
Abstract
Sepsis is a severe systemic infectious disease that often leads to multi-organ dysfunction. One of the common and serious complications of sepsis is renal injury. In this study, we aimed to investigate the potential mechanistic role of a novel compound called H-151 in septic kidney injury. We also examined its impact on renal function and mouse survival rates. Initially, we confirmed abnormal activation of the STING–TBK1 signaling pathway in the kidneys of septic mice. Subsequently, we treated the mice with H-151 and observed significant improvement in sepsis-induced renal dysfunction. This was evidenced by reductions in blood creatinine and urea nitrogen levels, as well as a marked decrease in inflammatory cytokine levels. Furthermore, H-151 substantially improved the seven-day survival rate of septic mice, indicating its therapeutic potential. Importantly, H-151 also exhibited an inhibitory effect on renal apoptosis levels, further highlighting its mechanism of protecting against septic kidney injury. These study findings not only offer new insights into the treatment of septic renal injury but also provide crucial clues for further investigations into the regulatory mechanisms of the STING–TBK1 signaling pathway and potential drug targets.
Graphical Abstract
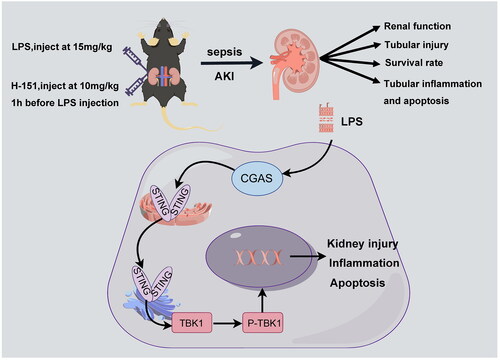
1. Introduction
Sepsis is a syndrome of organ dysfunction caused by a dysregulated host response to infection and can progress to multiple organ dysfunction syndrome if not promptly treated [Citation1,Citation2]. Sepsis-associated acute kidney injury (SA-AKI) is one of the most common complications of sepsis, with approximately half of severe sepsis patients eventually developing kidney injury [Citation3,Citation4]. Renal ischemia, cellular injury, and acute tubular necrosis are common pathogenic mechanisms of SA-AKI [Citation5]. Blood creatinine and blood urea nitrogen (BUN) are commonly used indicators for diagnosing acute kidney injury (AKI) in clinical practice [Citation6]. However, effective treatment options for SA-AKI are still lacking.
Stimulator of interferon genes (STING) is an adapter protein consisting of four transmembrane domains and a cytoplasmic C-terminal domain. It is primarily expressed in the endoplasmic reticulum of macrophages, dendritic cells, and T cells. Upon activation by cGAMP, the dimeric form of STING protein undergoes a conformational change and, with the assistance of the Golgi apparatus, is transported from the endoplasmic reticulum to perinuclear endosomes. Subsequently, it recruits TANK-binding kinase 1 (TBK1) to form a complex. TBK1 then phosphorylates nuclear transcription factors interferon regulatory factor 3 (IRF3) and inhibitor of nuclear factor kappa-B kinase (IKK), leading to the release of phosphorylated IRF3 and NF-KB into the nucleus. This stimulation promotes the production of IFN-I and related cytokines [Citation7,Citation8]. Recent studies have demonstrated that in septic cardiomyopathy, LPS can activate the cGAS–STING–TBK1 pathway through TLR4 receptors, exacerbating inflammation, apoptosis, and pyroptosis of cardiomyocytes [Citation9]. Additionally, in cisplatin-induced AKI, mitochondrial DNA (mtDNA) leakage triggers the activation of cGAS–STING, further aggravating renal dysfunction [Citation10]. However, the role of STING–TBK1 in SA-AKI remains poorly investigated.
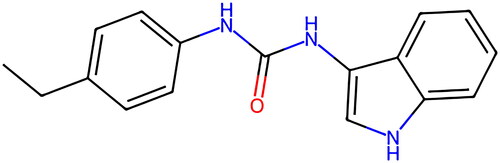
Two main categories of STING inhibitors currently exist. The first category comprises antagonists that occupy the cyclic dinucleotide (CDN) binding site and compete with STING. Examples of such antagonists include tetrahydroisoquinolines and astin C. The second category consists of antagonists that compete with STING by binding to Cys88 or Cys91 residues near the protein transmembrane domain. Inhibitors like C-176, C-178, and H-151 fall under this category [Citation11]. Notably, H-151 () is a novel selective inhibitor of STING that has been demonstrated to irreversibly reduce type I interferon response, inhibit TBK1 phosphorylation, and inhibit STING Cys91 palmitoylation, thereby suppressing downstream signaling activation [Citation12], thereby suppressing downstream signal activation.
