VEGF initiated the growth and proliferation of endothelial cells and mesangial cells, ultimately resulting in the extracellular matrix accumulation, ultrafiltration, and albuminuria [Citation63]. Other studies have also demonstrated the critical involvement of VEGF and NO in inducing tissue fibrosis in other organs such as heart and lungs. VEGF and NO were demonstrated to be key profibrotic mediators in bleomycin-induced pulmonary fibrosis [Citation64]. However, Chelo et al. reported the cardioprotective effects of NO and VEGF [Citation65]. The current evidence suggests a complex interaction between NO and VEGF, and that these may play different roles in different tissues.
3.4. VEGF and inflammatory response
Inflammatory response plays a central role in the occurrence and progression of renal fibrosis. Multiple inflammatory cells (such as T cells, macrophages, neutrophils) and inflammatory factors (interleukin-6, interleukin-8, monocyte chemotactic protein-1) are involved in inflammation, and VEGF is related to both fibrosis and inflammation.
VEGF is chemotactic for macrophages, and at the same time macrophages stimulate the proliferation of myofibroblasts during the fibrosis process. Increased levels of VEGF and macrophages are found in chronic kidney graft rejection, and we hypothesize that VEGF is associated with inflammation and the process of renal fibrosis [Citation66]. Available evidence suggests that VEGF acts as both a proinflammatory cytokine and an anti-inflammatory myokine. In a study, human umbilical cord mesenchymal stem cells were shown to secrete anti-inflammatory factors including VEGF via paracrine mechanism to inhibit inflammation and fibrosis [Citation67]. In a study by Jason et al., a construct of VEGF and elastin-like polypeptide was shown to induce a phenotypic shift of macrophages from M0 to M2 (an angiogenic phenotype), ameliorate interstitial fibrosis and vascular rarefaction, eventually slowing the progression of renal vascular disease to CKD [Citation68]. VEGF was shown to attenuate inflammation-mediated leukocyte–endothelial interactions through its downstream mediator NO [Citation69]. Blockade of VEGF induced an increase in P-selectin expression and leukocyte rolling [Citation70]. These findings point toward an anti-inflammatory role for VEGF. On the other hand, VEGF was found to exacerbate fibrotic deposition by facilitation of extravasation of macrophages to the area of injury [Citation71]. During inflammation, VEGF was found to stimulate the expression of vascular cell adhesion molecule-1, intercellular adhesion molecule-1, and E-selectin through activation of the NF-κB signaling pathway [Citation72]. VEGF inhibitors were found to downregulate the levels of glomerular macrophages and TNF-α [Citation73].
During progression of CKD, microvascular rarefaction is frequently accompanied by lymphangiogenesis. The role of lymphangiogenesis in renal fibrosis is also being explored in recent years. Chronic inflammation triggers the proliferation of lymphatic vessels and VEGF is the best characterized factor that regulates lymphangiogenesis. Lymphangiogenesis has been demonstrated to play a protective role in cardiac disease [Citation74]; however, whether lymphangiogenesis exacerbates or retards the progression of kidney disease is not clear. In a study, mice were administered recombinant human VEGF-C after UUO, and the proliferation of lymphatic vessels was observed; after 14 days, there was significant reduction in the expressions of various inflammatory factors and cells (including TFG-β and macrophages) along with suppression of renal interstitial fibrosis [Citation75]. This study suggested that VEGF-C-induced lymphangiogenesis may potentially have a beneficial effect on renal outcomes. In particular, SAR131675 (a selective VEGFR-3-tyrosine kinase inhibitor) was found to inhibit cell apoptosis, oxidative stress, and inflammatory cell infiltration by inhibition of lymphangiogenesis, resulting in decreased fibrosis in diabetic mice [Citation76]. Similar results were obtained in UUO mice. VEGF-C blockade reduced lymphatic vessel growth, but improved renal injury and fibrosis [Citation77]. These two studies suggest that VEGF-C may promote the proliferation of lymphatic vessels and development of renal fibrosis. There is insufficient evidence of the beneficial effect of lymphangiogenesis in the context of kidney diseases.
