
Abstract
Objective
The clinical characteristics, genetic mutation spectrum, treatment strategies and prognoses of 15 children with Dent disease were retrospectively analyzed to improve pediatricians’ awareness of and attention to this disease.
Methods
We analyzed the clinical and laboratory data of 15 Chinese children with Dent disease who were diagnosed and treated at our hospital between January 2017 and May 2023 and evaluated the expression of the CLCN5 and OCRL1 genes.
Results
All 15 patients were male and complained of proteinuria, and the incidence of low-molecular-weight proteinuria (LMWP) was 100.0% in both Dent disease 1 (DD1) and Dent disease 2 (DD2) patients. The incidence of hypercalciuria was 58.3% (7/12) and 66.7% (2/3) in DD1 and DD2 patients, respectively. Nephrocalcinosis and nephrolithiasis were found in 16.7% (2/12) and 8.3% (1/12) of DD1 patients, respectively. Renal biopsy revealed focal segmental glomerulosclerosis (FSGS) in 1 patient, minimal change lesion in 5 patients, and small focal acute tubular injury in 1 patient. A total of 11 mutations in the CLCN5 gene were detected, including 3 missense mutations (25.0%, c.1756C > T, c.1166T > G, and c.1618G > A), 5 frameshift mutations (41.7%, c.407delT, c.1702_c.1703insC, c.137delC, c.665_666delGGinsC, and c.2200delG), and 3 nonsense mutations (25.0%, c.776G > A, c.1609C > T, and c.1152G > A). There was no significant difference in age or clinical phenotype among patients with different mutation types (p > 0.05). All three mutations in the OCRL1 gene were missense mutations (c.1477C > T, c.952C > T, and c.198A > G).
Conclusion
Pediatric Dent disease is often misdiagnosed. Protein electrophoresis and genetic testing can help to provide an early and correct diagnosis.
Dent disease is a rare hereditary X-linked recessive proximal tubular disease. It is characterized by low-molecular-weight proteinuria (LMWP), hypercalciuria, nephrocalcinosis/nephrolithiasis, and progressive renal failure [Citation1]. Some patients might present with incomplete Fanconi syndrome, hypophosphatemic rickets or abnormal electrolyte balance [Citation2]. It is rare for Dent disease to develop into CKD in childhood, and only a few cases have been reported to date [Citation3]. Between 30 and 50 years of age, 30–80% of patients with Dent disease can develop end-stage renal disease (ESRD) [Citation4]. At present, there are no more than 1,000 families of reported cases worldwide. Dent disease was first reported in China in 2008, and most articles belong to case report or series case reports. There is only one multicenter study including 45 Chinese patients with Dent disease.
Dent disease is divided into Dent disease 1 (DD1) and Dent disease 2 (DD2) based on the mutated gene on the X chromosome [Citation5]. The CLCN5 gene is located at Xp11.22-p11.23 and encodes the protein ClC-5, a chloride channel protein. ClC-5 is mainly expressed in the kidney and participates in the endocytic reabsorption of LMWP in proximal tubular cells [Citation6]. The OCRL1 gene is located on chromosome Xq25 and has 24 exons. It is widely distributed in human tissues, especially the eyes, kidneys and brain. It encodes phosphatidylinositol 4,5-diphosphate 5-phosphatase, which hydrolyzes phosphatidylinositol 4,5-diphosphate (PIP2), which is a second messenger involved in vesicle transport [Citation7]. DD1 with mutations in the CLCN5 gene accounts for approximately 60% of patients with Dent disease, and DD2 with mutations in the OCRL1 gene accounts for approximately 15% of patients. The genetic origin of the remaining cases (25% of all cases) remains unknown [Citation8]. Dent disease almost exclusively affects hemizygous males in childhood. A small minority of affected females might exhibit a mild phenotype, such as LMWP or hypercalciuria, probably due to the random inactivation of one of the two X chromosomes [Citation9].
In the present study, the clinical characteristics, genetic mutation spectrum, treatment strategies and prognosis of 15 children with Dent disease were retrospectively analyzed to improve pediatricians’ awareness of and attention to this disease.
Subjects and methods
Subjects
The clinical data of 15 children with Dent disease (12 DD1 patients and 3 DD2 patients) who were admitted to the Department of Pediatric Nephrology and Rheumatology of Shandong Provincial Hospital from January 2017 to May 2023 were retrospectively analyzed. This study was approved by the Medical Ethics Committee of Shandong Provincial Hospital.
