
Abstract
Fabry disease, a lysosomal storage disease, is an uncommon X-linked recessive genetic disorder stemming from abnormalities in the alpha-galactosidase gene (GLA) that codes human alpha-Galactosidase A (α-Gal A). To date, over 800 GLA mutations have been found to cause Fabry disease (FD). Continued enhancement of the GLA mutation spectrum will contribute to a deeper recognition and underlying mechanisms of FD. In this study, a 27-year-old male proband exhibited a typical phenotype of Fabry disease. Subsequently, family screening for Fabry disease was conducted, and high-throughput sequencing was employed to identify the mutated gene. The three-level structure of the mutated protein was analyzed, and its subcellular localization and enzymatic activity were determined. Apoptosis was assessed in GLA mutant cell lines to confirm the functional effects. As a result, a new mutation, c.777_778del (p. Gly261Leufs*3), in the GLA gene was identified. The mutation caused a frameshift during translation and the premature appearance of a termination codon, which led to a partial deletion of the domain in C-terminal region and altered the protein’s tertiary structure. In vitro experiments revealed a significant reduction of the enzymatic activity in mutant cells. The expression was noticeably decreased at the mRNA and protein levels in mutant cell lines. Additionally, the subcellular localization of α-Gal A changed from a homogeneous distribution to punctate aggregation in the cytoplasm. GLA mutant cells exhibited significantly higher levels of apoptosis compared to wild-type cells.
Introduction
Fabry disease (FD) is an X-linked recessive genetic disorder stemming from abnormalities in the galactosidase alpha (GLA) gene. It is the second most prevalent lysosomal storage disease after Gaucher disease [Citation1]. The GLA gene is 12 kb long and located on X chromosome long-arm region 22 (Xq22). It contains seven exons that encode alpha-Galactosidase A (α-Gal A), a lysosomal enzyme, comprising 429 amino acids [Citation2]. α-Gal A is initially synthesized as a high-molecular-weight precursor. It undergoes a series of intricate processes within the endoplasmic reticulum, including proteolysis, glycosylation, and phosphorylation on mannose residues. Subsequently, it is transferred to lysosomal storage following the elimination of unstable variants by the quality control system [Citation3]. Within human podocytes, this process relies on the α-Gal A-interacting proteins, including mannose 6-phosphate/insulin-like growth factor II receptor (M6PR), megalin, and sortilin, which are critical endocytic receptors for α-Gal A transport [Citation4]. α-Gal A catalyzes the hydrolysis of α-galactosidic bonds in glycosphingolipids, polysaccharides, and glycoproteins [Citation5]. The deficiency of α-Gal A resulting from GLA abnormalities [Citation6–7] prevents the conversion of globotriaosylceramide (Gb3) to lactosylceramide [Citation8], which can cause the progressive accumulation of its main metabolic substrates, neutral glycosphingolipids, particularly globotriaosylceramide and galactosylceramide, within the lysosomes of cells across different systems and tissues. Over time, the progressive accumulation of these molecules disrupts cellular function and can ultimately trigger inflammation or fibrosis [Citation6]. Specifically, Gb3 accumulation causes grossly activated autophagy resulting from dysregulated mTOR activity and marked downregulation of AKT activity [Citation9–11]. Meanwhile, accumulation of Gb3 leads to enhanced profibrotic signaling through the activation of the Noch1 and TGF-β cascades [Citation12–14].
Male classical patients with FD present with various clinical manifestations, including acroparesthesia, angiokeratomas, corneal opacity during childhood and adolescence, hypohidrosis, and progressive vascular disease affecting the heart, kidney, and central nervous system [Citation15–16]. While the disease process initiates early, with evidence of storage occurring even during prenatal development, symptoms related to FD typically emerge in early childhood and tend to progress with growth [Citation6]. Notably, the correlation between genotype and phenotype in FD is significantly poor. Even in the same family, individuals with the same mutation can display distinct clinical phenotypes, including differences in disease severity and progression, age at onset, and involvement of specific systems [Citation17–18].
Patients with FD can be categorized into classic and nonclassic subtypes (mild/delayed) based on the biological activity of the mutant α-Gal A [Citation19]. The atypical type is generally associated with later onset and milder symptoms [Citation20–21]. Males are more likely than females to suffer classic subtypes with reduced or even full inactivation of α-Gal A activity. Classic-subtype male patients may even develop renal and cardiac involvement as well as cerebrovascular events [Citation19,Citation22]. Serious FD-related symptoms, including heart disease and stroke, are common in females with FD [Citation23–25]. Moreover, the Lyon effect, involving the random inactivation of a X chromosome, and skewed X-chromosome inactivation can cause variable symptom expression in female heterozygotes (carriers) of FD [Citation26–27].