3.5. VEGF and ER stress
Endoplasmic reticulum (ER) is an important organelle in eukaryotic cells involved in the synthesis, folding, and modification of secretory proteins. In various pathological conditions, there is accumulation of unfolded proteins in the ER, which induces ER stress [Citation78]. Unfolded protein response (UPR) is recognized as an adaptive response to degrade the unfolded proteins accumulated in the ER and promote tissue regeneration through three branches: PERK-eIF2α-ATF4, IRE1-XBP1, and ATF6 signaling pathways [Citation79]. In general, ER maintains protein homeostasis by UPR, but in the setting of prolonged and severe ER stress, UPR activates the apoptotic pathway to eliminate dysfunctional cells and tissues [Citation80]. Some studies have demonstrated the mechanism by which ER stress participates in various renal fibrosis processes, such as AKI, CKD, and AKI to CKD transition. Studies suggest that ER stress may play different roles in AKI and CKD [Citation81]. On one hand, transient ER stress was found to trigger adaptive UPR to protect kidney in AKI. On the other hand, upon exposure to persistent ER stress, UPR was found to initiate apoptotic pathways, inducing cell apoptosis and fibrosis in CKD. CHOP (downstream of the PERK and ATF6 pathway) and caspase-12 (downstream of IRE1α) are well recognized as robust signals of apoptosis. UUO was shown to activate persistent ER stress, which triggered the CHOP and caspase-12 apoptosis signaling pathway, eventually resulting in renal fibrosis [Citation82]. Knockdown of RTN1A expression in the UUO model inhibited cell apoptosis, and alleviated ER stress and renal fibrosis [Citation83]. This showed that RTN1A enhanced tubular cell apoptosis via interacting with PERK-CHOP signaling.
Nicolas assessed the expression of VEGF in a rat model of acute ischemic stress after activation of UPR and ER stress [Citation84], which unraveled the role of VEGF in UPR/ER stress. VEGF activated three UPR signaling pathways, but mainly stimulated PERK and ATF6 to promote cell survival and angiogenesis [Citation85]. In turn, PERK also stimulated VEGF/VEGFR system [Citation81]. UPR stimulated the expression of VEGF by ATF4 branch in human umbilical vein endothelial cells [Citation86]. VEGF antagonism showed an important protective function in neurons by suppressing ER stress-mediated caspase12 [Citation87]. A previous study on human retinal pigment epithelium showed that bevacizumab (an anti-VEGF drug) may block ER stress and reduce CHOP expression [Citation88]. In summary, ER stress has been shown to be associated with renal fibrosis, and VEGF and ER stress interact with each other. Therefore, we speculate that VEGF plays an important role during renal fibrosis via modulating ER stress signals.
4. VEGF-targeted therapy
VEGF is the most potent angiogenic signaling factor in the body. VEGF-targeted therapy has been commonly applied against tumors and ocular diseases. In the field of clinical oncology, bevacizumab, the first approved anti-angiogenesis agent by FDA, is an anti-VEGF monoclonal antibody. Currently, it is widely used in the treatment of ovarian cancer and colorectal cancer, but in the practical application, scientists found that it may cause thrombotic microangiopathy, triggering nephrotoxicity [Citation89,Citation90]. Furthermore, intravitreal injection of VEGF inhibitors was found to induce deterioration in renal function and uncontrolled hypertension [Citation91]. In general, drugs targeting the VEGF block its downstream signaling pathway, leading to kidney injury. Patients present with hypertension, proteinuria, renal impairment, and electrolyte disorders, among which, hypertension and proteinuria are the most frequent. The exact pathological mechanisms underlying proteinuria is yet to be unraveled, but there have been several hypotheses. Firstly, as discussed, podocyte-derived VEGF is critical for the functional integrity of the glomerular filtration barrier. Genetic knockout of VEGF, or pharmacological blockade induces the loss of endothelial fenestrations in glomerular capillaries leading to proteinuria [Citation92], and the reason is related to nephrin expression downregulation, which is an important cytoskeletal protein of glomerular slit diaphragm [Citation93]. Secondly, VEGF inhibitors induce the disruption of autophagy and podocyte loss via antagonizing PI3K/AKT/mTOR pathway [Citation94]. Some hypertensive patients receiving VEGF-targeted therapy were also accompanied by proteinuria. However, it was unknown whether hypertension was the main trigger of proteinuria or whether both of these represented toxic effects of VEGF inhibitors. Occurrence of hypertension was proposed to be related to NO. VEGF inhibitors block vasodilator (NO) production and increased vascular resistance; NO can inhibit sodium reabsorption in renal tubule. As a result, reduction of NO can also cause fluid accumulation, leading to hypertension [Citation95]. After VEGF-targeted therapies, the endothelin serum levels were upregulated. This might be linked to hypertension because endothelin can trigger the constriction of vessels [Citation96]. Interestingly, VEGF inhibitors were found to activate NADPH-oxidase to produce superoxide [Citation97], further decreasing NO expression, and superoxide has a detrimental effect on the glomerulus.