Methods
Clinical data collection
We used the electronic medical records system of the hospital to collect clinical data from the children’s medical records. The collected information included sex, age at diagnosis, clinical manifestations, routine blood test results, routine urine test results, the ratio of urinary calcium to creatinine (UC/Ucr), 24-h urinary calcium and protein content, the urinary protein to creatinine ratio (UP/Ucr), retinol-binding protein levels, α1 microglobulin levels, β2 microglobulin levels, microalbuminuria, hepatic and renal function test results, electrolyte levels, renal ultrasound, kidney biopsy, gene variations, treatment and prognosis.
The clinical diagnosis of Dent disease is based on the presence of at least two of the following criteria: i) LMWP: early renal injury index indicates that the low-molecular-weight protein concentrations in urine are at least 5 times greater than expected. The low-molecular-weight proteins used for monitoring include retinol-binding protein, α1 microglobulin, and β2 microglobulin. Urine protein electrophoresis shows that low-molecular-weight proteins account for more than 50% of urinary proteins. ii) Hypercalciuria: the 24-h urinary calcium value > 0.1 mmol/kg or a random increase in the UC/Ucr ratio, which varies greatly with age. iii) One of the following conditions: nephrocalcinosis, nephrolithiasis, hematuria, hypophosphatemia, or renal insufficiency. The identification of a mutation in either CLCN5 or OCRL1 confirms the diagnosis.
Kidney histopathology
Kidney biopsy was performed on six of 15 patients. Kidney tissue samples were obtained using a spring-loaded needle biopsy kit under the real-time guidance of a b-ultrasound detection system. After a percutaneous kidney puncture biopsy, the kidney tissue was stained [hematoxylin-eosin (HE) staining, periodic acid-Schiff (PAS) staining, hexamine silver (PASM) staining and Masson staining], and electron microscopy and immunofluorescence were used to detect IgA, IgG, IgM, Fib, C1q, and C3.
Genetic testing
Whole-exome sequencing (WES) was performed on the patients. Whole blood samples (2–5 mL) were collected from the affected children and their parents. DNA extraction was performed, and the whole exome was captured and sequenced with the IDT xGen Exome Research Panel v2.0 Full Exon Capture Chip. Variants were classified according to the three-factor classification system and the American College of Medical Genetics and Genomics (ACMG) gene variation classification system.
Statistical tests
The statistical software SPSS version 26.0 (IBM Corp., Armonk, NY, USA) was used for the statistical analyses. Quantitative data are expressed as the median with interquartile range (IQR). The Mann‒Whitney test and Kruskal‒Wallis H test were used for comparisons of two or three groups of nonnormally distributed sample data, respectively. The chi-square test was used to compare the rates between groups. p < 0.05 was considered to indicate statistical significance.
Results
Clinical phenotypes of DD1 and DD2
All 12 patients with DD1 were boys, with an age of onset ranging from 1.75 to 10.4 years (median 3.76 years) and an age at diagnosis ranging from 2.1 to 10.57 years (median 6.12 years). Three patients with DD2 were also boys, with an age of onset ranging from 2.50 to 6.29 years (median 5.8 years) and an age at diagnosis ranging from 2.67 to 12.8 years (median 6.87 years). The time from onset to clinical diagnosis was 0.25 ∼ 48 months (median 24 months) and 2 ∼ 84 months (median 7 months) in the DD1 and DD2 groups, respectively. There was no significant difference between the two groups (p > 0.05) ().
Table 1. Clinical features and gene mutations of patients with dent disease.
In the DD1 group, one patient was born prematurely at 32 weeks of gestation for unknown reasons and presented with delayed language development after birth (Patient 3). One patient was born prematurely at 35 weeks plus 5 days due to placenta previa (Patient 12). Three patients had a family history of kidney disease. One patient’s grandfather was diagnosed with ESRD, and his mother and elder brother had proteinuria (Patient 8). A younger brother of one patient presented with proteinuria (Patient 2). Pyelectasia was found in one patient’s father (Patient 7).
The main complaint of all patients with DD1 and DD2 was proteinuria. One patient was misdiagnosed with interstitial nephritis and was treated with prednisone for 1.5 years (Patient 7). One patient was misdiagnosed with glomerular nephritis and was given mycophenolate mofetil (MMF) for 2 months (Patient 9). One patient was diagnosed with nephrotic syndrome, and renal pathology revealed minimal change lesion. He was subsequently treated with prednisone for 2 months at a local hospital (Patient 13).
