
Abstract
Cisplatin is a particularly potent antineoplastic drug. However, its usefulness is restricted due to the induction of nephrotoxicity. More recent research has indicated that β-hydroxybutyrate (β-HB) protects against acute or chronic organ damage as an efficient healing agent. Nonetheless, the therapeutic mechanisms of β-HB in acute kidney damage caused by chemotherapeutic drugs remain unclear. Our study developed a model of cisplatin-induced acute kidney injury (AKI), which involved the administration of a ketogenic diet or β-HB. We analyzed blood urea nitrogen (BUN) and creatinine (Cr) levels in serum, and used western blotting and immunohistochemical staining to assess ferroptosis and the calcium/calmodulin-dependent kinase kinase 2 (Camkk2)/AMPK pathway. The mitochondrial morphology and function were examined. Additionally, we conducted in vivo and in vitro experiments using selective Camkk2 inhibitor or activator to investigate the protective mechanism of β-HB on cisplatin-induced AKI. Exogenous or endogenous β-HB effectively alleviated cisplatin-induced abnormally elevated levels of BUN and Cr and renal tubular necrosis in vivo. Additionally, β-HB reduced ferroptosis biomarkers and increased the levels of anti-ferroptosis biomarkers in the kidney. β-HB also improved mitochondrial morphology and function. Moreover, β-HB significantly attenuated cisplatin-induced cell ferroptosis and damage in vitro. Furthermore, western blotting and immunohistochemical staining indicated that β-HB may prevent kidney injury by regulating the Camkk2-AMPK pathway. The use of the Camkk2 inhibitor or activator verified the involvement of Camkk2 in the renal protection by β-HB. This study provided evidence of the protective effects of β-HB against cisplatin-induced nephrotoxicity and identified inhibited ferroptosis and Camkk2 as potential molecular mechanisms.
Graphical Abstract
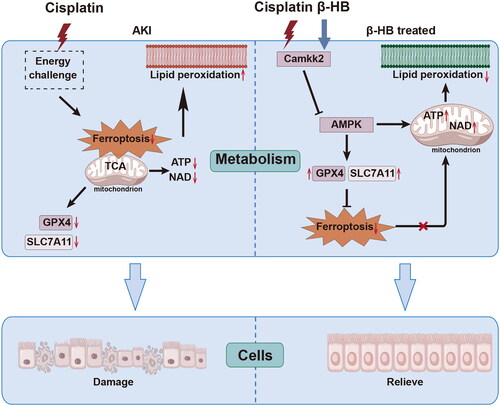
Cisplatin induces disruptions in mitochondrial energy metabolism and lipid peroxidation. Both endogenous and exogenous β-hydroxybutyrate can protected against cisplatin-induced renal damage by repairing mitochondrial energy homeostasis and preventing ferroptosis, and this protective effect might be attributed to Camkk2 regulation.
HIGHLIGHTS
β-HB protects against cisplatin-induced renal damage both in vivo and in vitro.
Moreover, β-HB is effective in attenuating cisplatin-induced lipid peroxidation and ferroptosis.
The regulation of energy metabolism, as well as the treatment involving β-HB, is associated with Camkk2.
1. Introduction
Acute kidney injury (AKI), a clinical condition frequently encountered in hospitalized patients, increases the probability of developing chronic kidney disease and end-stage renal disease [Citation1]. Nephrotoxicity is a serious and common complication of numerous potent chemotherapeutic agents, specifically cisplatin, is one of the contributing factors. The incidence of AKI in patients receiving cisplatin therapy is 30%. However, effective methods for alleviating cisplatin-induced nephrotoxicity are scarce [Citation2]. Recent studies have suggested a potential link between ferroptosis, a mode of programmed cell death triggered by iron-dependent lipid peroxidation, and cisplatin-induced nephrotoxicity. The administration of cisplatin leads to excessive lipid peroxidation, free iron release, and decreased expression of glutathione peroxidase-4 (GPX4) in mouse kidneys. This highlights the connection between ferroptosis and the pharmacological mechanisms of cisplatin-induced nephrotoxicity [Citation3]. Therefore, the administration of chemotherapeutic drugs to induce ferroptosis is a promising strategy in clinical practice. However, there are currently no effective drugs available for clinical application against ferroptosis.
Ketone bodies are small molecules synthesized by hepatocytes in the liver, with β-hydroxybutyrate (β-HB) being the major form in mammals. During periods of fasting, prolonged exercise, and limited carbohydrate metabolism, β-HB is produced and assimilated by the body to provide energy to vital organs. In 2015, Youm et al. [Citation4] reported that β-HB was capable of blocking inflammasome-mediated inflammatory disease. They also found that an increase in endogenous β-HB levels and exogenous β-HB supplementation could reduce renal ischemia-reperfusion (I/R) injury by inhibiting caspase-1 mediated pyroptosis in mice [Citation5]. Additionally, it has recently been reported that β-HB administration can alleviate ferroptosis-mediated acute pancreatic damage in vivo or in vitro [Citation6].
Calcium/calmodulin-dependent kinase 2 (Camkk2) is a crucial secondary messenger that regulates multiple biological activities through integrated functions [Citation7]. Extensive research has suggested that the Camkk2/AMPK signaling pathway contributes to both energy metabolism and cancer cell differentiation [Citation8,Citation9]. Additionally, it was recently found that Camkk2 is prominently activated during ferroptosis [Citation10]. β-HB plays a significant role in regulating energy metabolism. However, comprehensive analyses of the impact of β-HB on renal damage induced by cisplatin and the related molecular mechanisms are currently absent. Therefore, we speculated that β-HB alleviates cisplatin-induced AKI by regulating energy metabolism and ferroptosis, and that Camkk2 may be involved in this regulation. The present study investigated the pivotal pharmacological role of β-HB in limiting cisplatin-mediated lipid peroxidation and ferroptosis in cells and mice. Additionally, we explored the prospective involvement of the Camkk2/AMPK pathway, which could aid in ensuring the effective management of nephrotoxicity by utilizing β-HB.