Bioengineering fusion technique is an emerging therapy in medicine. ELP-VEGF construct was found to improve renal function, ameliorate capillary rarefaction and fibrosis in a porcine model of renal artery stenosis [Citation68]. Supplementation of VEGF had a potent effect in alleviating microvascular damage in thrombotic microangiopathy [Citation98], post-cyclosporine-mediated nephropathy [Citation99], and remnant kidney [Citation25]. Surprisingly, exogenous administration of VEGF relieved partial tubulointerstitial damage in the remnant kidney model, but glomerulosclerosis was not remarkably affected [Citation25]. Supplementation of VEGF following ischemic injury effectively protected renal microvascular structure and prevented its progression to CKD, but the protective effect was lost at a later stage [Citation100]. In conclusion, VEGF appears to have renoprotective effects. Nevertheless, some studies have yielded contradictory evidence. Exacerbation of microvascular rarefaction and renal injury was observed after VEGF supplementation in neonatal UUO [Citation101]. Overexpression of VEGF caused nodular glomerulosclerosis in eNOS null mice and type 1 diabetes [Citation102,Citation103]. Thus, it is a distinct possibility that VEGF may be beneficial at a particular concentration. At early stages of diabetic nephropathy, VEGF activity was elevated, and administration of VEGF inhibitor was found to block disease progression [Citation104]. However, VEGF is responsible for maintenance of vascular homeostasis, and excessive inhibition may produce nonfunctional or leaky vessels, further aggravating inflammation and fibrosis [Citation105]. The available evidence suggests complex nature of VEGF function. It exerts different effects in different diseases and/or at different concentrations. Thus, further in-depth studies are required to address these issues.
5. Conclusion
VEGF plays a role in renal microcirculation and fibrosis through the regulation of EMT, NO metabolism, inflammation, and ER stress. It represents a critical target for the treatment of renal fibrosis. Based on the available evidence, it is debatable whether VEGF exacerbates or inhibits the process of renal fibrosis. Excessive blockade or promotion of the level of VEGF may be advantageous or disadvantageous. This implies that the effect of VEGF is possibly dependent on the type of disease models, the specific area or cell type within the kidney, disease stage, timing of VEGF administration, and even its concentration. Additionally, HIF responds to ischemia/hypoxia by driving activation of VEGF. Roxadustat, an HIF stabilizer, can improve renal tubular injury and reduce the release of inflammatory cells, suggesting that it inhibits inflammation by inducing VEGF-driven angiogenesis. This indicates that upstream regulatory signals affect VEGF functions [Citation106
]. Therefore, more rigorous large-scale clinical studies are required for better characterization of the role of VEGF in renal fibrosis.
Abbreviation
Author contributions
Changxiu Miao: drafted the manuscript; Yujun Du: revised the manuscript; Xiaoyu Zhu and Xuejiao Wei: contributed to the conception and design of the study; Mengtuan Long designed the figure; Lili Jiang, Chenhao Li and Die Jin: organized the database. All authors have read and agreed to the published version of the manuscript.
Disclosure statement
The authors declare no conflict of interest for this study.
Additional information
Funding
References
- Jager KJ, Kovesdy C, Langham R, et al. A single number for advocacy and communication-worldwide more than 850 million individuals have kidney diseases. Kidney Int. 2019;96(5):881–1050.
- Liyanage T, Ninomiya T, Jha V, et al. Worldwide access to treatment for end-stage kidney disease: a systematic review. Lancet. 2015;385(9981):1975–1982.
- Farris AB, Colvin RB. Renal interstitial fibrosis: mechanisms and evaluation. Curr Opin Nephrol Hypertens. 2012;21(3):289–300.
- Guerci P, Ergin B, Ince C. The macro- and microcirculation of the kidney. Best Pract Res Clin Anaesthesiol. 2017;31(3):315–329.
- Lindenmeyer MT, Kretzler M, Boucherot A, et al. Interstitial vascular rarefaction and reduced VEGF-A expression in human diabetic nephropathy. J Am Soc Nephrol. 2007;18(6):1765–1776.