The global incidence rate of FD in male newborns is approximately 1 in 40,000 to 1 in 117,000(Citation28). However, the actual incidence of FD is likely underestimated because of the challenge of diagnosing non-classic FD. In fact, researchers investigating newborn screening for FD have found a significantly high incidence of non-classical FD [Citation29–31].
To date, over 800 GLA mutations have been found to cause FD, including point mutations and complex rearrangements, according to the Human Gene Mutation Database (http://www.hgmd.org) and the Fabry Mutants List (http://fabry-database.org/mutants/) [Citation29]. Approximately 60% of these are missense mutations, which alter the α-Gal A protein by one amino acid [Citation32–33].
In this study, a new nonsense mutation, c.777_778del (p. Gly261Leufs*3), was reported in the Chinese Han family with FD. With the aim of contributing to a greater understanding of the FD mutation spectrum and pathogenesis of this disease, we analyzed the function of the pathogenic mutations and the corresponding clinical phenotype.
Materials and methods
Patients
The FD patient organization helped to recruit the Chinese Han family, which was located in Guangdong Province, China. Physical and experimental examinations were performed together with the assessment and evaluation of patients’ medical records. The diagnosis of FD was established through a combination of clinical presentation and examination results.
Sequencing analysis
The genomic DNA extraction was conducted from peripheral blood samples obtained from the proband and his family members (II-2, ) using the standardized phenol/chloroform extraction method in the Suzhou PerkinElmer Medical Laboratory (Suzhou, China) [Citation34]. Long-range polymerase chain reaction (PCR) was employed to amplify the entire genomic sequence of the GLA gene. The purified amplicons were processed to generate paired-end libraries, after which high-throughput sequencing was carried out on an Illumina Novaseq platform. After conducting quality control using the fastp software, the Burrows–Wheeler Aligner software (version 0.59) [Citation35] was then used to align the sequencing reads with the GRCh37.p10 human reference genome. The Burrows-Wheeler-aligned reads underwent local realignment and base quality recalibration through the GATK IndelRealigner and GATK Base Recalibrator, respectively [Citation36]. The GATK HaplotypeCaller was used to identify single-nucleotide variants and small insertions or deletions (indel). Variants were annotated with snpEff software (version 4.0).
Figure 1. Pedigree and bioinformatics analysis of the GLA mutant gene c.777_778del (p.Gly261Leufs*3). (a) Family pedigree and Sanger sequencing results. The proband is indicated by an arrow. Sanger sequencing results of the hemizygous mutation c.777_778del in the GLA gene. (b) Evolutionary conservation analysis of the mutated amino acid residues across different species. Arrow indicates the starting sites of the altered amino acid sequence of the mutant protein. (c–d) 3D structures of the wild-type (WT) (c) and mutant (d) α-Gal A proteins predicted by I-TASSER. WT and mutant monomers are colored in white; and amino acid sequences close to the starting site of the mutation are in other colors. The first letter denotes the amino acid abbreviation, and the number denotes the number of amino acid residue sequences. “A” indicates that the site is located in the first domain.
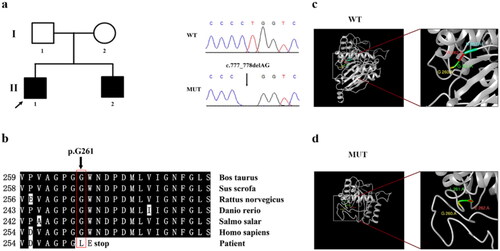
Sanger sequencing was then used to verify FD-associated mutations in GLA gene exon 5 () using the peripheral blood of the proband. Following the analysis of the PCR products using agarose gel electrophoresis, Sanger sequencing was conducted.
Bioinformatics
The three-dimensional (3D) protein structures of both wild-type (WT) and mutant α-Gal A were predicted using the I-TASSER (Iterative Threading ASSEmbly Refinement, http://zhanglab.ccmb.med.umich.edu/I-TASSER/) tool. The MutationTaster (http://www.mutationtaster.org/) tool was applied to predict the probable changes in protein structure and function owing to the amino acid change. The Basic Local Alignment Search Tool (http://blast.st-va.ncbi.nlm.nih.gov/Blast.cgi) helped with sequence alignments and conservation analysis.
Plasmid constructs and mutagenesis
The total RNA extraction was conducted with Trizol reagent (Invitrogen, Carlsbad, CA, USA). Following the manufacturer’s instructions of the HiScript II 1st Strand cDNA Synthesis Kit (Vazyme, Jiangsu, China), The first-strand cDNA was synthesized. The primers constructed with EcoRI and XhoI (New England Biolabs, Beijing, China) restriction sites were used in the amplification of the GLA coding sequence, as follows.