2. Materials and methods
2.1. Animal and experimental protocol
Thirty-five male C57BL/6J mice, weighing 20 to 22 g, were purchased from Changzhou Cavens Laboratory Animal Co., LTD. They were subsequently kept under standard conditions and provided with free access to food and tap water. All experimental procedures were in compliance with the Principles of Laboratory Animal Care and approved by the Animal Studies Committee of Shanxi Medical University (Taiyuan, China).
To investigate the impact of β-HB on cisplatin-induced nephrotoxicity, a cohort of mice were randomly assigned to one of the following groups (n = 7): Normal control group (administered intraperitoneal (i.p.) injection of saline daily), β-HB group (received 10 mmol/kg β-HB i.p. injection daily for eight days), Cisplatin (Cis) group (received a single i.p. injection of cisplatin, 20 mg/kg), Cis + β-HB group (received a single i.p. injection of cisplatin and were pretreated with β-HB at a dose of 10 mmol/kg for eight consecutive days). Cis + KD group (received a single i.p. injection of cisplatin and were fed a ketogenic diet according to Ref. [Citation11] for eight consecutive days). The experimental procedure is shown in . Ketogenic diet was supplied by Jiangsu XietongBio Co., Ltd (XTKD01). It contained 90% fat, 10% protein, 0% carbohydrate in kcal. The control diet contained 11% fat, 22% protein, and 67% carbohydrate in kcal (1010001). At 6th day, saline or cisplatin was injected. After 72 h of saline or cisplatin challenge, all groups of mice were sacrificed via isoflurane anesthesia followed by cervical dislocation.
2.2. Cell culture and experimental protocol
The human proximal tubular cell (HK-2) was purchased from ShareBio (Shanghai, China) and cultured in Dulbecco’s Modified Eagle Medium (DMEM) (KEYGen Bio, Jiangsu, China), supplemented with 10% fetal bovine serum (FBS) (Abwbio, Germany). The cells were incubated in5% CO2 atmosphere in a humidified chamber at 37 °C.
To assess the cytotoxicity of cisplatin (Sigma, St. Louis, MO, USA) and identify the potential protective effects of β-HB in HK-2 cells, cultured cells were subjected to 20 µM cisplatin and 30 μM β-HB for 24 h. Additionally, we established a control group of normal cells treated with β-HB to gauge its cytotoxicity in vitro.
To verify the essential functions of Camkk2 in the protective effects of β-HB against cisplatin-induced AKI, we set Cis + STO609 (1 μg/mL, Camkk2 inhibitor, MCE, HY-19805). Additionally, a β-HB + cisplatin group was created, followed by the inclusion of the Camkk2 activator methyl cinnamate (100 μM, β-HB + Cis + Meth group, MCE, HY-W017212).
2.3. Renal function monitoring
At the conclusion of the experiment, the serum samples of mice were obtained underwent centrifugation at 5000 rpm for 20 min. Blood urea nitrogen (BUN, C013-2-1, JianchengBio, China) and creatinine (Cr, C011-2-1, JianchengBio) levels were determined using commercial kits as per the manufacturer’s instructions.
2.4. HE and PAS staining
Kidney tissues were fixed using 4% paraformaldehyde in phosphate-buffered saline (PBS) and subsequently embedded in paraffin wax before being sliced. The ensuing slices were stained using hematoxylin-eosin (HE) and periodic acid-schiff (PAS). Tubular injury was assessed on PAS-stained sections. The samples were evaluated based on the score of tubular injury, determined by assessing tubular epithelial cell loss, tubular necrosis, accumulation of cellular debris, and tubular cast formation, was scored according to the percentage of affected tubules under high-power microscopy. The percentage of tubules affected was assigned a score: no damage = 0; <25% = 1; <50% = 2; <75% = 3; and >75% = 4 according to Ref. [Citation12].
2.5. Measurement of tissue iron levels
Iron ion concentrations in renal cortical tissue homogenates were determined using a tissue iron assay kit (A039-2-1, JianchengBio), in accordance with the manufacturer’s instructions. In brief, kidney tissues retrieved from a −80 °C freezer and were sonicated in sample diluent (200 μl) to produce a 10% homogenate (20 mg). The homogenates were centrifuged at 3000 rpm for 15 min at 4 °C. Iron ion levels were determined by measuring the optical density (OD) at 520 nm, and iron levels were normalized to the total protein levels.
2.6. β-HB and lipid peroxidation detection
β-HB and lipid peroxidation concentrations in renal cortical tissue homogenates, and serum were determined using an enzyme-linked immunosorbent assay (ELISA) kit (E-BC-K785-M, Elabscience, China), Glutathione (GSH, A006-2-1, JianchengBio), Glutathione peroxidase 4 (GPX4, H545-1-1, JianchengBio), Lipid hydroperoxide (LPO, A106-1-3, JianchengBio) and Malondialdehyde (MDA, A003-4-1, JianchengBio), in accordance with the manufacturer’s instructions. In brief, kidney tissues retrieved from a −80 °C freezer and were sonicated in sample diluent (200 μl) to produce a 10% homogenate (20 mg). The homogenates were centrifuged at 3000 rpm for 15 min at 4 °C. The resulting supernatant was employed as ELISA samples following the instructions for detection.
Subsequently, serum samples were collected and centrifuged at 5000 rpm for 20 min as ELISA samples in accordance with the detection protocol. The optical density (OD) was then measured with a continuous-wavelength multifunctional enzyme analyzer (Eppendorf, Germany) at a wavelength of 450 nm. Finally, the content of β-HB and lipid peroxidation was determined using standard curves derived from provided calibrators.
2.7. Cell viability assay
The HK-2 cells (3 × 103/well) in 96-well plates were pretreated with different concentrations (10 μM, 20 μM, 30 μM) of β-HB (52017, Sigma, USA) for 24 h. Subsequently, 20 μM cisplatin (1134357, Sigma, USA) was added, while the control group was exposed to PBS. Following a 3 h, 6 h, 12 h, or 24 h incubation, CCK-8 solution (40203ES60, Yeasen, China) was introduced to the cell medium and incubated for an hour at 37 °C. The microplate reader determined the absorbance values at 450 nm (Thermo Scientific, USA). Cell viability = OD450/0h OD450.