Proteinuria was observed in all DD1 and DD2 patients. Albustix testing yielded results of 1 ∼ 3+ and 2 ∼ 3+ in the DD1 and DD2 groups, respectively. Fourteen patients underwent urinary protein electrophoresis, and the results suggested LMWP, with low-molecular-weight proteins accounting for 54.4 ∼ 73.9% and 57.6 ∼ 62.3% of the total urinary protein in the DD1 and DD2 groups, respectively (p > 0.05). The 24-h urinary protein concentrations were 13.3 ∼ 53.66 mg/kg (median 34.29 mg/kg) in the DD1 group and 32.05 ∼ 67.39 mg/kg (median 65.83 mg/kg) in the DD2 group (p > 0.05). The UP/Ucr ratio ranged from 0.82 mg/mg to 5.35 mg/mg (mean 2.15) and 2.48 to 4.97 mg/mg (mean 3.46) in the two groups, respectively (p > 0.05) ().
The UC/Ucr ratios were 0.078 ∼ 0.69 mg/mg and 0.14 ∼ 0.58 mg/mg in the DD1 and DD2 groups, respectively (p > 0.05). The 24-h urinary calcium concentration ranged from 0.04 to 0.29 mmol/kg and 0.11 to 0.13 mmol/kg in the two groups (p > 0.05). Hypercalciuria was observed in 58.3% (7/12) and 66.7% (2/3) of DD1 and DD2 patients, respectively (p > 0.05) ().
Metabolic acidosis was observed in 5 patients (41.7%), glucosuria in 1 patient (8.3%), backward language development in 1 patient (8.3%) and enuresis in 1 patient (8.3%) in the DD1 group. Hypophosphatemia was observed in 2 patients (66.7%), metabolic acidosis in 3 patients (100%), growth deficiency in 1 patient (33.3%), and glucosuria in 1 patient (33.3%) in the DD2 group. The incidence of hypophosphatemia, metabolic acidosis, and growth deficiency was lower in the DD1 group than in the DD2 group (p < 0.05). Renal ultrasound revealed slightly enhanced renal cone echo in 2 patients (16.7%) and double renal medullary sponge kidney in 1 patient (8.3%) in the DD1 group ().
Five patients with DD1 and one patient with DD2 underwent renal biopsy. Biopsy revealed focal segmental glomerulosclerosis (FSGS) in 1 patient, minimal change lesion in 5 patients, and small focal acute tubular injury in 1 patient ().
Novel and recurrent mutations
DNA sequence analysis revealed that seven of the CLCN5 and OCRL1 mutations have not previously been described (c.1166T > G, c.407delT, c.1702_c.1703insC, c.137delC, c.665_666delGGinsC, and c.2200delG in the CLCN5 gene and c.198A > G in the OCRL1 gene). The other seven mutation sites were previously described ().
Of the 11 mutations in the CLCN5 gene, 3 were missense mutations (25.0%, c.1756C > T, c.1166T > G, and c.1618G > A), 5 were frameshift mutations (41.7%, c.407delT, c.1702_c.1703insC, c.137delC, c.665_666delGGinsC, and c.2200delG), and 3 were nonsense mutations (25.0%, c.776G > A, c.1609C > T, and c.1152G > A). There was no significant difference in age of onset, age of diagnosis or clinical phenotype among patients with different mutation types (p > 0.05). We identified 3 mutations in the OCRL1 gene, all of which were missense mutations (c.1477C > T, c.952C > T, and c.198A > G) ().
Bioinformatics analysis
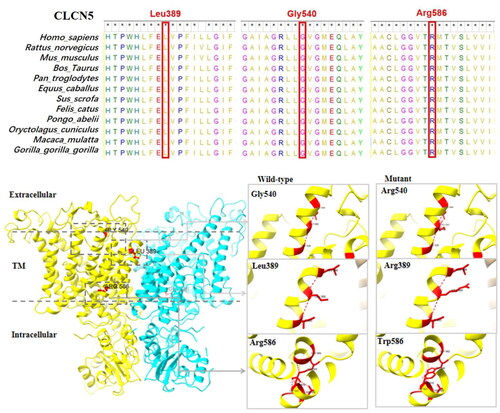
The three missense variants of CLCN5 in this cohort were p.Leu389Arg, p.Gly540Arg, and p.Arg586Trp. These amino acid sequences are highly conserved according to multiple-species sequence alignment with MEGA 7 software. The two ClC-5 monomers form dimers, and all three missense mutations are located on helices involved in the formation of the dimer interface. There was no significant change in hydrogen bonds for p.Leu389Arg and p.Gly540Arg. The proximity of p.Gly540Arg to the transporter interface affects the stability of the helix and thus prevents proper folding of the monomer. p.Arg586Trp leads to the disappearance of hydrogen bond interactions with Gly583, Ala619, Asp620, and Gly623, forming a new hydrogen bond with Glu625; a mutation in this position may have an effect on the protein structure and thus affect protein function ().