- Ohashi R, Kitamura H, Yamanaka N. Peritubular capillary injury during the progression of experimental glomerulonephritis in rats. JASN. 2000;11(1):47–56.
- Ohashi R, Shimizu A, Masuda Y, et al. Peritubular capillary regression during the progression of experimental obstructive nephropathy. J Am Soc Nephrol. 2002;13(7):1795–1805.
- Ishii Y, Sawada T, Kubota K, et al. Injury and progressive loss of peritubular capillaries in the development of chronic allograft nephropathy. Kidney Int. 2005;67(1):321–332.
- Kida Y, Tchao BN, Yamaguchi I. Peritubular capillary rarefaction: a new therapeutic target in chronic kidney disease. Pediatr Nephrol. 2014;29(3):333–342.
- Tammela T, Enholm B, Alitalo K, et al. The biology of vascular endothelial growth factors. Cardiovasc Res. 2005;65(3):550–563.
- Park JE, Keller GA, Ferrara N. The vascular endothelial growth factor (VEGF) isoforms: differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell. 1993;4(12):1317–1326.
- Brown LF, Berse B, Tognazzi K, et al. Vascular permeability factor mRNA and protein expression in human kidney. Kidney Int. 1992;42(6):1457–1461.
- Dimke H, Sparks MA, Thomson BR, et al. Tubulovascular cross-talk by vascular endothelial growth factor A maintains peritubular microvasculature in kidney. J Am Soc Nephrol. 2015;26(5):1027–1038.
- Basile DP, Friedrich JL, Spahic J, et al. Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am J Physiol Renal Physiol. 2011;300(3):F721–33.
- Iliescu R, Fernandez SR, Kelsen S, et al. Role of renal microcirculation in experimental renovascular disease. Nephrol Dial Transplant. 2010;25(4):1079–1087.
- Hohenstein B, Colin M, Foellmer C, et al. Autocrine VEGF-VEGF-R loop on podocytes during glomerulonephritis in humans. Nephrol Dial Transplant. 2010;25(10):3170–3180.
- Eremina V, Quaggin SE. The role of VEGF-A in glomerular development and function. Curr Opin Nephrol Hypertens. 2004;13(1):9–15.
- Sison K, Eremina V, Baelde H, et al. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol. 2010;21(10):1691–1701.
- Vaughan MR, Quaggin SE. How do mesangial and endothelial cells form the glomerular tuft? J Am Soc Nephrol. 2008;19(1):24–33.
- Eremina V, Cui S, Gerber H, et al. Vascular endothelial growth factor a signaling in the podocyte-endothelial compartment is required for mesangial cell migration and survival. J Am Soc Nephrol. 2006;17(3):724–735.
- Baelde HJ, Eikmans M, Doran PP, et al. Gene expression profiling in glomeruli from human kidneys with diabetic nephropathy. Am J Kidney Dis. 2004;43(4):636–650.
- Cooper ME, Vranes D, Youssef S, et al. Increased renal expression of vascular endothelial growth factor (VEGF) and its receptor VEGFR-2 in experimental diabetes. Diabetes. 1999;48(11):2229–2239.
- Sivaskandarajah GA, Jeansson M, Maezawa Y, et al. Vegfa protects the glomerular microvasculature in diabetes. Diabetes. 2012;61(11):2958–2966.
- Kang DH, Anderson S, Kim YG, et al. Impaired angiogenesis in the aging kidney: vascular endothelial growth factor and thrombospondin-1 in renal disease. Am J Kidney Dis. 2001;37(3):601–611.
- Kang DH, Hughes J, Mazzali M, et al. Impaired angiogenesis in the remnant kidney model: II. Vascular endothelial growth factor administration reduces renal fibrosis and stabilizes renal function. JASN. 2001;12(7):1448–1457.
- Marquez-Exposito L, Lavoz C, Rodrigues-Diez RR, et al. Gremlin regulates tubular epithelial to mesenchymal transition via VEGFR2: Potential role in renal fibrosis. Front Pharmacol. 2018;9:1195.
- Rosenberger C, Mandriota S, Jürgensen JS, et al. Expression of hypoxia-inducible factor-1 alpha and -2alpha in hypoxic and ischemic rat kidneys. J Am Soc Nephrol. 2002;13(7):1721–1732.
- Xu Y, Kong X, Li J, et al. Mild hypoxia enhances the expression of HIF and VEGF and triggers the response to injury in rat kidneys. Front Physiol. 2021;12:690496.