Figure 1. Chemical structure of H-151.
The aim of this study was to investigate the activation of the STING–TBK1 pathway in lipopolysaccharide-induced AKI and the protective effects of H-151 in inhibiting the activation of STING–TBK1 and its impact on renal function.
2. Methods
2.1. Animals
C57BL/6 male mice (6–8 weeks old) free of pathogens were obtained from Huafukang Biotechnology Co., Ltd. (Beijing, China). All mice were housed under standard conditions with a humidity of 50% and a temperature of 20–22 °C, following a 12-h light/dark cycle. They were provided with ad libitum access to water and standard rodent chow. All protocols were approved by the Institutional Animal Care and Use Committee of Tianjin Medical University (Ethical permit number: IRB2021-DW-52).
2.2. Animals experiment
LPS (Sigma, St. Louis, MO, L2630, 0111:B4) was purchased and prepared into a solution with a concentration of 1 mg/ml following the provided instructions. The mice were randomly divided into three groups, each consisting of six mice: sham operation group, LPS group, and LPS + H-151 group. The septic AKI mouse model was induced by intraperitoneal injection of LPS at a dose of 15 mg/kg. The sham operation group received the same dose of normal saline via intraperitoneal injection. The H-151 inhibitor group, purchased from MedChemExpress (Monmouth Junction, NJ), received an intraperitoneal injection of 10 mg/kg 1 h prior to LPS administration [Citation13,Citation14]. After 12 h, all mice in each group were anesthetized, and blood samples were collected from the heart while kidney tissue was perfused and collected. The fresh blood was centrifuged at 4 °C at 3000 rpm for 15 min, and the supernatant was used for ELISA detection of serum creatinine (CRE), urea nitrogen, and serum inflammatory factors. The left kidney was fixed with 4% paraformaldehyde, and the right kidney tissue was immediately stored in liquid nitrogen.
2.3. Survival rate
The mice were divided into four groups: the control group (blank), the H-151 group, the LPS group, and the LPS + H-151 group. The dosages, timing, and administration methods were consistent with the previous description. Under the same conditions, the survival of the mice was monitored for seven days, and the survival percentage was calculated.
2.4. Renal function assessment and histological analysis
The levels of blood creatinine and BUN in each group of mice were measured using an automated biochemical analyzer for animal use (Mindray BS-240VET). Twelve hours after all intraperitoneal injections were performed, we gathered kidney tissue samples and immediately fixed them in a 10% formalin solution. Then, kidney tissues were dehydrated in graded ethanol, embedded in paraffin, cut into 5 μm thick slices, and stained with hematoxylin and eosin (H&E) reagent. Images were obtained with an optical microscope (3DHISTECH Pannoramic MIDI, Budapest, Hungary). Without knowledge of the assigned group, the pathologist evaluating the images randomly selected four different regions to observe in each sample. The degree of renal histopathological damage after sepsis was assessed according to renal interstitial congestion and edema, intraluminal cell swelling, cell necrosis, and protein casts. The renal tubulointerstitial injury score ranged from 0 to 4. Based on the proportion of damaged renal tubules, the scoring criteria were as follows: none (0); <25% (1); 25–50% (2); 50–75% (3); >75% (4). Scoring was performed on four randomly selected fields, and all assessments were conducted in a blinded manner [Citation15].
2.5. Measurement of serum inflammatory cytokines by ELISA
The inflammatory cytokines examined in this study included interleukin-6 (IL-6), interleukin-1β (IL-1β), and tumor necrosis factor alpha (TNF-α). ELISA assay kits obtained from Abclonal (Woburn, MA) were used for detection, following the manufacturer’s instructions.