Figure 1. Structural diagram of the protein structure of three missense variants of the CLCN5 gene (p.Gly540Arg, p.Leu389Arg, and p.Arg586Trp).
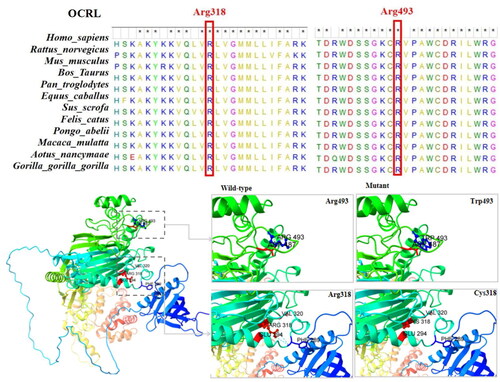
The two missense variants of OCRL1 in this cohort were p.Arg318Cys and p.Arg493Trp. These amino acid sequences are highly conserved according to multiple-species sequence alignment with MEGA 7 software. Arg318Cys has one hydrogen bond with Glu294 and Val320 and two hydrogen bonds with Phe289. After mutation to Cys, these four hydrogen bond interactions disappear, which may have affect the protein structure and thus the protein function. p.Arg318Cys affects the 5-phosphatase domain of OCRL1. Arg493Trp has one hydrogen bond with Asp487, and no hydrogen bond changes occur after mutation to Trp ().
Figure 2. Structural diagram of the protein structure of two missense variants of the OCRL1 gene (p.Arg493Trp, p.Arg318Cys).
Treatment and prognosis
All 15 patients received oral benazepril or perindopril. Six patients were given oral potassium citrate, and two patients were treated with dihydrochlorothiazide (HCTZ). Two children were lost to follow-up, and the follow-up time of the remaining 13 patients ranged from 0.75 to 111.2 months (median 46.9 months). At the last follow-up visit, the patients’ urinary protein levels remained positive. Renal function was normal, and serum creatinine levels were within the normal range. Hypercalciuria was observed in three patients with DD1 (Patients 1, 4, 9). Three patients were found to have medullary calcium deposition (Patients 1, 3, 5); one child developed a left kidney stone (Patient 5), and one child had a stroke with moyamoya disease (Patient 9) in the DD1 group.
Discussion
Dent disease is a rare hereditary X-linked recessive renal tubular disease characterized by LMWP and hypercalciuria with or without nephrocalcinosis. Other signs include kidney stones, hematuria, hypophosphatemia, and chronic kidney disease (CKD), which occur at variable frequencies. Occasionally, rickets, osteomalacia, and short stature are observed [Citation10]. Dent disease mainly occurs in male patients, with relatively mild manifestations in female patients.
In our study, all 15 patients were male and complained of proteinuria, and the incidence of LMWP was 100.0% in both DD1 and DD2 patients. The incidence of hypercalciuria was 58.3% (7/12) and 66.7% (2/3) in DD1 and DD2 patients, respectively. Nephrocalcinosis and nephrolithiasis were observed in 16.7% (2/12) and 8.3% (1/12) of DD1 patients, respectively. Hypophosphatemia, metabolic acidosis, glucosuria, growth deficiency, delayed language development, and enuresis were also observed in this study. However, no rickets, microscopic hematuria, or renal impairments were observed in DD1 or DD2 patients. In one national multicenter study in China enrolling 32 DD1 and 13 DD2 patients [Citation11], the incidence of LMWP was 100.0% in both the DD1 and DD2 populations; the incidence of hypercalciuria was 65.6% (21/32) and 92.3% (12/13), that of nephrocalcinosis was 43.8% (14/32) and 23.1% (3/13), that of abnormal renal function was 12.5% (4/32) and 7.7% (1/13), that of nephrolithiasis was 9.4% (3/32) and 15.4% (2/13), and that of rickets was 9.4% (3/32) and 15.4% (2/13) in DD1 and DD2 patients, respectively. The incidences of LMWP, hypercalciuria, nephrocalcinosis, abnormal renal function, nephrolithiasis and rickets were not significantly different from those in our study (p > 0.05). A 46-item web-based cross-sectional survey from the European Rare Kidney Disease Reference Network (ERKNet) identified 163 male patients with genetically confirmed DD1[Citation12]. During follow-up, all patients had LMWP, 66.4% had nephrocalcinosis, 44.4% had hypercalciuria, and 26.4% had nephrolithiasis. These findings were consistent with those of previous studies of European and American populations and Asian populations [Citation13]. However, there was significant intra- and interfamilial variability in clinical presentations [Citation14]. An increasing number of reports have described subjects carrying CLCN5 or OCRL1 mutations who exhibit an incomplete phenotype. This diversity of phenotypes can lead to missed diagnoses, misdiagnosis, inappropriate treatment, or even unnecessary kidney biopsies.