The pEGFP-C1 plasmid: 5′-CCGCTCGAGTAATGCAGCTGAGGAACCCAGA-3′ (forward) and 5′-GGAATTCTTAAAGTAAGTCTTTTAATGACATCTGCA-3′ (reverse). The pcDNA3.1-3xFLAG plasmid: 5′-GGAATTCTATGCAGCTGAGGAACCCAGA-3′ (forward) and 5′-CCGCTCGAGTTAAAGTAAGTCTTTTAATGACATCTGCA-3′ (reverse).
The encoding region of the WT GLA gene was cloned into the XhoI and EcoRI sites of pcDNA3.1-3xFLAG vector (GENEWIZ, NJ, USA) and the pEGFP-C1 vector (GENEWIZ, NJ, USA) through double digestion. PCR-based site-directed mutagenesis was used to introduce the mutation into the WT GLA plasmid. The primer used were as follows: 5′-TGATGTTGCTGGACCGGGGTTGGAATGACC-3′(forward) and 5′-GGTCATTCCAACCCCGGTCCAGCAACATCA-3′(reverse). The Plasmid Miniprep Kit (Axygen, New York, United States) was used to purify the recombinant plasmids and eliminated random mutations after sequencing.
Cell culture and transient transfection
The cultivation of human embryonic kidney (HEK) 293 T cells was performed using standard culture conditions at 37 °C and 5% CO2. Dulbecco’s modified Eagle medium (DMEM; Gibco, NY, USA) was mixed with 100 mg/mL streptomycin, 100 U/mL penicillin, and 10% fetal bovine serum (FBS) (Gibco). The transfection reagents were replaced with fresh 10% FBS DMEM following incubation under standard cultivation conditions for 5 h. The transfected cell culture, in six-well plates, was allowed to stabilize over 8 h. The transfection was carried out using an identical number of cells and a consistent quantity of plasmid DNA across all experimental conditions to maintain uniform transfection efficiency.
RNA analysis
Using polyethylenimine, the transfection of HEK293T cells was performed with the pEGFP-C1 vector (1 μg) or the recombinant plasmids, including either WT or mutant GLA genes. The total RNA was isolated from the cell lysate using Trizol reagent (Invitrogen) after a 36-h incubation period. Using the PrimeScript RT reagent kit (Takara, Dalian, China), reverse transcription was performed. The expression of GLA genes mRNA in WT and mutant cells was compared via Real-time PCR using 2 × RealStar Green Fast Mixture (GenStar, Beijing, China). The internal reference was set as Glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The primers used were as follows:
GLA: 5′-GTTGGATGGCTCCCCAAAGA-3′(forward) and 5′-AGCAAAGGTCTGGGCATCAA-3′(reverse).
GAPDH: 5′-GTGAAGGTCGGAGTCAAGG-3′(forward) and 5′-TGAGGTCAATGAAGGGGTC-3′(reverse).
To ensure reliability of the results, The (2–ΔΔCT) method was repeated three times to evaluate gene transcription levels.
Western blotting analysis
The expression differences of α-Gal A were compared in cells that expressed the WT or mutant GLA genes via western blotting. The resuspension of cells was conducted with 1% phenylmethanesulfonyl fluoride (PMSF, Beyotime Biotechnology) in cell lysis buffer (Beyotime Biotechnology, Shanghai, China) after two times of cold PBS washing (Sigma-Aldrich, Inc., St. Louis, MO, USA). Before transferring onto a polyvinylidene fluoride (PVDF) membrane (MilliporeSigma, Burlington, MA, USA), The protein samples were separated via 10% sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE), followed by standard procedures. The analysis of protein bands’ intensities was performed by ImageJ software. Western blotting was repeated three times for each test. The internal reference was set as GAPDH. The antibodies used were as follows: GAPDH mouse monoclonal antibody (Proteintech, 60004-1-Ig), goat anti-rabbit IgG (Proteintech, SA00001-2), and GFP-tagged rabbit polyclonal antibody (Proteintech, 50430-2-AP).
Cellular localization and transfection efficiency analysis
Immunological fluorescence assays were used to detect the subcellular location of α-Gal A and the transfection efficiency of the WT and mutant GLA genes was analyzed. After 1.5 days of cultivation post-transfection, cells transfected with pEGPF-C1 or the recombinant plasmids, including either WT or mutant GLA genes, were fixed using 4% paraformaldehyde (Sigma-Aldrich) for 30 min. After removing the paraformaldehyde, 0.1% Triton X-100 solution (Thermo Fisher Scientific, Waltham, MA, USA) was incubated to enhance cytomembrane permeability. The cells were imaged with a confocal fluorescence microscope (LSM 880; Carl Zeiss AG, Jena, Germany). Next, the proportion of fluorescent cells in the immunofluorescence images was compared between WT and mutant GLA plasmid-transfected cells by ImageJ software.