- Liu LX, Lu H, Luo Y, et al. Stabilization of vascular endothelial growth factor mRNA by hypoxia-inducible factor 1. Biochem Biophys Res Commun. 2002;291(4):908–914.
- de Almeida LF, Coimbra TM. Neonatal hyperoxia: effects on nephrogenesis and the key role of Klotho as an antioxidant factor. J Matern Fetal Neonatal Med. 2020;1–3.DOI: 10.1080/14767058.2020.1801624.
- Zhang J, Liu A, Hou R, et al. Salidroside protects cardiomyocyte against hypoxia-induced death: a HIF-1alpha-activated and VEGF-mediated pathway. Eur J Pharmacol. 2009;607(1-3):6–14.
- Rudnicki M, Perco P, Enrich J, et al. Hypoxia response and VEGF-A expression in human proximal tubular epithelial cells in stable and progressive renal disease. Lab Invest. 2009;89(3):337–346.
- Mayer G. Capillary rarefaction, hypoxia, VEGF and angiogenesis in chronic renal disease. Nephrol Dial Transplant. 2011;26(4):1132–1137.
- Lian YG, Zhou QG, Zhang YJ, et al. VEGF ameliorates tubulointerstitial fibrosis in unilateral ureteral obstruction mice via inhibition of epithelial-mesenchymal transition. Acta Pharmacol Sin. 2011;32(12):1513–1521.
- Hong JP, Li XM, Li MX, et al. VEGF suppresses epithelial-mesenchymal transition by inhibiting the expression of Smad3 and miR-192, a Smad3-dependent microRNA. Int J Mol Med. 2013;31(6):1436–1442.
- Xu Z, Jia K, Wang H, et al. METTL14-regulated PI3K/Akt signaling pathway via PTEN affects HDAC5-mediated epithelial-mesenchymal transition of renal tubular cells in diabetic kidney disease. Cell Death Dis. 2021;12(1):32.
- Lee KS, Park SJ, Kim SR, et al. Inhibition of VEGF blocks TGF-beta1 production through a PI3K/Akt signalling pathway. Eur Respir J. 2008;31(3):523–531. DOI: 10.1183/09031936.00125007.
- Wang Y, Lu YH, Tang C, et al. Calcium dobesilate restores autophagy by inhibiting the VEGF/PI3K/AKT/mTOR signaling pathway. Front Pharmacol. 2019;10:886.
- Zeisberg M, Hanai J, Sugimoto H, et al. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9(7):964–968.
- Li D, Yuan D, Shen H, et al. Gremlin-1: an endogenous BMP antagonist induces epithelial-mesenchymal transition and interferes with redifferentiation in fetal RPE cells with repeated wounds. Mol Vis. 2019;25:625–635.
- Chen L, Lin G, Chen K, et al. VEGF promotes migration and invasion by regulating EMT AND mmps in nasopharyngeal carcinoma. J Cancer. 2020;11(24):7291–7301. DOI: 10.7150/jca.46429.
- Ardura JA, Rayego-Mateos S, Rámila D, et al. Parathyroid hormone-related protein promotes epithelial-mesenchymal transition. J Am Soc Nephrol. 2010;21(2):237–248. doi: 10.1681/ASN.2009050462.
- Sun YB, Qu X, Caruana G, et al. The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation. 2016;92(3):102–107.
- Kida Y, Duffield JS. Pivotal role of pericytes in kidney fibrosis. Clin Exp Pharmacol Physiol. 2011;38(7):467–473.
- Lin SL, Chang FC, Schrimpf C, et al. Targeting endothelium-pericyte cross talk by inhibiting VEGF receptor signaling attenuates kidney microvascular rarefaction and fibrosis. Am J Pathol. 2011;178(2):911–923.
- Nathan C, Xie QW. Nitric oxide synthases: roles, tolls, and controls. Cell. 1994;78(6):915–918.
- Moncada S. Nitric oxide in the vasculature: physiology and pathophysiology. Ann N Y Acad Sci. 1997;811:60–67. discussion 7–9.
- Kang DH, Nakagawa T, Feng L, et al. Nitric oxide modulates vascular disease in the remnant kidney model. Am J Pathol. 2002;161(1):239–248.
- Shashar M, Chernichovski T, Pasvolsky O, et al. Vascular endothelial growth factor augments arginine transport and nitric oxide generation via a KDR receptor signaling pathway. Kidney Blood Press Res. 2017;42(2):201–208.