2.6. Western blot assay
Protein immunoblot analysis was performed to examine protein expression in the renal tissues of each group. First, renal tissues from each group were collected and added to RIPA lysis buffer containing phosphatase inhibitors and protease inhibitors. The tissues were homogenized using a grinder at 4 °C to extract total proteins. Protein concentration was determined using the BCA assay. Then, 25 μg of protein was loaded onto an SDS-PAGE gel for electrophoresis and subsequently transferred to a polyvinylidene fluoride (PVDF) membrane. The membrane was blocked with 5% skim milk in TBS containing 0.1% Tween-20 for 1 h at room temperature. After overnight incubation with the primary antibody at 4 °C, STING antibody (A3262, dilute 1:1000), TBK1 antibody (A3458, dilute 1:1000), P-TBK1 (AP1026, dilute 1:1000), Bcl-2 (A19693, dilute 1:1000), BAX (A18642, dilute 1:1000), and GAPDH (A19056, dilute 1:80,000) were purchased from Abclonal (Woburn, MA); the membrane was washed three times and incubated with rabbit anti-HRP secondary antibody (AS014, Abclonal, Woburn, MA, dilute 1:8000) for 1 h. Finally, the protein bands were visualized using the BLT Gelview 6000 Pro chemiluminescence imaging system. The relative band intensities were quantified using Image J software (Bethesda, MD).
2.7. Real-time PCR
Quantitative real-time polymerase chain reaction (qPCR) was performed to measure the expression levels of inflammatory cytokines in renal tissues. Total RNA was extracted from the renal tissues using the AGbio RNA extraction kit. The concentration and purity of the total RNA were quantified and determined at 260 nm and 280 nm using spectrophotometry. Subsequently, the RNA was reverse transcribed into cDNA using the AGbio reverse transcription kit. The mRNA expression was measured using the CFX 96 Deep Well Dx ORM instrument (Bio-Rad, Hercules, CA). The obtained data were analyzed using the 2−ΔΔCt method, with the transcript levels of the target genes normalized to GAPDH. The primers used in this study are listed in .
Table 1. Primers in real-time PCR.
2.8. Immunofluorescence staining
The paraffin sections were dewaxed and placed in EDTA antigen retrieval buffer for antigen retrieval. They were then washed three times with PBS. Goat serum was added dropwise to evenly cover the tissue, and the sections were blocked at room temperature for 30 min. After blocking, anti-STING (1:100, A3262, Abclonal, Woburn, MA) was applied to the sections and left to block overnight at 4 °C. A goat anti-rabbit secondary antibody IgG/FITC (1:100, ZF-0311, ZSGB-BIO, Beijing, China) was used at room temperature for 1 h. Cell nuclei were counterstained using DAPI nuclear staining solution (C0065, Solarbio, Beijing, China). Finally, the expression of STING protein was observed using a panoramic scanner (3DHISTECH, Budapest, Hungary).
2.9. TUNEL staining
The tissue TUNEL staining kit (MedChemExpress, Monmouth Junction, NJ, HY-K1078) was used to detect the level of apoptosis in kidney tissue. Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) was performed according to the manufacturer’s instructions. The apoptosis index was quantified by determining the proportion of TUNEL-positive cells.
2.10. Statistical analysis
All data were analyzed using GraphPad Prism 9 software (San Diego, CA) and presented as mean ± standard error (SE). Statistical analysis was performed using Student’s t-test or ANOVA, with p < .05 considered statistically significant.
3. Results
3.1. STING–TBK1 pathway is activated in LPS-induced acute kidney injury
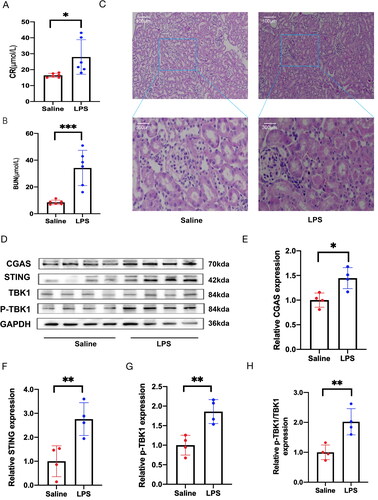
Serum creatinine and BUN are commonly used indicators for evaluating renal function. In C57 BL/6 mice, we observed a significant increase in the levels of BUN and CRE. Furthermore, HE staining revealed clear necrosis of the renal tubules in the mice in the LPS group, confirming the successful establishment of the sepsis-induced renal injury model (). Subsequently, we confirmed the activation of the cGAS–STING pathway in the kidney tissue of mice with LPS-induced AKI. demonstrates that the protein levels of cGAS, STING, and p-TBK1 were significantly elevated in LPS-treated mice compared to those receiving intraperitoneal saline injection (), indicating the activation of the cGAS–STING–TBK1 pathway in SA-AKI.