α-Gal A enzyme activity assay
The transfection of HEK293T cells was performed with pEGPF-C1 or the recombinant plasmids, including either WT or mutant GLA genes. Cells were collected via centrifugation after 48 h, resuspended with extraction buffer, and sonicated. The α-Gal A enzyme activity in supernatants was assessed in accordance with the manufacturer’s instructions (QIYBO, QYS-03233M, Shanghai, China). As per the manufacturer’s instructions, the assay reagents were added to transfected cells, followed by thorough mixing. The absorbance of the samples at 400 nm was calculated as the difference between the measurement value and the control value. A standard curve was created for numerical conversion between concentration and absorbance. An enzyme activity unit was defined as the production of 1 nmol of p-nitrophenol per hour per 10,000 cells. Following the calculation method given by the supplier, α-Gal A activity was determined.
Apoptosis assays
The transfection of HEK293T cells was performed with the pcDNA3.1-3xFLAG vector or the recombinant plasmids, including either WT or mutant GLA genes, followed by a 48-h incubation period. The resuspension of cells was conducted with binding buffer after disposal of the samples, following the manufacturer’s instructions (YEASEN, 40302ES60). The samples were incubated at 25 °C for 15 min, protected from light exposure, after adding Annexin V-FITC and propidium iodide (PI). Flow cytometry was applied to analyze the cell subsets by fluorescence detection. Briefly, the cells were divided into three subsets: the viable cells with weak background fluorescence, the early-stage apoptotic cells with green fluorescence (Annexin V-FITC staining), and the late-stage apoptotic cells with both green and red fluorescence (both Annexin V-FITC and PI staining).
Statistical analysis
Statistical analysis was conducted with GraphPad Prism software 8.0 (GraphPad Software Inc., San Diego, CA, USA). The comparison of the measured data was conducted using an independent sample two-sided t-test, which were presented as the mean ± standard deviation (SD). The significance level was set at 0.05, with NS denoting results that were not statistically different.
Results
Clinical phenotype and genetic analysis
A 27-year-old Chinese man (II-1, ) was referred to Peking University Shenzhen Hospital in 2021 due to proteinuria. Upon admission, the physical examination disclosed angiokeratomas, supraorbital ridge protrusion, and lip thickening, which are characteristic facial deformities associated with FD [Citation37]. Laboratory results () indicated an estimated glomerular filtration rate (eGFR) of 131.5 mL/min, a serum creatinine level of 63 μmol/L, and 24-h proteinuria of 877 mg. Furthermore, routine urine tests revealed a proteinuria level of 2+. A review of the proband’s medical history unveiled recurrent unexplained fever and acroparesthesia since the age of 11 years. Over time, the proband also began to encounter intermittent pain in the extremities and hypohydrosis. Notably, the proband suffered cerebral infarctions and subsequent hemiplegia in 2015 and 2021. Additionally, foamy urine was observed in 2018 concurrent with a 2+ urinary protein finding in routine urine tests. Furthermore, the proband exhibited paralytic strabismus in the left eye and bilateral inflammatory pseudotumors. Considering the possibility of the proband suffering from FD, activity measurement of α-Gal A in peripheral blood leukocytes of the patient revealed a significant reduction (0.45 μmol/L/h; normal, 2.40–17.65 μmol/L/h). Moreover, plasma globotriaosylsphingosine (LysoGb3) was elevated (84.84 ng/mL; normal, <1–11 ng/mL) (). A review of his family history uncovered that his younger brother (II-2) had FD with a milder clinical phenotype. II-2 presented with a small extent of angiokeratinoma, intermittent pain in the extremities, normal eGFR, and a urine result that suggested proteinuria (2+). His parents and other close relatives were healthy, with no symptoms of FD.
Table 1. Clinical examination results of the proband.
To identify potential pathogenic mutations, sufficient genomic DNA was extracted from the proband and his sibling (II-1 and II-2) using peripheral blood. The long-range PCR was employed to amplify genomic sequence of the GLA gene, followed by high-throughput sequencing. Subsequently, Sanger sequencing was performed. Sequencing results revealed that both patients carried hemizygous mutations in the GLA gene, specifically the c.777_778del mutation ().
Sequence and structure analysis
Sequence alignment revealed that the c.777_778del mutation caused a translation frame shift (Figure S1a) and introduced a stop codon at codon 263 (p.Gly261Leufs*3). Consequently, this led to the deletion of a portion of the first domain (262–330 amino acid residues) and the entire second domain (331–429 amino acid residues) of the encoded protein (Figure S1b). Evolutionary conservation analysis revealed that damaged residues in the truncated protein were highly conserved during the evolution of α-Gal A in various species, suggesting that the mutation was pathogenic to FD (). Further analysis of I-TASSER revealed that the mutation led to changes in the protein’s tertiary structure. (); accordingly, corresponding changes in protein function may occur.