In our study, DNA sequence analysis revealed 11 different mutations in the CLCN5 gene and three mutations in the OCRL1 gene. These mutations were classified as frameshift mutations (8 patients), nonsense mutations (3 patients), or missense mutations (3 patients). We compared the clinical data of DD1 patients according to CLCN5 mutation type and did not observe obvious differences in age of onset, age of diagnosis or clinical phenotype. Previous reports have shown that the distribution of CLCN5 mutation types is consistent in European and American populations and Asian populations. There was no obvious relationship between mutation types and clinical phenotypes in different populations [Citation13].
Although Dent disease is a proximal tubular disease, steroid-resistant nephrotic syndrome (SRNS), FSGS, and minimal change lesion are the most common clinical misdiagnoses before a molecular diagnosis of Dent disease is made in children [Citation15]. In our study, three patients were misdiagnosed with interstitial nephritis, glomerular nephritis or nephrotic syndrome and were treated with prednisone and/or immunosuppressive agents.
The main reason for misdiagnosis was the lack of urine protein electrophoresis performed to detect the composition of urinary protein. The ratios of low-molecular-weight proteins, including retinol-binding protein, α1 microglobulin, and β2 microglobulin, are among the most important criteria for the diagnosis of Dent disease. Nephrotic-range proteinuria or mixed proteinuria in Dent disease patients is often secondary to renal tubular lesions, so the early detection of Dent disease is particularly important. Dent disease is usually detected by asymptomatic proteinuria during urine screening, such as that routinely performed at age 3 or at school in Japan [Citation16]. Therefore, it is suggested that urine screening can aid in early detection of urinary protein concentrations and analysis of the composition of urinary protein, which can identify Dent disease early and reduce the occurrence of misdiagnosis.
A variety of histopathological findings have been reported in renal biopsies from DD1 and DD2 patients. Patients may have normal glomerular and/or tubular compartments or glomerulosclerosis with or without tubular atrophy or interstitial fibrosis [Citation17,Citation18]. In our study, six patients underwent renal biopsy. FSGS was found in 1 patient, minimal change lesion was found in 5 patients, and small focal acute tubular injury was found in one patient. In recent years, glomerular involvement in Dent disease has been hypothesized. Since the discovery that CLCN5 and OCRL1 are expressed in the glomerular compartment, a new theory has emerged suggesting that the loss of function of these two proteins leads to primary glomerular cell damage [Citation19]. Glomerular injury is responsible for nephrotic-range proteinuria in more than 30% of patients with Dent disease. However, no correlations have emerged between the type of CLCN5/OCRL1 mutations and histopathological findings. Renal biopsy is not helpful in the diagnosis of Dent disease because the histological findings are nonspecific; six of our patients underwent a kidney biopsy, but the results did not help to establish a definitive diagnosis.
The screening of patients with Dent disease by next-generation sequencing (NGS) may be a good method for identifying unknown, atypical or mixed phenotypes of other known genetic nephropathies. Genetic screening of males with LMWP and at least one typical associated clinical sign (hypercalciuria, nephrocalcinosis, renal stones, hematuria, and/or hypophosphatemia) led to a Dent disease detection rate ranging from 66 to 92% [Citation20]. To date, approximately 200 CLCN5 mutations that cause Dent disease have been reported [Citation21]. The main mutation types include missense mutations and frameshift mutations; the other mutation types are nonsense mutations, splicing mutations and large fragment deletions. The frameshift deletion-insertion, donor splice site and nonsense mutations are predicted to result in truncated or absent ClC-5 proteins, which would lead to complete loss of antiporter function and chloride conductance [Citation22]. Missense and synonymous variants can also have severe effects on the function and expression of a protein. The three missense variants of CLCN5 in this cohort were p.Leu389Arg, p.Gly540Arg, and p.Arg586Trp, which are located on helices involved in the formation of the dimer interface. p.Leu389Arg is located on helix J, and the nearby Leu394Arg mutation has been reported to interfere with channel function and prevent transport to the plasma membrane [Citation23]. The p.Gly540Arg mutation close to the transporter interface has been reported to affect the stability of the helix and thus prevent proper folding of the monomer [Citation21]. In addition, p.Arg586Trp has been reported to cause ClC-5 retention in the endoplasmic reticulum [Citation24].