2.8. ATP and NAD+/NADH assay
Commercial kits were used to homogenize the cells in RIPA lysis buffer (P0013C, BeyotimeBio, China) and collect samples for ATP (MAK190, Sigma) and NAD+/NADH (S0175, BeyotimeBio) assays, following the manufacturer’s protocols.
2.9. Apoptosis and mitochondrial membrane potential assay
Apoptosis was assessed by flow cytometry (BD, United States) using the Annexin V-Alexa Fluor 488/propidium iodide (PI) Apoptosis Detection Kit (SB-Y6002, ShareBio, China), in accordance with the instructions provided. The mitochondrial membrane potential (MMP) was evaluated by flow cytometry using the TMRE MMP Kit (ab113852, Abcam) according to the instruction.
2.10. Western blotting
Cells and kidney tissues were homogenized in RIPA lysis buffer containing a protease inhibitor cocktail (P1005, Beyotime). Subsequently, the protein concentrations were determined using the BCA kit (P0012S, Beyotime). The protein extracts were separated on 7.5% or 12.5% SDS-PAGE gels (120 V, 90 min) and further electro-transferred to FVDF membranes (80 V, 90 min). After being blocked with 5% bovine serum albumin, the membranes were incubated at 4 °C overnight with the appropriate primary antibodies (Camkk2, 11549-1-AP, Proteintech; p-Camkk2, 12818, CST; AMPK, 2532, CST; p-AMPK, 2535, CST; GPX4, ab125066, Abcam; SLC7A11, 11549-1-AP, Proteintech;). Following this, the membranes were washed with TBST for 5 × 5 min and subsequently incubated with HRP-conjugated secondary antibody (LF102, Epizyme, Shanghai) for 1 h at room temperature. Finally, the bands on the membranes were visualized and analyzed by electrochemiluminescence. The expression of β-actin (4970, CST) was utilized as a control.
2.11. Immunofluorescence and immunohistochemical staining
Slides containing tissues were immersed in xylene followed by decreasing concentrations of alcohol (100%, 95%, 90%, 85%, and 75%) and finally phosphate buffer solution (PBS). The slides were incubated in citrate buffer to retrieve the epitope for 20 min at 95 °C, treated with 3% hydrogen peroxide for 10 min, and then exposed to 10% goat serum for 60 min at room temperature.
For immunohistochemical staining, primary antibodies (HAVCR1 (KIM-1), Proteintech, 30948) were added overnight at 4 °C.slides were washed with PBS for immunohistochemical staining and subsequently treated with the MaxVision TM HRP-Polymer anti-Rabbit IHC Kit (MaixinBio, KIT-5004) for 15 min at room temperature, then washed with PBS. Sections were counterstained with DAB for 10 min then washed with PBS. After treatment with hematoxylin for five minutes and washing with PBS, microscope photographs were taken.
For immunofluorescent staining, primary antibodies (GPX4, ab125066, Abcam; SLC7A11, 11549-1-AP, Proteintech; p-Camkk2, 12818, CST; p-AMPK, 2535, CST) were added overnight at 4 °C.
Afterwards, all slides were washed with PBS and treated with AF488-conjugated anti-rabbit antibody (ab150077, Abcam) for 60 min at room temperature, followed by additional washing with PBS. Finally, the sections were counterstained with DAPI and photographed using a fluorescence microscope.
2.12. Transmission electron microscopy
The renal cortex was gathered and preserved using 2.5% glutaraldehyde. Subsequent to rinsing with PBS, the preserved tissues underwent dehydration utilizing a sequence of graded ethanol and were sliced into ultrathin sections. The sections were subsequently colored with a 1% osmium solution for an hour, followed by fixation and staining with a 2% uranium acetate aquatic solution for 30 min. Ultimately, the sections were analyzed using an HT-7500 transmission electron microscope.
3. Statistic analysis
Data are presented as mean ± standard deviation (SD). To identify differences between groups, one-way ANOVA was conducted, followed by Bonferroni multiple tests. A p value of less than 0.05 was considered as statistically significant. Statistical analyses were carried out using GraphPad Prism 9 software.
4. Results
4.1. β-HB alleviated cisplatin-induced lipid peroxidation in vitro
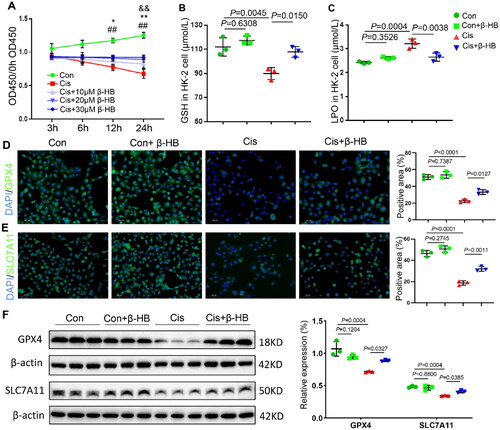
To investigate the mechanism underlying cisplatin induced nephrotoxicity, lipid peroxidation and ferroptosis biomarkers were measured in HK-2 cells. As shown in , cell viability was determined using the CCK-8 assay to assess the extent of damage. Cis-treated cells showed a strongly decreased optical density (OD) value, while cells treated with different concentrations of β-HB showed an increased OD value, indicating that β-HB protected against cisplatin-mediated damage. According to the results of the CCK-8 assay, a concentration of 30 µM β-HB and a time-point of 24 h were used chosen for use in the cell assay. The results of GSH and LPO showed that the addition of β-HB alleviated cisplatin-induced lipid peroxidation, and β-HB-treated normal cells showed no changes (). Immunofluorescence staining of cells was also performed to detect the biomarkers of anti-ferroptosis GPX4 and SLC7A11. The results showed that cisplatin resulted in the lower expression, but the β-HB intervention increased the expression levels of GPX4 and SLC7A11, indicating the effects of β-HB on anti-lipid peroxidation in vitro (). Western blotting was also used to determine the expression of GPX4 and SLC7A11, and the results were consistent with those of the immunofluorescence staining in HK-2 cells ().