Variant GLA causes abnormalities in enzymatic activity
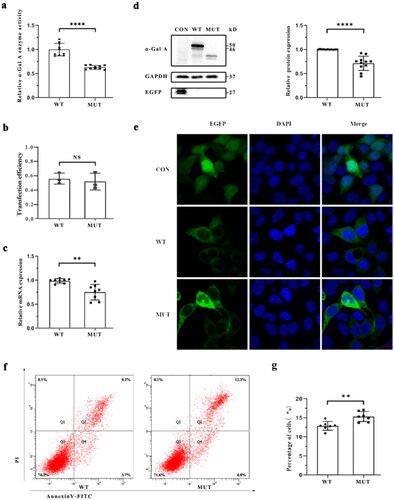
To determine whether the mutant α-Gal A retained residual activity, enzymatic activity of α-Gal A in HEK293T cells transfected with EGPF-GLA-WT and EGFP-GLA-mutant plasmids was determined. Compared to that in WT cells, enzymatic activity was significantly reduced (n = 7, t-test, p < 0.0001) in the mutant cells ().
Figure 2. Functional test of the mutant α-Gal A protein. (a) α-Gal A protease activity of the wild-type (WT) and mutant HEK293T cells. (b) Comparison of transfection efficiency of the recombinant plasmids between WT and mutant HEK293T cells. The transfection efficiency of WT and mutant recombinant plasmids was relatively consistent (n = 3, t-test, p = 0.6260). (c) Comparison of mRNA expression levels of GLA between WT and mutant HEK293T cells. Significant differences were observed between two groups (p < 0.01). (d) Western blot assay of GLA expression in WT and mutant HEK293T cells. Compared with those in WT cells, α-Gal A protein levels were decreased in mutant cells (p < 0.0001). (e) Subcellular localization of WT and mutant α-Gal A proteins in HEK293T cells. The WT α-Gal A protein was uniformly expressed in the cytoplasm; however, the mutant α-Gal A protein presented punctate aggregation in the cytoplasm. (f) Proportion of apoptotic cells was determined in WT cells and mutant HEK293T cells via flow cytometry. (g) Differences in apoptosis between WT and mutant HEK293T cells. Compared with those in WT HEK293T cells, the levels of apoptosis in mutant HEK293T cells was significantly higher (p < 0.01).
Functional analysis
Functional analysis of GLA mutations was performed using HEK293T cells. The transfection efficiency of WT and mutant plasmids was similar (n = 3, t-test, p = 0.6260) (). Compared to the WT cells, The expression of GLA was significantly reduced at mRNA (n = 8, t-test, p = 0.0014) and protein (n = 11, t-test, p < 0.0001) level in mutant cells (). Significant differences in the subcellular localization of α-Gal A were observed between WT and mutant cells. The mutant α-Gal A exhibited punctate aggregations in the cytoplasm, whereas the WT α-Gal A was uniformly expressed in the cytoplasm (). In addition, the level of apoptosis in GLA WT cells was significantly lower than mutant cells (n = 7, t-test, p = 0.0033) ().
Discussion
In this study, a previously unreported nonsense mutation of hemizygous GLA, c.777_778del (p.Gly261Leufs*3), was identified by high-throughput sequencing in a proband with classical FD. This mutation is pathogenic and is one of three highly mutated fragments (amino acids 162–172, 215–231, and 258–269) in the protein sequence of α-Gal A [Citation38].
As a homodimer, each monomer of mature α-Gal A is composed of two domains. The active site comprises residues W47, D92, D93, Y134, C142, C172, K168, D170, E203, L206, Y207, R227, D231, D266, and M267 in domain 1 [Citation5, Citation39]. The 15 side chains that form the active site are highly conserved and determine substrate specificity.
Typically, mutations associated with severe FD phenotypes are predominantly located within the hydrophobic core of α-Gal A, attributable to impaired enzyme folding resulting from alterations in this region [Citation38]. In this study, the deletion of the active site residues D266/M267 directly impacted the formation of the enzyme’s active site and its specific binding to the substrate. Additionally, mutations affecting G261, a buried residue near the active sites D266 and M267 (Fabry database, http://fabry-database.org/), along with the deletion of domain 2, can result in protein-folding defects. The presence of W262 is crucial in forming the hydrophobic core and maintaining the conformational stability of α-Gal A [Citation38]. Consequently, a mutation in W262 could damage the protein hydrophobic core, resulting in the inability to form α-Gal A properly under the acidic conditions of lysosomes, ultimately affecting enzyme stability or transport. The decrease in mRNA and protein expression of GLA reported herein () may be due to reduced mRNA stability, which has been attributed to nonsense-mediated decay for mutations generating premature termination codons [Citation40]. Additionally, surprising punctate aggregations () were observed in the cytoplasm of mutant cells, possibly as a result of α-Gal A C-terminal deletion, which requires further investigation for confirmation.