- Bussolati B, Dunk C, Grohman M, et al. Vascular endothelial growth factor receptor-1 modulates vascular endothelial growth factor-mediated angiogenesis via nitric oxide. Am J Pathol. 2001;159(3):993–1008.
- Dulak J, Józkowicz A. Regulation of vascular endothelial growth factor synthesis by nitric oxide: facts and controversies. Antioxid Redox Signal. 2003;5(1):123–132.
- Feliers D, Chen X, Akis N, et al. VEGF regulation of endothelial nitric oxide synthase in glomerular endothelial cells. Kidney Int. 2005;68(4):1648–1659.
- Schinner E, Schramm A, Kees F, et al. The cyclic GMP-dependent protein kinase Iα suppresses kidney fibrosis. Kidney Int. 2013;84(6):1198–1206.
- Tharaux PL, Chatziantoniou C, Casellas D, et al. Vascular endothelin-1 gene expression and synthesis and effect on renal type I collagen synthesis and nephroangiosclerosis during nitric oxide synthase inhibition in rats. Circulation. 1999;99(16):2185–2191.
- Hsu YC, Lee PH, Lei CC, et al. Nitric oxide donors rescue diabetic nephropathy through oxidative-stress-and nitrosative-stress-mediated Wnt signaling pathways. J Diabetes Investig. 2015;6(1):24–34.
- Jadot I, Colombaro V, Martin B, et al. Restored nitric oxide bioavailability reduces the severity of acute-to-chronic transition in a mouse model of aristolochic acid nephropathy. PLoS ONE. 2017;12(8):e0183604.
- Sun D, Wang Y, Liu C, et al. Effects of nitric oxide on renal interstitial fibrosis in rats with unilateral ureteral obstruction. Life Sci. 2012;90(23-24):900–909.
- Matsumoto Y, Ueda S, Yamagishi S, et al. Dimethylarginine dimethylaminohydrolase prevents progression of renal dysfunction by inhibiting loss of peritubular capillaries and tubulointerstitial fibrosis in a rat model of chronic kidney disease. J Am Soc Nephrol. 2007;18(5):1525–1533.
- Yang JW, Han ST, Kim YS, et al. Effects of a cGMP-specific phosphodiesterase inhibitor on expression of endothelial nitric oxide synthase and vascular endothelial growth factor in rats with cyclosporine-induced nephrotoxicity. Transplant Proc. 2010;42(10):4625–4632.
- Shihab FS, Bennett WM, Yi H, et al. Expression of vascular endothelial growth factor and its receptors Flt-1 and KDR/Flk-1 in chronic cyclosporine nephrotoxicity. Transplantation. 2001;72(1):164–168.
- Shihab FS, Yi H, Bennett WM, et al. Effect of nitric oxide modulation on TGF-beta1 and matrix proteins in chronic cyclosporine nephrotoxicity. Kidney Int. 2000;58(3):1174–1185.
- Shihab FS, Bennett WM, Isaac J, et al. Nitric oxide modulates vascular endothelial growth factor and receptors in chronic cyclosporine nephrotoxicity. Kidney Int. 2003;63(2):522–533.
- Nakagawa T. Uncoupling of the VEGF-endothelial nitric oxide axis in diabetic nephropathy: an explanation for the paradoxical effects of VEGF in renal disease. Am J Physiol Renal Physiol. 2007;292(6):F1665–72.
- Iyer AK, Ramesh V, Castro CA, et al. Nitric oxide mediates bleomycin-induced angiogenesis and pulmonary fibrosis via regulation of VEGF. J Cell Biochem. 2015;116(11):2484–2493.
- Park CW, Kim HW, Lim JH, et al. Vascular endothelial growth factor inhibition by dRK6 causes endothelial apoptosis, fibrosis, and inflammation in the heart via the Akt/eNOS axis in db/db mice. Diabetes. 2009;58(11):2666–2676.
- Pilmore HL, Eris JM, Painter DM, et al. Vascular endothelial growth factor expression in human chronic renal allograft rejection. Transplantation. 1999;67(6):929–933.
- Xiang E, Han B, Zhang Q, et al. Human umbilical cord-derived mesenchymal stem cells prevent the progression of early diabetic nephropathy through inhibiting inflammation and fibrosis. Stem Cell Res Ther. 2020;11(1):336.