Figure 1. β-HB reduced cisplatin-induced lipid peroxidation in vitro. A Cell viability of HK-2 cells exposed to 20 µM Cis without or with 10 µM, 20 µM or 30 µM β-HB for 3h, 6h, 12h and 24h, between Con, Cis and Cis + β-HB groups (n = 3, &&p < 0.01 vs Cis + 10µM β-HB group, *p < 0.05 vs Cis + 20µM β-HB group, **p < 0.01 vs Cis + 20µM β-HB group, ##p < 0.01 vs Cis + 30µM β-HB group); B, C GSH and LPO levels in HK-2 cells was quantified between different groups including Con, Con + β-HB, Cis and Cis + β-HB, cells were treated with 20 µM Cis and/or 30 µM β-HB for 24h (n = 3); D, E Representative confocal images of the expression of GPX4 (green), SLC7A11 (green) and DAPI (blue) in HK-2 cells; F GPX4 and SLC7A11 in HK-2 cells were detected by western blotting. β-actin was used as a loading control. Data were analyzed using one-way ANOVA with Tukey’s test and expressed as the means ± SD.
4.2. β-HB inhibited cisplatin-induced mitochondrial dysfunction in vitro
Since lipid peroxidation is mainly caused by mitochondrial dysfunction, we examined the effect of β-HB on mitochondrial function in vitro. As shown in , cisplatin-treated cells showed lower levels of ATP and NAD+/NADH than the control (Con) group, whereas the ATP and NAD+/NADH levels were significantly increased after the addition of β-HB. Cisplatin-treated cells had lower differences in mitochondrial membrane potential (MMP) compared to the Con group, and β-HB intervention reversed the changes in MMP ().
Figure 2. β-HB inhibited cisplatin-induced mitochondrial dysfunction in vitro. A, B Quantitative analysis of the results of ATP and NAD+/NADH in HK-2 cells between different groups including Con, Con + β-HB, Cis and Cis + β-HB, cells were treated with 20 µM Cis and/or 30 µM β-HB for 24h (n = 3); C, D Representative flow cytometric data of MMP and Annexin V/PI staining. Data were analyzed using one-way ANOVA with Tukey’s test and expressed as the means ± SD.
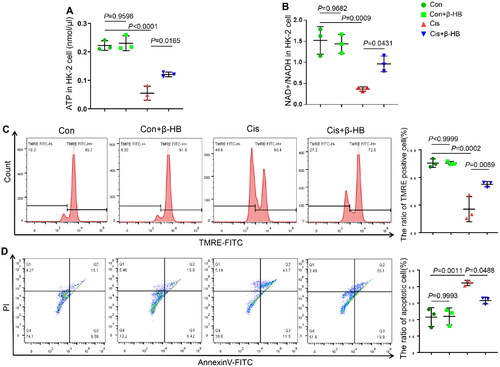
In addition, cisplatin-treated cells showed more positive staining for both annexin V and PI, indicating more cell injury than in the Con group. However, treatment with 30 µM β-HB ameliorated cell apoptosis (). Moreover, β-HB had little effect on normal cells. These results suggest that β-HB prevented cisplatin-induced HK-2 cell injury by regulating mitochondrial energy metabolism.
4.3. β-HB decreased cisplatin-induced renal injury in vivo
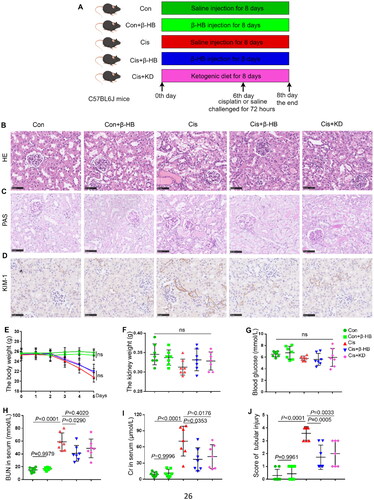
Next, we investigated the efficacy of β-HB in mice. The experimental flowchart is shown in . HE and PAS staining revealed that renal tubules in the Cisplatin group were significantly necrotic, with structural damage and necrotic disintegration of the lumen. However, supplementation with exogenous or endogenous β-HB resulted in relatively intact tubular structures, and the damage was somewhat reduced, as shown by a significant decrease in pathological damage scores (p < 0.05, ). We also examined kidney injury molecule-1 (KIM-1) using immumohistochemical staining, which showed that KIM-1 expression was significantly increased in kidney tissue in AKI mice and that endogenous or exogenous β-HB significantly alleviated kidney injury (). Changes in body weight indicate severe injury caused by cisplatin, but mice in the Con or Con + β-HB group did not experience significant changes in body weight (). Moreover, there were no significant differences in kidney weight and blood glucose levels between the groups (). Common biochemical indicators of renal injury, such as serum BUN and Cr, were significantly higher in the Cis group than in the control group. After intervention with exogenous β-HB, there was a significant decrease in BUN and Cr levels (p < 0.05); however, treatment with a ketogenic diet (KD) elicited a significant decrease in Cr but not in BUN levels. Furthermore, there were no noticeable changes in BUN and Cr levels in the Con + β-HB group when compared to the Con group ().
Figure 3. β-HB decreased cisplatin-induced renal injury in vivo. A Schematic of the experimental design (male, n = 7, by Figdraw); B, C Representative HE-stained and PAS-stained pathological images of kidney from different groups, including Con, Con + β-HB, Cis, Cis + β-HB and Cis + KD, the mice were treated with 20 mg/kg cisplatin, 10 mmol/kg β-HB or ketogenic diet (n = 7); D Representative immunohistochemical staining of KIM-1 from different groups; E, F The determination of body weight and renal weight in mice within the various groups; G-I Statistical analysis of blood glucose, blood urea nitrogen (BUN), and creatinine (Cr) in serum; J Pathological score of tubular injury. Data were analyzed using one-way ANOVA with Tukey’s test and expressed as the means ± SD.