Figure 2. The activation of the STING–TBK1 pathway in kidney tissue of LPS-treated mice. (A) The levels of creatinine in mouse serum were measured (n = 6). (B) Blood urea nitrogen levels were assessed (n = 6). (C) Kidney tissue sections were stained with HE and observed under a ×200 microscope 12 h after intraperitoneal injection of LPS (n = 3). (D) Protein levels of cGAS, STING, TBK1, and P-TBK1 were analyzed after injecting each group with normal saline or LPS (n = 4). (E–H) Quantitative analysis of STING, P-TBK1, and the ratio of P-TBK1 to TBK1 proteins, normalized with GAPDH. All data are presented as mean ± SD. *p < .05, **p < .01, and ***p < .001.
3.2. H-151 inhibits the expression of STING and the phosphorylation of TBK1
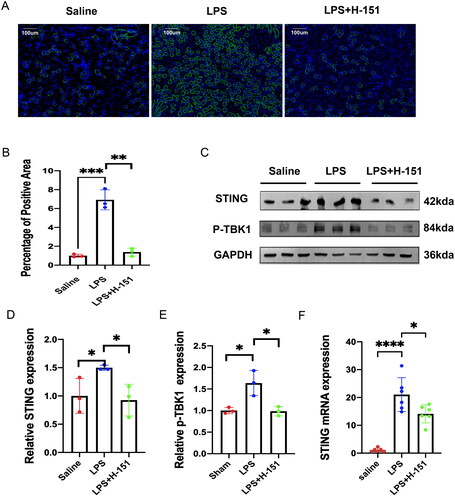
To investigate the impact of H-151 on the STING–TBK1 pathway in mouse kidney tissue, we conducted immunofluorescence analysis. Our results showed that the immunofluorescence intensity of STING in the kidney of mice in the LPS group was significantly higher compared to the sham operation group. However, when we administered the H-151 inhibitor, we observed a reversal of this trend (). Furthermore, Western blot analysis revealed that the expression levels of STING and p-TBK1 were significantly decreased in the kidneys of mice injected with the H-151 inhibitor (). Additionally, qPCR results confirmed that the H-151 inhibitor reduced the mRNA level of STING (). In conclusion, these findings provide evidence that H-151 effectively inhibits the activation of the STING–TBK1 pathway in SA-AKI.
Figure 3. H-151 inhibits the activation of the STING–TBK1 signaling pathway. (A, B) Immunofluorescence staining was performed to assess the positive area percentage of STING protein in kidney tissue sections. (C–E) Western blot analysis was conducted to evaluate the expression of STING and p-TBK1 in mouse kidneys after intraperitoneal injection of H-151, with normalization to GAPDH. (F) Quantitative real-time PCR analysis was carried out to determine the gene expression of STING in kidney tissue. *p < .05, **p < .01, and ***p < .001.
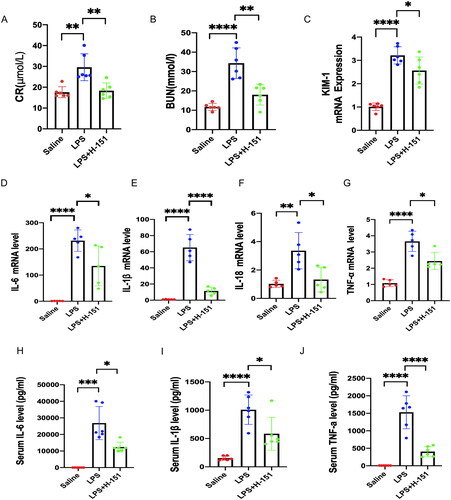
3.3. H-151 attenuates LPS-induced renal dysfunction and inflammatory response
To investigate the impact of H-151 on LPS-induced renal dysfunction and inflammatory factors, we evaluated several parameters including levels of CRE, BUN, IL-6, IL-1β, interleukin-18 (IL-18), and TNF-α in both kidney and serum. Notably, the LPS + H-151 group exhibited significantly lower levels of BUN and CRE compared to the LPS group (). Additionally, the H-151 + LPS group showed reduced mRNA levels of KIM-1, a novel marker for kidney injury, suggesting that H-151 administration provided protection against kidney tissue damage (). Real-time quantitative PCR (QPCR) analysis further revealed significantly lower levels of inflammatory indicators, including IL-6, IL-1β, IL-18, and TNF-α, in the H-151 + LPS group compared to the LPS group (). Moreover, ELISA analysis of inflammatory factors in serum demonstrated that H-151 significantly reduced the levels of IL-6, IL-1β, and TNF-α in serum (). These findings indicate that H-151 effectively attenuates renal dysfunction and inflammatory response in LPS-treated mice, suggesting its potential protective role.