- Engel JE, Williams E, Williams ML, et al. Targeted VEGF (vascular endothelial growth factor) therapy induces long-term renal recovery in chronic kidney disease via macrophage polarization. Hypertension. 2019;74(5):1113–1123.
- Scalia R, Booth G, Lefer DJ. Vascular endothelial growth factor attenuates leukocyte-endothelium interaction during acute endothelial dysfunction: essential role of endothelium-derived nitric oxide. FASEB J. 1999;13(9):1039–1046.
- Walshe TE, Dole VS, Maharaj AS, et al. Inhibition of VEGF or TGF-{beta} signaling activates endothelium and increases leukocyte rolling. Arterioscler Thromb Vasc Biol. 2009;29(8):1185–1192.
- Gewin L, Zent R, Pozzi A. Progression of chronic kidney disease: too much cellular talk causes damage. Kidney Int. 2017;91(3):552–560.
- Kim I, Moon SO, Kim SH, et al. Vascular endothelial growth factor expression of intercellular adhesion molecule 1 (ICAM-1), vascular cell adhesion molecule 1 (VCAM-1), and E-selectin through nuclear factor-kappa B activation in endothelial cells. J Biol Chem. 2001;276(10):7614–7620.
- Bus P, Scharpfenecker M, Van Der Wilk P, et al. The VEGF-A inhibitor sFLT-1 improves renal function by reducing endothelial activation and inflammation in a mouse model of type 1 diabetes. Diabetologia. 2017;60(9):1813–1821.
- Henri O, Pouehe C, Houssari M, et al. Selective stimulation of cardiac lymphangiogenesis reduces myocardial edema and fibrosis leading to improved cardiac function following myocardial infarction. Circulation. 2016;133(15):1484–1497. discussion 97.
- Hasegawa S, Nakano T, Torisu K, et al. Vascular endothelial growth factor-C ameliorates renal interstitial fibrosis through lymphangiogenesis in mouse unilateral ureteral obstruction. Lab Invest. 2017;97(12):1439–1452.
- Hwang SD, Song JH, Kim Y, et al. Inhibition of lymphatic proliferation by the selective VEGFR-3 inhibitor SAR131675 ameliorates diabetic nephropathy in db/db mice. Cell Death Dis. 2019;10(3):219.
- Guo YC, Zhang M, Wang FX, et al. Macrophages regulate unilateral ureteral Obstruction-Induced renal lymphangiogenesis through C-C motif chemokine receptor 2-dependent phosphatidylinositol 3-Kinase-AKT-mechanistic target of rapamycin signaling and hypoxia-inducible factor-1α/vascular endothelial growth factor-C expression. Am J Pathol. 2017;187(8):1736–1749.
- Shen X, Zhang K, Kaufman RJ. The unfolded protein response-a stress signaling pathway of the endoplasmic reticulum. J Chem Neuroanat. 2004;28(1-2):79–92.
- Sovolyova N, Healy S, Samali A, et al. Stressed to death – mechanisms of ER stress-induced cell death. Biol Chem. 2014;395(1):1–13.
- Yan M, Shu S, Guo C, et al. Endoplasmic reticulum stress in ischemic and nephrotoxic acute kidney injury. Ann Med. 2018;50(5):381–390.
- Ricciardi CA, Gnudi L. The endoplasmic reticulum stress and the unfolded protein response in kidney disease: implications for vascular growth factors. J Cell Mol Med. 2020;24(22):12910–12919.
- Chiang CK, Hsu SP, Wu CT, et al. Endoplasmic reticulum stress implicated in the development of renal fibrosis. Mol Med. 2011;17(11-12):1295–1305.
- Fan Y, Xiao W, Li Z, et al. RTN1 mediates progression of kidney disease by inducing ER stress. Nat Commun. 2015;6:7841.
- Bouvier N, Fougeray S, Beaune P, et al. The unfolded protein response regulates an angiogenic response by the kidney epithelium during ischemic stress. J Biol Chem. 2012;287(18):14557–14568.
- Karali E, Bellou S, Stellas D, et al. VEGF signals through ATF6 and PERK to promote endothelial cell survival and angiogenesis in the absence of ER stress. Mol Cell. 2014;54(4):559–572.
- Oskolkova OV, Afonyushkin T, Leitner A, et al. ATF4-dependent transcription is a key mechanism in VEGF up-regulation by oxidized phospholipids: critical role of oxidized sn-2 residues in activation of unfolded protein response. Blood. 2008;112(2):330–339.