4.4. β-HB restrained cisplatin-induced ferroptosis in vivo
First, we observed elevated levels of β-HB in the serum and kidneys due to exogenous β-HB supplementation (p < 0.05, ). However, the levels of β-HB significantly increased after endogenous β-HB supplementation only in the blood, not in the kidneys (). This difference may be related to the amount of food consumed by the mice. Iron overload is a crucial factor for ferroptosis. Therefore, we analyzed the iron ion content of kidney tissues in each group. These results revealed that cisplatin caused iron overload in the renal tissues of mice. However, endogenous or exogenous β-HB treatment significantly reduced the iron ion content. It is worth noting that exogenous β-HB did not cause any changes in iron ions in the renal tissues of normal mice (). Biomarkers indicating anti-lipid peroxidation, namely GSH and GPX4, showed a significant decline following exposure to cisplatin. However, the supplementation of either exogenous or endogenous β-HB effectively increased their levels, even though renal GPX4 levels in the Cis + KD group were not statistically significant (). LPO and MDA, which are products of lipid peroxidation, showed an increase in the Cis group, while β-HB reduced the levels of LPO and MDA, even though the ketogenic diet did not significantly reduce MDA levels. These results indicate an anti-ferroptotic effect of β-HB (). We found no significant alterations in lipid peroxidation in β-HB treated normal mice. To further verify the anti-ferroptosis effectiveness of β-HB, immunofluorescent staining was conducted to evaluate the expression of GPX4 and SLC7A11, and the findings were consistent with previously reported anti-ferroptosis outcomes. ( and ). Normal nuclear and mitochondrial morphologies were observed in the Con and Con + β-HB groups. In contrast, cisplatin caused vacuolization and constringency in the mitochondria, but the ultrastructural damage in the kidneys was significantly improved by endogenous or exogenous β-HB pretreatment ().
Figure 4. β-HB inhibited cisplatin-induced ferroptosis in vivo. A, B Quantitative analysis of β-HB in serum and in kidney tissue between different groups, including Con, Con + β-HB, Cis, Cis + β-HB and Cis + KD, the mice were treated with 20 mg/kg cisplatin, 10 mmol/kg β-HB or ketogenic diet (n = 7); C. Quantitative analysis of iron ion in kidney tissue between different groups; D-G Quantitative analysis of GSH, GPX4, LPO, and MDA in kidney tissue between different groups; H, I Representative confocal images of the expression of GPX4 (green) and SLC7A11 (green) and DAPI (blue) in kidney tissue; J Images for transimission electron micrographs of kidney mitochondria. Data were analyzed using one-way ANOVA with Tukey’s test and expressed as the means ± SD.
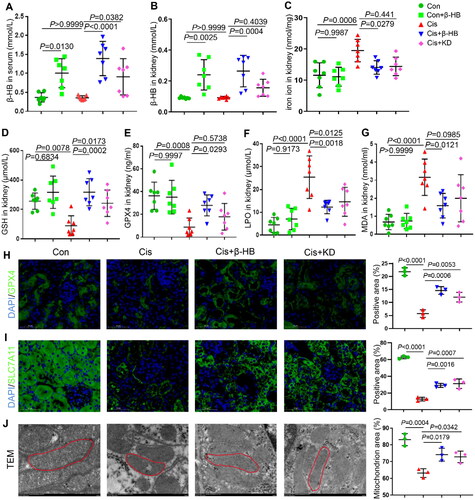
4.5. β-HB regulated Camkk2 expression
To investigate the pharmacological effects of β-HB in regulating Camkk2 and ferroptosis, immunofluorescence staining and western blotting were performed in HK-2 cells. The results of kidney immunofluorescence staining showed that the levels of p-Camkk2 and p-AMPK were significantly increased. The p-AMPK levels decreased in the Cisplatin group. In the Cisplatin + β-HB and Cis + KD groups, the expression levels of p-Camkk2 decreased, and those of p-AMPK significantly increased (). The results of the immunofluorescence staining of HK-2 cells were consistent with the above results (). Western blotting of kidney tissues and HK-2 cells also showed that β-HB could regulate the Camkk2/AMPK pathway in cisplatin-induced kidney injury (). This suggest that β-HB may exert its effects by regulating the Camkk2/AMPK pathway.
Figure 5. β-HB regulated Camkk2 expression. A, B Representative confocal images of the expression of p-Camkk2 (green) and p-AMPK (green), and DAPI (blue) in kidney tissue between different groups, including Con, Cis, Cis + β-HB and Cis + KD, the mice were treated with 20 mg/kg cisplatin, 10 mmol/kg β-HB or ketogenic diet (n = 3); C, D Representative confocal images of the expression of p-Camkk2 (green) and p-AMPK (green), and DAPI (blue) in HK-2 cells between different groups, including Con, Con + β-HB, Cis and Cis + β-HB, cells were treated with 20 µM Cis and/or 30 µM β-HB for 24h (n = 3); E, F Camkk2/p-Camkk2 and AMPK/p-AMPK in kidney tissue and HK-2 cells were detected by western blotting. β-Actin was used as a loading control (n = 3). Data were analyzed using one-way ANOVA with Tukey’s test and expressed as the means ± SD.
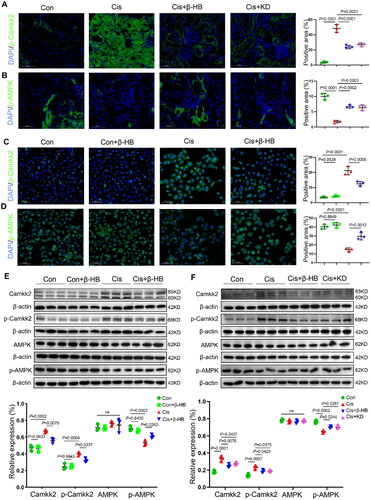
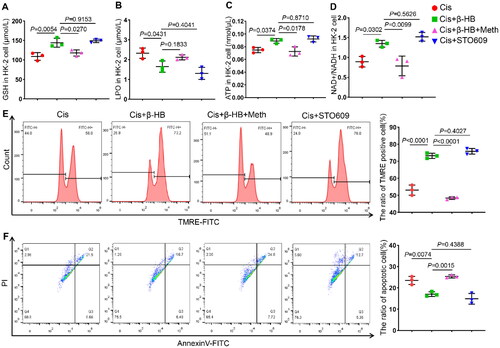
4.6. Camkk2 is involved in the regulation of mitochondrial dysfunction in vitro
Finally, to verify the key role of Camkk2 in the treatment of cisplatin-induced kidney injury by β-HB, we added the STO609, a selective Camkk2 inhibitor, and methyl cinnamate, a selective Camkk2 activator. Inhibiting the activation of Camkk2 resulted in an improvement in the GSH levels, with the same efficiency observed in β-HB treatment. Furthermore, the Camkk2 activator offset these anti-ferroptosis effects (). The results of the lipid peroxidation biomarker LPO also supported this conclusion (). Next, ATP and NAD+/NADH levels in HK-2 cells were tested, and the results showed that the decrease in ATP and NAD+/NADH caused by cisplatin could be alleviated by STO609, while activated Camkk2 could offset this effect (). The results of MMP and apoptosis of cells showed that STO609 had the same effects in increasing MMP and decreasing the proportion of apoptosis with β-HB intervention. When Camkk2 activator is added, the protected efficacy of β-HB can be reversed ().