Figure 4. H-151 improves LPS-induced renal dysfunction and inflammatory response. (A, B) Levels of serum creatinine and blood urea nitrogen in mice in each group (n = 6). (C) Detection of kidney injury marker KIM-1 (n = 6). (D–G) Detection on the mRNA levels of IL-6, IL-1β, IL-18, and TNF-α in kidney tissue of saline group, LPS group, and H-151 + LPS group (n = 5). (H–J) Determination of IL-6, IL-1β, and TNF-α levels in serum (n = 6). *p < .05, **p < .01, ***p < .01, and ****p < .0001.
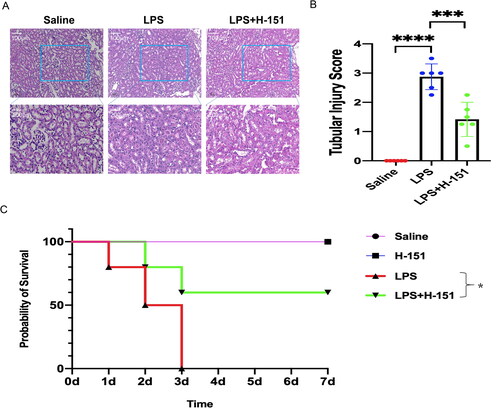
3.4. H-151 improves renal histopathological abnormalities and survival in mice
Kidney sections were used for HE staining to evaluate renal histopathological changes. In the saline group, there were minimal pathological changes observed in the kidneys of the mice. However, the renal tubules of the mice in the LPS group exhibited noticeable swelling, necrosis, and protein casts, accompanied by inflammatory exudation. On the other hand, the renal tubules of the mice in the H-151 group showed significant improvement (). The renal tubular injury score indicated that the H-151 inhibitor effectively alleviated the LPS-induced histopathological changes in the mouse kidneys (). Additionally, we examined the seven-day survival rate of the H-151 group after LPS treatment (). We compared the seven-day survival rate among four groups: saline group, H-151 group, LPS group, and LPS + H-151 group. The seven-day survival rate of the saline group and the H-151 group was nearly 100%, while all mice in the LPS group died on the third day (p < .05 relative to the saline group). Notably, the survival percentage of mice in the H-151 + LPS group increased to 60% compared to the LPS group (p < .05 relative to the LPS group).
Figure 5. The effects of H-151 administration on renal tissue function and survival rate in mice injected with LPS. (A) Kidney tissue sections were obtained and stained with HE 12 h after intraperitoneal injection of normal saline, LPS, and H-151. The kidney morphology of mice in different groups was observed under a ×200 microscope (n = 6). (B) Tubular injury was assessed using the tubular injury scoring system, with four individual samples from each group (n = 6) evaluated at ×200 magnification. (C) The seven-day survival rate of mice after intraperitoneal injection of normal saline, LPS, and H-151 + LPS was recorded, with each group consisting of 10 mice (n = 10). ***p < .001 and ****p < .0001.