The proband was a patient with classic-type FD, characterized by extremely low residual enzyme activity leading to multi-system deposition of Gb3, changes in cell metabolism, and oxidative stress, which ultimately results in apoptosis [Citation41]. Apoptosis was observed in a lower proportion of WT cells compared to mutant cells (). In addition, both the proband and his younger brother (II-1 and II-2) presented with symptoms of angiokeratoma, which may be related to the deposition of Gb3 in dermal vascular endothelial cells. The proband had cerebrovascular involvement, which may be associated with the deposition of Gb3 in cerebrovascular endothelial cells and cerebrovascular smooth muscle cells [Citation42].
Kidney involvement in male patients with FD is almost universal. FD nephropathy is a metabolic proteinuric nephropathy that initially manifests as microalbuminuria and proteinuria and is characterized by renal fibrosis [Citation43–44]. Notably, the two patients with FD in this family had already exhibited characteristic proteinuria with preserved renal function, and glomerulosclerosis and interstitial fibrosis would inevitably appear with disease progression.
Enzyme replacement therapy has been utilized for the treatment of Fabry disease since 2001 [Citation45], and over the long term, this treatment can remove Gb3 inclusion bodies from the glomerular mesangium and glomerular endothelial cells [Citation46]. Enzyme replacement therapy requires early and lifelong application. Once severe organ damage has occurred, it is difficult to achieve the expected effect [Citation47]. In addition, long-term and repeated use of recombinant enzyme preparations can induce the development of anti-α-Gal A antibodies [Citation48]. Following treatment with agalsidase α, the frequency of fever was significantly reduced, and extremity pain was significantly relieved in the proband. The pharmaceutical chaperone migalastat is also used to treat patients with FD, but only in patients with amenable mutations, which stabilizes the conformation of a specific α-Gal A mutant, thereby promoting its transport to lysosomes [Citation33].
In the family described herein, the two patients with FD (II-1 and II-2) shared the same genotype; however, there were substantial differences in clinical manifestations. The phenotypic heterogeneity of FD may be partially attributed to epigenetic mechanisms, including histone modifications and DNA methylation [Citation49]. Moreover, The individual inflammatory response resulting from Gb3 deposition can affect the clinical feature and the outcome [Citation50].
In summary, a novel pathogenic mutation, c.777_778del (p.Gly261Leufs*3), was identified in FD, which enriches the mutation spectrum of FD, improves our understanding of the pathological process and genotype–phenotype differences of FD, and provides a basis for the precise diagnosis and therapy.
Authors’ contributions
Conceptualization: FX, HX, and YM; Data curation: YP and ZS; Formal analysis: YP, MP, and YW; Supervision: JX; Funding acquisition: YM; Writing – original draft: YP and YW.
Ethics statement
This study received approval from the Ethics Committee of Peking University Shenzhen Hospital, an affiliation of Peking University (2022-177). The research was conducted on the basis of the World Medical Association Declaration of Helsinki, and written informed consent was given in all subjects.
Supplemental Material
Download TIFF Image (37 MB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Upon reasonable request, the relevant authors will make the data available.
Additional information
Funding
References
- Tuttolomondo A, Pecoraro R, Simonetta I, et al. Anderson-Fabry disease: a multiorgan disease. Curr Pharm Des. 2013;19(33):1–10. doi: 10.2174/13816128113199990352.
- Kertész AB, Édes I. Fabry disease cardiomyopathy: from genes to clinical manifestations. Curr Pharm Biotechnol. 2012;13(13):2477–2484. doi: 10.2174/138920112804583069.
- Monticelli M, Liguori L, Allocca M, et al. Drug repositioning for Fabry disease: acetylsalicylic acid potentiates the stabilization of lysosomal alpha-galactosidase by pharmacological chaperones. Int J Mol Sci. 2022;23(9):5105. doi: 10.3390/ijms23095105.
- Prabakaran T, Nielsen R, Larsen JV, et al. Receptor-mediated endocytosis of α-galactosidase a in human podocytes in Fabry disease. PLoS One. 2011;6(9):e25065. doi: 10.1371/journal.pone.0025065.
- Guce AI, Clark NE, Salgado EN, et al. Catalytic mechanism of human alpha-galactosidase. J Biol Chem. 2010;285(6):3625–3632. doi: 10.1074/jbc.M109.060145.
- Zarate YA, Hopkin RJ. Fabry’s disease. Lancet. 2008;372(9647):1427–1435. doi: 10.1016/S0140-6736(08)61589-5.
- Brady RO, Gal AE, Bradley RM, et al. Enzymatic defect in Fabry’s disease. Ceramidetrihexosidase deficiency. N Engl J Med. 1967;276(21):1163–1167. doi: 10.1056/NEJM196705252762101.
- Bernstein HS, Bishop DF, Astrin KH, et al. Fabry disease: six gene rearrangements and an exonic point mutation in the alpha-galactosidase gene. J Clin Invest. 1989;83(4):1390–1399.