More than 140 mutations in the OCRL1 gene, including insertions, deletions, splicing and missense mutations, have been reported to cause Dent disease. Most DD2 mutations are deletions in exons, while the proportions of truncating variants (nonsense and frameshift) and nontruncating variants (missense) are similar. Truncating variants were found in the PH and linker domains, while missense variants were found in the inositol-5-phosphatase domain and only occasionally in the ASH-RhoGAP module [Citation25]. The two missense variants of OCRL1 in this cohort were p.Arg318Cys and p.Arg493Trp. These 2 variants are recurrent variants and have been reported several times. p.Arg318Cys affects the 5-phosphatase domain of OCRL1 and is considered a mutational hotspot in the OCRL1 gene [Citation26]. The Arg493Trp mutation may have an effect on protein structure and thus protein function. Another variant at the same position of Arg493, R493A, has been reported to eliminate 5-phosphatase activity [Citation27].
The clinical management of patients with Dent disease is focused on symptoms rather than any disease-specific treatment [Citation28]. Proteinuria is a key feature of the DD1 and DD2 phenotypes. Angiotensin-converting enzyme inhibitors (ACEis) and angiotensin II receptor blockers (ARBs) have been shown to be effective in reducing proteinuria in Dent disease patients [Citation29]. ACEis/ARBs are well tolerated in children with Dent disease. Thiazide diuretics are commonly used to reduce hypercalciuria in patients with Dent disease. Kidney stones are usually controlled by citrate supplementation (mainly potassium citrate). Citrates are thought to improve acidosis and slow the progression of CKD [Citation30]. In our study, all 15 patients received benazepril or perindopril orally. Six patients received potassium citrate orally, and two patients were treated with HCTZ. Only 13 patients were followed up from 0.75 to 111.2 months (median 46.9 months). At the last follow-up visit, the urinary protein levels remained positive. Three patients developed hypercalciuria. Three patients were found to have medullary calcium deposition, and one child presented with a left kidney stone. The incidence of nephrolithiasis and nephrocalcinosis increases with increasing age. A survey by ERKNet revealed that hypercalciuria was more common in pediatric patients than in adults, while nephrolithiasis, nephrocalcinosis and renal dysfunction were more common in adults [Citation12].
In summary, pediatric Dent disease is often misdiagnosed. Protein electrophoresis and genetic testing can help to achieve an early and correct diagnosis.
Disclosure statement
No potential conflicts of interest are reported by the authors.
Data availability statement
The data used and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Wang Y, Xu L, Zhang Y, et al. Dent disease 1-linked novel CLCN5 mutations result in aberrant location and reduced ion currents. Int J Biol Macromol. 2024;257(Pt 2):1. doi: 10.1016/j.ijbiomac.2023.128564.
- Gianesello L, Del Prete D, Anglani F, et al. Genetics and phenotypic heterogeneity of dent disease: the dark side of the moon. Hum Genet. 2021;140(3):401–10. doi: 10.1007/s00439-020-02219-2.
- Wong W, Poke G, Stack M, et al. Phenotypic variability of dent disease in a large New Zealand kindred. Pediatr Nephrol. 2017;32(2):365–369. doi: 10.1007/s00467-016-3472-8.
- Jin YY, Huang LM, Quan XF, et al. Dent disease: classification, heterogeneity and diagnosis. World J Pediatr. 2021;17(1):52–57. doi: 10.1007/s12519-020-00357-1.
- Sakakibara N, Nagano C, Ishiko S, et al. Comparison of clinical and genetic characteristics between dent disease 1 and dent disease 2. Pediatr Nephrol. 2020;35(12):2319–2326. doi: 10.1007/s00467-020-04701-5.