Figure 6. Camkk2 is involved in the regulation of mitochondrial dysfunction in vitro. A-D Quantitative analysis of GSH, LPO, ATP, and NAD+/NADH in HK-2 cells between different groups, including Cis, Cis + β-HB, Cis + STO609 and Cis + β-HB + Meth, cells were treated with 20 µM Cis and/or 30 µM β-HB for 24h (n = 3); E-F Representative flow cytometric data of MMP and Annexin V/PI staining. Data were analyzed using one-way ANOVA with Tukey’s test and expressed as the means ± SD.
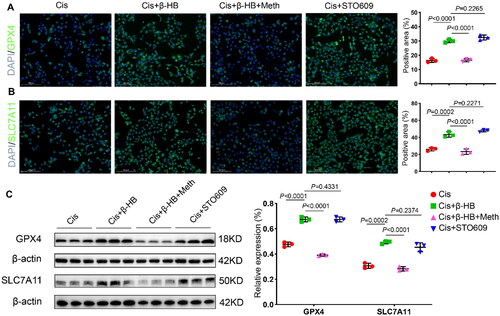
4.7. Camkk2 is involved in the regulation of ferroptosis in vitro
Finally, immunofluorescence staining of HK-2 cells was performed to detect the anti-ferroptosis biomarkers, GPX4 and SLC7A11. The results showed that STO609 had the same effect as β-HB in increasing the levels of GPX4 and SLC7A11. When the Camkk2 activator was added, the anti-ferroptosis efficacy of β-HB vanished ( and ). Moreover, western blotting was used to detect GPX4 and SLC7A11 to validate the critical role of Camkk2 in cisplatin-induced ferroptosis. The results indicated that Camkk2 is involved in the regulation of ferroptosis ().
5. Discussion
Cisplatin, recommended as a first-line chemotherapeutic agent, passes freely through the glomerular filter and is heavily reabsorbed in the proximal tubules; this accumulation can lead to nephrotoxicity. Accumulating evidence suggests that there is a close relationship between metabolism and ferroptosis. The present results showed that cisplatin disrupted mitochondrial homeostasis, and induced the accumulation of lipid peroxidation and loss of GPX4, resulting in ferroptosis of renal tubular epithelial cells and AKI. Endogenous or exogenous β-HB treatment could inhibit cisplatin-induced ferroptosis both in vivo and in vitro. In addition, the anti-ferroptosis effects of β-HB may be related to the regulation of the Camkk2/AMKP pathway. STO609, a selective Camkk2 inhibitor, rescued mitochondrial damage, lipid peroxidation and ferroptosis, thereby ameliorating cisplatin-induced cellular damage. Importantly, the addition of the Camkk2 activator neutralized the efficacy of β-HB to protect against cisplatin-induced mitochondrial injury and lipid peroxidation, suggesting a potential interference with the protective mechanism. This suggests an important role for Camkk2 in cisplatin-induced renal injury. Mechanistically, β-HB protects against cisplatin-induced AKI by regulating Camkk2 and restoring mitochondrial energy control and ferroptosis, revealing new pharmacological mechanisms of β-HB, and targeted regulation of ferroptosis could become a promising strategy for treating various diseases. The mechanism of cisplatin-induced renal toxicity mainly includes inhibition of DNA synthesis, the destruction of mitochondrial membrane potential, and the promotion of cell apoptosis, inflammatory responses and oxidative stress [Citation13], and the association between ferroptosis and cisplatin-induced AKI has been recently recognized. Studies have shown that ferrostatin-1 (Fer-1), an inhibitor of ferroptosis, preserves renal function and reduces histological injury, oxidative stress, and tubular cell death in a folic acid-induced AKI mouse model [Citation14]. In addition, the loss of ferroptosis suppressor protein 1 (Fsp1) or the active center of the selenoprotein GPX4 has been shown to result in acute tubular necrosis and ferroptosis [Citation15]. The results of our study support these findings. Cisplatin paralyzed GPX4 expression and mitochondrial function, resulting in severe lipid peroxidation and ferroptosis ( and ).
Scientific evidence and rationale have also been presented for the administration of various types of exogenous and endogenous ketone sources as a supplement to the restrictive diet protocol or as an alternative to the diet. Intraperitoneal administration of low doses of β-HB can reduce tubular cell apoptosis by upregulating FOXO3 expression, thereby reducing inflammatory damage caused by renal I/R injury in mice [Citation5]. Previous studies have shown that β-HB is an endogenous inhibitor of the NLRP3 inflammasome and inhibits inflammation by downregulating NF-κB activity and NLRP3 expression [Citation16]. In addition, β-HB alleviates oxidative stress, reduces apoptosis and attenuates renal injury caused by paraquat poisoning in rats, in which Nrf2 activation plays an important role [Citation17]. Supplementation with endogenous β-HB at low concentrations has been associated with epigenetic gene regulation, which in turn leads to changes in mitochondrial ATP metabolism that can alleviate oxidative stress in vivo [Citation18,Citation19]. Thus, the protective effects of β-HB may be mediated through metabolic signals by enhancing mitochondrial function for ATP generation. However, one report found that hepatocyte injury and apoptosis in mice with acute inflammation models induced by LPS and D-galactosamine were further exacerbated by β-HB administration [Citation20]. The specific reasons may be related to the characteristics of different disease models, the mode of β-HB treatment and the therapeutic dose of β-HB, and further experimental investigation is required.