- Pintos-Morell G. Dysregulated autophagy contributes to podocyte damage in Fabry’s disease. Current medical literature. Lysosomal Storage Diseases with Focus on Fabry Disease. 2013;11(4):132.
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. doi: 10.1016/j.cell.2011.10.026.
- Braun F, Blomberg L, Brodesser S, et al. Enzyme replacement therapy clears Gb3 deposits from a podocyte cell culture model of Fabry disease but fails to restore altered cellular signaling. Cell Physiol Biochem. 2019;52(5):1139–1150. doi: 10.33594/000000077.
- Sirin Y, Susztak K. Notch in the kidney: development and disease. J Pathol. 2012;226(2):394–403.
- Niranjan T, Bielesz B, Gruenwald A, et al. The notch pathway in podocytes plays a role in the development of glomerular disease. Nat Med. 2008;14(3):290–298. doi: 10.1038/nm1731.
- Bielesz B, Sirin Y, Si H, et al. Epithelial notch signaling regulates interstitial fibrosis development in the kidneys of mice and humans. J Clin Invest. 2010;120(11):4040–4054. doi: 10.1172/JCI43025.
- Schiffmann R, Warnock DG, Banikazemi M, et al. Fabry disease: progression of nephropathy, and prevalence of cardiac and cerebrovascular events before enzyme replacement therapy. Nephrol Dial Transplant. 2009;24(7):2102–2111. doi: 10.1093/ndt/gfp031.
- Tuttolomondo A, Duro G, Pecoraro R, et al. A family with various symptomatology suggestive of Anderson-Fabry disease and a genetic polymorphism of alpha galactosidase a gene. Clin Biochem. 2015;48(1-2):55–62.
- Verovnik F, Benko D, Vujkovac B, et al. Remarkable variability in renal disease in a large Slovenian family with Fabry disease. Eur J Hum Genet. 2004;12(8):678–681. doi: 10.1038/sj.ejhg.5201184.
- Hopkin RJ, Bissler J, Grabowski GA. Comparative evaluation of alpha-galactosidase a infusions for treatment of Fabry disease. Genet Med. 2003;5(3):144–153. doi: 10.1097/01.GIM.0000069509.57929.CD.
- Desnick RJ, Brady R, Barranger J, et al. Fabry disease, an under-recognized multisystemic disorder: expert recommendations for diagnosis, management, and enzyme replacement therapy. Ann Intern Med. 2003;138(4):338–346. doi: 10.7326/0003-4819-138-4-200302180-00014.
- von Scheidt W, Eng CM, Fitzmaurice TF, et al. An atypical variant of Fabry’s disease with manifestations confined to the myocardium. N Engl J Med. 1991;324(6):395–399. doi: 10.1056/NEJM199102073240607.
- Nakao S, Kodama C, Takenaka T, et al. Fabry disease: detection of undiagnosed hemodialysis patients and identification of a “renal variant” phenotype. Kidney Int. 2003;64(3):801–807.
- Eng CM, Germain DP, Banikazemi M, et al. Fabry disease: guidelines for the evaluation and management of multi-organ system involvement. Genet Med. 2006;8(9):539–548. doi: 10.1097/01.gim.0000237866.70357.c6.
- Wilcox WR, Oliveira JP, Hopkin RJ, et al. Females with Fabry disease frequently have major organ involvement: lessons from the Fabry registry. Mol Genet Metab. 2008;93(2):112–128.
- Simonetta I, Riolo R, Todaro F, et al. Case report: de novo mutation of a-galactosidase a in a female patient with end-stage renal disease: report of a case of late diagnosis of Anderson-Fabry disease. Front Genet. 2023;14:1122893. doi: 10.3389/fgene.2023.1122893.
- Tuttolomondo A, Duro G, Miceli S, et al. Novel alpha-galactosidase a mutation in a female with recurrent strokes. Clin Biochem. 2012;45(16-17):1525–1530.
- Echevarria L, Benistan K, Toussaint A, et al. X-chromosome inactivation in female patients with Fabry disease. Clin Genet. 2016;89(1):44–54. doi: 10.1111/cge.12613.
- Linthorst GE, Poorthuis BJ, Hollak CE. Enzyme activity for determination of presence of Fabry disease in women results in 40% false-negative results. J Am Coll Cardiol. 2008;51(21):2082; author reply 2082–3. doi: 10.1016/j.jacc.2008.02.050.
- Hoffmann B, Mayatepek E. Fabry disease-often seen, rarely diagnosed. Dtsch Arztebl Int. 2009;106(26):440–447. doi: 10.3238/arztebl.2009.0440.
- van der Tol L, Smid BE, Poorthuis BJHM, et al. A systematic review on screening for Fabry disease: prevalence of individuals with genetic variants of unknown significance. J Med Genet. 2014;51(1):1–9. doi: 10.1136/jmedgenet-2013-101857.