- Picollo A, Pusch M. Chloride/proton antiporter activity of mammalian CLC proteins ClC-4 and ClC-5. Nature. 2005; 436(7049):420–423. doi: 10.1038/nature03720.
- De Matteis MA, Staiano L, Emma F, et al. The 5-phosphatase OCRL in lowe syndrome and dent disease 2. Nat Rev Nephrol. 2017;13(8):455–470. doi: 10.1038/nrneph.2017.83.
- Ehlayel AM, Copelovitch L. Update on dent disease. Pediatr Clin North Am. 2019;66(1):169–178. doi: 10.1016/j.pcl.2018.09.003.
- Claverie-Martín F, Ramos-Trujillo E, García-Nieto V. Dent’s disease: clinical features and molecular basis. Pediatr Nephrol. 2011;26(5):693–704. doi: 10.1007/s00467-010-1657-0.
- Drosataki E, Maragkou S, Dermitzaki K, et al. Dent-2 disease with a bartter-like phenotype caused by the Asp631Glu mutation in the OCRL gene. BMC Nephrol. 2022;23(1):182. doi: 10.1186/s12882-022-02812-9.
- Ye Q, Shen Q, Rao J et al. Multicenter study of the clinical features and mutation gene spectrum of Chinese children with dent disease. Clin Genet. 2020;97(3):407–417. doi: 10.1111/cge.13663.
- Burballa C, Cantero-Recasens G, Prikhodina L, et al. Clinical and genetic characteristics of dent’s disease type 1 in Europe. Nephrol Dial Transplant. 2023;38(6):1497–1507. doi: 10.1093/ndt/gfac310.
- Zhang Y, Fang XY, Xu H, et al. Five case series of dent-1 disease and literature review. Chin J Evid-Based Pediatr. 2016;11(5):382–387. doi: 10.3969/j.issn.1673-5501.2016.05.014.
- Leite de Sousa L, Pimenta G, Veríssimo R, et al. Dent’s disease: an unusual cause of kidney failure. Clin Nephrol Case Stud. 2023;11(1):1–5. doi: 10.5414/CNCS110975.
- Trautmann A, Lipska-Ziętkiewicz BS, Schaefer F. Exploring the clinical and genetic spectrum of steroid resistant nephrotic syndrome: the PodoNet registry. Front Pediatr. 2018;6:200. doi: 10.3389/fped.2018.00200.
- Sekine T, Komoda F, Miura K, et al. Japanese dent disease has a wider clinical spectrum than dent disease in Europe/USA: genetic and clinical studies of 86 unrelated patients with low-molecular-weight proteinuria. Nephrol Dial Transplant. 2014;29(2):376–384. doi: 10.1093/ndt/gft394.
- Bezdíčka M, Langer J, Háček J, et al. Dent disease type 2 as a cause of focal segmental glomerulosclerosis in a 6-Year-Old boy: a case report. Front Pediatr. 2020;8:583230. doi: 10.3389/fped.2020.583230.
- Wang X, Anglani F, Beara-Lasic L, et al. Glomerular pathology in dent disease and its association with kidney function. Clin J Am Soc Nephrol. 2016;11(12):2168–2176. doi: 10.2215/CJN.03710416.
- Huang LM, Mao JH. Glomerular podocyte dysfunction in inherited renal tubular disease. World J Pediatr. 2021;17(3):227–233. doi: 10.1007/s12519-021-00417-0.
- Zhang Y, Fang X, Xu H, et al. Genetic analysis of dent’s disease and functional research of CLCN5 mutations. DNA Cell Biol. 2017;36(12):1151–1158. doi: 10.1089/dna.2017.3731.
- Mansour-Hendili L, Blanchard A, Le Pottier N, et al. Mutation update of the CLCN5 gene responsible for dent disease 1. Hum Mutat. 2015;36(8):743–752. doi: 10.1002/humu.22804.
- Wu F, Reed AA, Williams SE, et al. Mutational analysis of CLC-5, cofilin and CLC-4 in patients with dent’s disease. Nephron Physiol. 2009;112(4):p53–62. doi: 10.1159/000225944.
- Ludwig M, Doroszewicz J, Seyberth HW, et al. Functional evaluation of dent’s disease-causing mutations: implications for ClC-5 channel trafficking and internalization. Hum Genet. 2005;117(2-3):228–237. doi: 10.1007/s00439-005-1303-2.