- Feng SQ, Zong SY, Liu JX, et al. VEGF antagonism attenuates cerebral ischemia/reperfusion-induced injury via inhibiting endoplasmic reticulum stress-mediated apoptosis. Biol Pharm Bull. 2019;42(5):692–702.
- Park JH, Kim M, Oh JH. Effects of bevacizumab on endoplasmic reticulum stress in hypoxic retinal pigment epithelial cells. PLoS ONE. 2017;12(6):e0179048.
- Morimoto M, Arai T, Matsuura M, et al. Bevacizumab-associated glomerular microangiopathy that occurred after postoperative chemotherapy for ovarian cancer. CEN Case Rep. 2021;10(1):6–11.
- Ozawa M, Ohtani H, Komatsuda A, et al. VEGF-VEGFR2 inhibitor-associated hyaline occlusive glomerular microangiopathy: a Japanese single-center experience. Clin Exp Nephrol. 2021;25(11):1193–1202.
- Ahmed M, Alouch N, Ahmed A, et al. Worsening of renal function and uncontrolled hypertension from intravitreal bevacizumab injections. Proc (Bayl Univ Med Cent). 2021;34(4):527–529.
- Izzedine H, Massard C, Spano JP, et al. VEGF signalling inhibition-induced proteinuria: Mechanisms, significance and management. Eur J Cancer. 2010;46(2):439–448.
- Sugimoto H, Hamano Y, Charytan D, et al. Neutralization of circulating vascular endothelial growth factor (VEGF) by anti-VEGF antibodies and soluble VEGF receptor 1 (sFlt-1) induces proteinuria. J Biol Chem. 2003;278(15):12605–12608.
- Van Wynsberghe M, Flejeo J, Sakhi H, et al. Nephrotoxicity of anti-angiogenic therapies. Diagnostics. 2021;11(4):640.
- Carlström M. Nitric oxide signalling in kidney regulation and cardiometabolic health. Nat Rev Nephrol. 2021;17(9):575–590.
- Kappers MH, van Esch JH, Sluiter W, et al. Hypertension induced by the tyrosine kinase inhibitor sunitinib is associated with increased circulating endothelin-1 levels. Hypertension. 2010;56(4):675–681.
- Neves KB, Rios FJ, van der Mey L, et al. VEGFR (vascular endothelial growth factor receptor) inhibition induces cardiovascular damage via Redox-Sensitive processes. Hypertension. 2018;71(4):638–647.
- Kim YG, Suga SI, Kang DH, et al. Vascular endothelial growth factor accelerates renal recovery in experimental thrombotic microangiopathy. Kidney Int. 2000;58(6):2390–2399.
- Kang DH, Kim YG, Andoh TF, et al. Post-cyclosporine-mediated hypertension and nephropathy: amelioration by vascular endothelial growth factor. Am J Physiol Renal Physiol. 2001;280(4):F727–36.
- Leonard EC, Friedrich JL, Basile DP. VEGF-121 preserves renal microvessel structure and ameliorates secondary renal disease following acute kidney injury. Am J Physiol Renal Physiol. 2008;295(6):F1648–57.
- Burt LE, Forbes MS, Thornhill BA, et al. Renal vascular endothelial growth factor in neonatal obstructive nephropathy. II. Exogenous VEGF. Am J Physiol Renal Physiol. 2007;292(1):F168–74.
- Veron D, Aggarwal PK, Velazquez H, et al. Podocyte-specific VEGF-a gain of function induces nodular glomerulosclerosis in eNOS null mice. J Am Soc Nephrol. 2014;25(8):1814–1824.
- Veron D, Bertuccio CA, Marlier A, et al. Podocyte vascular endothelial growth factor (Vegf164) overexpression causes severe nodular glomerulosclerosis in a mouse model of type 1 diabetes. Diabetologia. 2011;54(5):1227–1241.
- Vriese AS, Tilton RG, Elger M, et al. Antibodies against vascular endothelial growth factor improve early renal dysfunction in experimental diabetes. J Am Soc Nephrol. 2001;12(5):993–1000.
- Thurston G. Complementary actions of VEGF and angiopoietin-1 on blood vessel growth and leakage. J Anat. 2002;200(6):575–580.
- Miao AF, Liang JX, Yao L, et al. Hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) protects against renal ischemia/reperfusion injury by inhibiting inflammation. Ren Fail. 2021;43(1):803–810.