Camkk2 is also involved in tumorigenesis, energy metabolism and inflammation. In 2011, Camkk2 mRNA expression in tissue samples was correlated with prostate cancer and disease progression in clinical cohorts [Citation21]. Camkk2 activity is also known to be regulated by a variety of phosphorylation events and is therefore primarily involved in the regulation of the cellular oxidative respiratory chain. For example, cyclin-dependent kinase 5 (CDK5) and glycogen synthase kinase 3 (GSK3) phosphorylate and regulate the activity and stability of Camkk2 during neurodevelopment [Citation22]. Camkk2 has also been shown to regulate appetite through the phosphorylation and activation of AMP-dependent protein kinase (AMPK) in the rodent hypothalamus, activation of AMPK by Camkk2 is important for the maintenance of different cellular and physiological processes. Loss of Camkk2 affects the formation of hippocampus-dependent long-term memory [Citation23]. Furthermore, Camkk2 contributed to the regulation of energy balance by regulating the hypothalamic production of the orexigenic hormone neuropeptide Y (NPY), thus regulating the functions of AMPK kinase. In addition, a loss of Camkk2 protected mice from high-fat diet-induced obesity, insulin resistance, and glucose intolerance [Citation24]. When Camkk2 was deleted in the livers of mice, slower weight gain was observed, and the mice showed lower blood glucose levels and improved glucose tolerance compared to wild-type mice. In addition, primary hepatocytes isolated from Camkk2 knockout mice showed decreased mRNA levels of peroxisome proliferator-activated receptor γ coactivator 1-α and gluconeogenic enzymes, such as glucose-6-phosphatase, resulting in decreased phosphorylation of histone deacetylase 5 [Citation25].
Camkk2 improves glucose tolerance by regulating GAPDH to promote glucose metabolism and PEX3 to blunt peroxisomal fatty acid catabolism in the liver, which may be a new molecular mechanism [Citation26]. Camkk2 is also a key regulator of mitochondrial function, and Camkk2 deletion suppressed cellular respiration and shifted the metabolic phenotype toward aerobic glycolysis by controlling succinate dehydrogenase expression, post-translational modification and megacomplex assembly [Citation27]. Normal cells require an adequate supply of nutrients and energy to maintain basic function. Energy stress is characterized by the depletion of intracellular ATP and a corresponding increase in intracellular AMP levels. AMPK, a critical sensor of cellular energy status, has been implicated in the regulation of ferroptosis-related diseases such as sepsis, diabetic kidney disease and tumors in mouse models [Citation28–30]. In our study, cisplatin-induced AKI or HK-2 cell injury showed increased phosphorylated AMPK and ATP loss, indicating a state of energy stress, while endogenous and exogenous β-HB inhibited the activation of AMPK () and restored energy metabolism homeostasis (ATP and NAD+, ). However, a recent study also reported the promoting role of AMPK in the regulation of ferroptosis, which is contrary to the findings described here. Under prolonged and severe energy stress with excessive ATP depletion, adaptive responses are induced, leading to the activation of AMPK, which inhibits renal I/R injury [Citation31]. In contrast to these findings, we observed an anti-ferroptosis effect in cisplatin-induced AKI, as opposed to using the ischemia-reperfusion injury mouse model, which is likely due to the difference in energy adaptive response patterns.
Figure 7. Camkk2 is involved in the regulation of ferroptosis in vitro. A, B Representative confocal images of the expression of GPX4 (green), SLC7A11 (green)and DAPI (blue) in HK-2 cells between different groups, cells were treated with 20 µM Cis and/or 30 µM β-HB for 24h (n = 3); C GPX4 and SLC7A11 in HK-2 cells were detected by western blotting. β-Actin was used as a loading control. Data were analyzed using one-way ANOVA with Tukey’s test and expressed as the means ± SD.
This study through the use of high-dose cisplatin renal injury model, along with small-dose β-HB intervention, we found that β-HB can effectively alleviate cisplatin-induced renal injury, inhibiting ferroptosis at the time of injury. We also investigated the potential mechanism and found that the Camkk2/AMPK pathway was involved in the protective effects of β-HB in AKI. However, this mechanism was investigated using Camkk2 inhibitors and activators, which is a limitation of this study. Future studies should delve deeper into the Camkk2/AMPK pathway using genetically modified mice or cells.
The involvement of energy metabolism in the onset and development of AKI suggests that β-HB is a promising therapeutic agent for mitigating cisplatin nephrotoxicity and is worthy of further investigation.
Author contributions
Xiaoshuang Zhou and Rongshan Li conceived and designed the research. Material preparation and data collection were performed by Ruixue Tian and Shuqin Tang. Data analysis was performed by Ruixue Tian, Shuqin Tang, Jingyu Zhao, Yajie Hao, Limei Zhao, Xiutao Han, Xingru Wang, and Lijun Zhang. The manuscript was drafted by Ruixue Tian and Shuqin Tang and revised by Xiaoshuang Zhou. All authors read and approved the final version of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The published preprint is available on request from the corresponding author at https://data.mendeley.com/drafts/8cd77m8k22.
Additional information
Funding
References
- Wang Z, Zhang C. From AKI to CKD: maladaptive repair and the underlying mechanisms. Int J Mol Sci. 2022;23(18):10880. doi: 10.3390/ijms231810880.
- Tang C, Livingston MJ, Safirstein R, et al. Cisplatin nephrotoxicity: new insights and therapeutic implications. Nat Rev Nephrol. 2023;19(1):1–13. doi: 10.1038/s41581-022-00631-7.
- Ikeda Y, Hamano H, Horinouchi Y, et al. Role of ferroptosis in cisplatin-induced acute nephrotoxicity in mice. J Trace Elem Med Biol. 2021;67:126798.
- Youm YH, Nguyen KY, Grant RW, et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med. 2015;21(3):263–269. doi: 10.1038/nm.3804.
- Tajima T, Yoshifuji A, Matsui A, et al. Beta-hydroxybutyrate attenuates renal ischemia-reperfusion injury through its anti-pyroptotic effects. Kidney Int. 2019;95(5):1120–1137.
- Zheng Y, Sun W, Shan C, et al. Beta-hydroxybutyrate inhibits ferroptosis-mediated pancreatic damage in acute liver failure through the increase of H3K9bhb. Cell Rep. 2022;41(12):111847.