- Smith AJ, Reed AA, Loh NY, et al. Characterization of dent’s disease mutations of CLC-5 reveals a correlation between functional and cell biological consequences and protein structure. Am J Physiol Renal Physiol. 2009;296(2):F390–7. doi: 10.1152/ajprenal.90526.2008.
- Gianesello L, Arroyo J, Del Prete D, et al. Genotype phenotype correlation in dent disease 2 and review of the literature: OCRL gene pleiotropism or extreme phenotypic variability of lowe syndrome? Genes . 2021;12(10):1597. doi: 10.3390/genes12101597.
- Mura-Escorche G, Perdomo-Ramírez A, Ramos-Trujillo E, et al. Characterization of pre-mRNA splicing defects caused by CLCN5 and OCRL mutations and identification of novel variants associated with dent disease. Biomedicines. 2023;11(11):3082. doi: 10.3390/biomedicines11113082.
- van Rahden VA, Brand K, Najm J, et al. The 5-phosphatase OCRL mediates retrograde transport of the mannose 6-phosphate receptor by regulating a Rac1-cofilin signalling module. Hum Mol Genet. 2012;21(23):5019–5038. doi: 10.1093/hmg/dds343.
- Zaniew M, Bökenkamp A, Kolbuc M, et al. Long-term renal outcome in children with OCRL mutations: retrospective analysis of a large international cohort. Nephrol Dial Transplant. 2018;33(1):85–94. doi: 10.1093/ndt/gfw350.
- Blanchard A, Curis E, Guyon-Roger T, et al. Observations of a large dent disease cohort. Kidney Int. 2016;90(2):430–439. doi: 10.1016/j.kint.2016.04.022.
- Zaniew M, Mizerska-Wasiak M, Załuska-Leśniewska I, et al. Dent disease in Poland: what we have learned so far? Int Urol Nephrol. 2017;49(11):2005–2017. doi: 10.1007/s11255-017-1676-x.
- Akuta N, Lloyd SE, Igarashi T, et al. Mutations of CLCN5 in Japanese children with idiopathic low molecular weight proteinuria, hypercalciuria and nephrocalcinosis. Kidney Int. 1997;52(4):911–916. doi: 10.1038/ki.1997.412.
- Hoopes RR, Jr, Raja KM, Koich A, et al. Evidence for genetic heterogeneity in dent’s disease. Kidney Int. 2004;65(5):1615–1620. doi: 10.1111/j.1523-1755.2004.00571.x.
- Lourdel S, Grand T, Burgos J, et al. ClC-5 mutations associated with dent’s disease: a major role of the dimer interface. Pflugers Arch. 2012;463(2):247–256. doi: 10.1007/s00424-011-1052-0.
- Domingo-Gallego A, Pybus M, Bullich G, et al. Clinical utility of genetic testing in early-onset kidney disease: seven genes are the main players. Nephrol Dial Transplant. 2022;37(4):687–696. doi: 10.1093/ndt/gfab019.
- Ranawaka R, Sirisena ND, Dayasiri KC, et al. The first Sri Lankan family with dent disease-1 due to a pathogenic variant in the CLCN5 gene: a case report. BMC Res Notes. 2017;10(1):539. doi: 10.1186/s13104-017-2881-5.
- Gambaro G, Naticchia A, Ferraro PM, et al. Living kidney donation in a type 1 dent’s disease patient from his mother. Kidney Blood Press Res. 2019;44(5):1306–1312. doi: 10.1159/000503301.
- Ramos-Trujillo E, González-Acosta H, Flores C, et al. A missense mutation in the chloride/proton ClC-5 antiporter gene results in increased expression of an alternative mRNA form that lacks exons 10 and 11. Identification of seven new CLCN5 mutations in patients with dent’s disease. J Hum Genet. 2007;52(3):255–261. doi: 10.1007/s10038-007-0112-y.
- Utsch B, Bökenkamp A, Benz MR, et al. Novel OCRL1 mutations in patients with the phenotype of dent disease. Am J Kidney Dis. 2006;48(6):942.e1-14–942.14. Dec doi: 10.1053/j.ajkd.2006.08.018.
- Hoopes RR, Jr, Shrimpton AE, Knohl SJ, et al. Dent disease with mutations in OCRL1. Am J Hum Genet. 2005;76(2):260–267. doi: 10.1086/427887.
- Hichri H, Rendu J, Monnier N, et al. From lowe syndrome to dent disease: correlations between mutations of the OCRL1 gene and clinical and biochemical phenotypes. Hum Mutat. 2011;32(4):379–388. doi: 10.1002/humu.21391.