- Spada M, Pagliardini S, Yasuda M, et al. High incidence of later-onset Fabry disease revealed by newborn screening. Am J Hum Genet. 2006;79(1):31–40.
- Lin H-Y, Chong K-W, Hsu J-H, et al. High incidence of the cardiac variant of Fabry disease revealed by newborn screening in the Taiwan Chinese population. Circ Cardiovasc Genet. 2009;2(5):450–456. doi: 10.1161/CIRCGENETICS.109.862920.
- Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5(1):30. doi: 10.1186/1750-1172-5-30.
- Benjamin ER, Della VM, Wu X, et al. The validation of pharmacogenetics for the identification of Fabry patients to be treated with migalastat. Genet Med. 2017;19(4):430–438.
- Santos EM, Paula JFR, Motta PMC, et al. Comparison of three methods of DNA extraction from peripheral blood mononuclear cells and lung fragments of equines. Genet Mol Res. 2010;9(3):1591–1598. doi: 10.4238/vol9-3gmr818.
- Zheng Y, Xu J, Liang S, et al. Whole exome sequencing identified a novel heterozygous mutation in HMBS gene in a Chinese patient with acute intermittent porphyria with rare type of mild anemia. Front Genet. 2018;9:129. doi: 10.3389/fgene.2018.00129.
- Zhang R, Chen S, Han P, et al. Whole exome sequencing identified a homozygous novel variant inCEP290 gene causes Meckel syndrome. J Cell Mol Med. 2020;24(2):1906–1916. doi: 10.1111/jcmm.14887.
- Ries M, Moore DF, Robinson CJ, et al. Quantitative dysmorphology assessment in Fabry disease. Genet Med. 2006;8(2):96–101. doi: 10.1097/01.gim.0000200950.25118.dd.
- Garman SC, Garboczi DN. Structural basis of Fabry disease. Mol Genet Metab. 2002;77(1-2):3–11. doi: 10.1016/s1096-7192(02)00151-8.
- Garman SC, Garboczi DN. The molecular defect leading to Fabry disease: structure of human alpha-galactosidase. J Mol Biol. 2004;337(2):319–335. doi: 10.1016/j.jmb.2004.01.035.
- Lindeboom R, Vermeulen M, Lehner B, et al. The impact of nonsense-mediated mRNA decay on genetic disease, gene editing and cancer immunotherapy. Nat Genet. 2019;51(11):1645–1651. doi: 10.1038/s41588-019-0517-5.
- De Francesco PN, Mucci JM, Ceci R, et al. Higher apoptotic state in fabry disease peripheral blood mononuclear cells.: effect of globotriaosylceramide. Mol Genet Metab. 2011;104(3):319–324.
- Viana-Baptista M. Stroke and Fabry disease. J Neurol. 2012;259(6):1019–1028. doi: 10.1007/s00415-011-6278-4.
- Weidemann F, Sanchez-Niño MD, Politei J, et al. Fibrosis: a key feature of Fabry disease with potential therapeutic implications. Orphanet J Rare Dis. 2013;8(1):116. doi: 10.1186/1750-1172-8-116.
- Tøndel C, Bostad L, Hirth A, et al. Renal biopsy findings in children and adolescents with Fabry disease and minimal albuminuria. Am J Kidney Dis. 2008;51(5):767–776. doi: 10.1053/j.ajkd.2007.12.032.
- Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase a replacement therapy in Fabry’s disease. N Engl J Med. 2001;345(1):9–16. doi: 10.1056/NEJM200107053450102.
- Tøndel C, Bostad L, Larsen KK, et al. Agalsidase benefits renal histology in young patients with Fabry disease. J Am Soc Nephrol. 2013;24(1):137–148. doi: 10.1681/ASN.2012030316.
- Weidemann F, Niemann M, Breunig F, et al. Long-term effects of enzyme replacement therapy on Fabry cardiomyopathy: evidence for a better outcome with early treatment. Circulation. 2009;119(4):524–529. doi: 10.1161/CIRCULATIONAHA.108.794529.
- Wilcox WR, Linthorst GE, Germain DP, et al. Anti-α-galactosidase a antibody response to agalsidase beta treatment: data from the Fabry registry. Mol Genet Metab. 2012;105(3):443–449. doi: 10.1016/j.ymgme.2011.12.006.
- Hassan S, Sidransky E, Tayebi N. The role of epigenetics in lysosomal storage disorders: uncharted territory. Mol Genet Metab. 2017;122(3):10–18. doi: 10.1016/j.ymgme.2017.07.012.
- Feriozzi S, Rozenfeld P. Pathology and pathogenic pathways in Fabry nephropathy. Clin Exp Nephrol. 2021;25(9):925–934. doi: 10.1007/s10157-021-02058-z.