- Marcelo KL, Means AR, York B. The Ca(2+)/calmodulin/CaMKK2 axis: nature’s metabolic CaMshaft. Trends Endocrinol Metab. 2016;27(10):706–718. doi: 10.1016/j.tem.2016.06.001.
- Pulliam TL, Goli P, Awad D, et al. Regulation and role of CAMKK2 in prostate cancer. Nat Rev Urol. 2022;19(6):367–380. doi: 10.1038/s41585-022-00588-z.
- Tomaszewski WH, Waibl-Polania J, Chakraborty M, et al. Neuronal CaMKK2 promotes immunosuppression and checkpoint blockade resistance in glioblastoma. Nat Commun. 2022;13(1):6483. doi: 10.1038/s41467-022-34175-y.
- Wang S, Yi X, Wu Z, et al. CAMKK2 defines ferroptosis sensitivity of melanoma cells by regulating AMPK–NRF2 pathway. J Invest Dermatol. 2022;142(1):189–200.
- Ang QY, Alexander M, Newman JC, et al. Ketogenic diets alter the gut microbiome resulting in decreased intestinal Th17 cells. Cell. 2020;181(6):1263–1275. doi: 10.1016/j.cell.2020.04.027.
- Song N, Xu Y, Paust HJ, et al. IKK1 aggravates ischemia-reperfusion kidney injury by promoting the differentiation of effector T cells. Cell Mol Life Sci. 2023;80(5):125. doi: 10.1007/s00018-023-04763-2.
- Zhang J, Ye ZW, Tew KD, et al. Cisplatin chemotherapy and renal function. Adv Cancer Res. 2021;152:305–327.
- Martin-Sanchez D, Ruiz-Andres O, Poveda J, et al. Ferroptosis, but not necroptosis, is important in nephrotoxic folic acid-induced AKI. J Am Soc Nephrol. 2017;28(1):218–229.
- Tonnus W, Meyer C, Steinebach C, et al. Dysfunction of the key ferroptosis-surveilling systems hypersensitizes mice to tubular necrosis during acute kidney injury. Nat Commun. 2021;12(1):4402. doi: 10.1038/s41467-021-24712-6.
- Yamanashi T, Iwata M, Kamiya N, et al. Beta-hydroxybutyrate, an endogenic NLRP3 inflammasome inhibitor, attenuates stress-induced behavioral and inflammatory responses. Sci Rep. 2017;7(1):7677. doi: 10.1038/s41598-017-08055-1.
- Wei T, Tian W, Liu F, et al. Protective effects of exogenous beta-hydroxybutyrate on paraquat toxicity in rat kidney. Biochem Biophys Res Commun. 2014;447(4):666–671.
- Shimazu T, Hirschey MD, Newman J, et al. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science. 2013;339(6116):211–214. doi: 10.1126/science.1227166.
- Cheng B, Lu H, Bai B, et al. d-beta-Hydroxybutyrate inhibited the apoptosis of PC12 cells induced by H2O2 via inhibiting oxidative stress. Neurochem Int. 2013;62(5):620–625.
- Yang Y, Shao R, Jiang R, et al. beta-Hydroxybutyrate exacerbates lipopolysaccharide/d-galactosamine-induced inflammatory response and hepatocyte apoptosis in mice. J Biochem Mol Toxicol. 2019;33(9):e22372.
- Karacosta LG, Foster BA, Azabdaftari G, et al. A regulatory feedback loop between Ca2+/calmodulin-dependent protein kinase kinase 2 (CaMKK2) and the androgen receptor in prostate cancer progression. J Biol Chem. 2012;287(29):24832–24843. doi: 10.1074/jbc.M112.370783.
- Green MF, Scott JW, Steel R, et al. Ca2+/calmodulin-dependent protein kinase kinase beta is regulated by multisite phosphorylation. J Biol Chem. 2011;286(32):28066–28079. doi: 10.1074/jbc.M111.251504.
- Peters M, Mizuno K, Ris L, et al. Loss of Ca2+/calmodulin kinase kinase beta affects the formation of some, but not all, types of hippocampus-dependent long-term memory. J Neurosci. 2003;23(30):9752–9760. doi: 10.1523/JNEUROSCI.23-30-09752.2003.
- Anderson KA, Ribar TJ, Lin F, et al. Hypothalamic CaMKK2 contributes to the regulation of energy balance. Cell Metab. 2008;7(5):377–388. doi: 10.1016/j.cmet.2008.02.011.
- Anderson KA, Lin F, Ribar TJ, et al. Deletion of CaMKK2 from the liver lowers blood glucose and improves whole-body glucose tolerance in the mouse. Mol Endocrinol. 2012;26(2):281–291. doi: 10.1210/me.2011-1299.
- Stork BA, Dean A, Ortiz AR, et al. Calcium/calmodulin-dependent protein kinase kinase 2 regulates hepatic fuel metabolism. Mol Metab. 2022;62:101513. doi: 10.1016/j.molmet.2022.101513.
- Sabbir MG, Taylor CG, Zahradka P. CAMKK2 regulates mitochondrial function by controlling succinate dehydrogenase expression, post-translational modification, megacomplex assembly, and activity in a cell-type-specific manner. Cell Commun Signal. 2021;19(1):98. doi: 10.1186/s12964-021-00778-z.
- Zhou B, Zhang J, Chen Y, et al. Puerarin protects against sepsis-induced myocardial injury through AMPK-mediated ferroptosis signaling. Aging. 2022;14(8):3617–3632.). doi: 10.18632/aging.204033.
- Lu Q, Yang L, Xiao JJ, et al. Empagliflozin attenuates the renal tubular ferroptosis in diabetic kidney disease through AMPK/NRF2 pathway. Free Radic Biol Med. 2023;195:89–102.
- Liu Y, Huang P, Li Z, et al. Vitamin C sensitizes pancreatic cancer cells to Erastin-Induced ferroptosis by activating the AMPK/Nrf2/HMOX1 pathway. Oxid Med Cell Longev. 2022;2022:5361241.
- Lee H, Zandkarimi F, Zhang Y, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol. 2020;22(2):225–234. doi: 10.1038/s41556-020-0461-8.