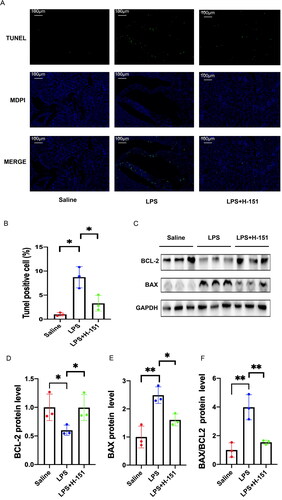
3.5. Anti-apoptotic effect of H-151 on SA-SKI
Apoptosis of renal tubular epithelial cells is the primary characteristic of renal injury in sepsis. To assess the level of apoptosis in kidney cells, we performed TdT-mediated dUTP nick-end labeling (TUNEL) staining on kidney tissues from each group. In the saline group, only a small number of green fluorescently labeled TUNEL-positive cells were observed in the kidney tissue. However, the number of positive cells significantly increased in the LPS group. Comparatively, the proportion of TUNEL-positive cells in the H-151 + LPS group was significantly lower than that in the LPS group, indicating a significant anti-apoptotic effect of H-151 (). The expression of the anti-apoptotic protein BCL-2 and the pro-apoptotic protein BAX in the kidney was assessed at the protein level. The findings were consistent with the results of the TUNEL staining assay. In the H-151 + LPS group, there was a significant decrease in the BAX/Bcl-2 ratio compared to the LPS group (). These results suggest that STING–TBK1 contributes to apoptosis in LPS-induced renal injury, while H-151 has a protective effect against renal apoptosis.
Figure 6. H151 alleviated apoptosis of renal tubular cells induced by LPS. (A) Representative images of TUNEL. Scale bars = 100 μm. (B) Quantification of TUNEL-positive cells (n = 3). (C) Representative blots of BAX and BCL-2. (D–F) Relative densitometry of BAX, BCL-2, and BAX/BCL-2 normalized to GAPDH (n =3). *p < .05 and **p < .01.
4. Discussion
Acute kidney injury is a commonly observed complication of sepsis, characterized by inflammation and apoptosis, which play significant roles in SA-AKI. Inflammatory cytokines are closely associated with both AKI and chronic kidney disease [Citation16]. The level of inflammatory cytokines has a strong correlation with the development of SA-AKI. Pathogens trigger the activation of inflammatory genes in renal tubular epithelial cells through pattern recognition receptors. This activation leads to an increase in the expression and upregulation of inflammatory factors [Citation17]. The apoptosis of renal tubular epithelial cells in cases of AKI has been widely acknowledged. Studies have shown that the targeted removal of the pro-apoptotic protein BAX can effectively reduce apoptosis in ischemia–reperfusion AKI [Citation18]. Thus, mitigating renal inflammation and apoptosis plays a crucial role in preventing septic renal injury and its progression.
The initial observation demonstrated that STING has the ability to relocate from the endoplasmic reticulum to the Golgi apparatus when there is a viral infection. This relocation leads to the recruitment of TBK1 and subsequent phosphorylation of TBK1. The phosphorylation of TBK1 activates IRF3, initiating the interferon pathway, which plays a crucial role in exerting antiviral effects and regulating the expression of various inflammatory factors [Citation7,Citation19,Citation20]. Recent research has highlighted the significant role of STING in inflammatory diseases. STING–IRF3 signaling has been implicated in chronic inflammation and insulin resistance in adipose tissue associated with obesity [Citation21]. Clearing mtDNA using DNase I has been shown to inhibit the activation of cGAS–STING and reduce intestinal damage in sepsis [Citation22]. Additionally, the STING antagonist C-176 has been found to significantly improve joint damage in rheumatoid arthritis (RA) [Citation23]. In this study, it was observed that the cGAS–STING–TBK1 pathway was activated in the AKI model of LPS-induced sepsis and played a role in the pathological process of kidney injury.
H-151, a selective small molecule antagonist of STING, exhibits good biological activity in mice and inhibits STING in human embryonic kidney 293 cells. This provides potential for clinical research [Citation12]. The mechanism of action of H-151 involves inhibiting STING palmitoylation and phosphorylation of TBK1 [Citation24,Citation25]. In the case of STING-mediated psoriasis, H-151 demonstrates significant reduction in the expression of STING protein, thereby improving psoriasis [Citation26]. Our study reveals that H-151 not only inhibits the activation of the STING–TBK1 pathway, but also reduces the mRNA level of STING protein.
The SA-AKI model was established by injecting LPS intraperitoneally. This injection resulted in a notable rise in CRE and BUN levels. The histopathology of the kidneys provided further evidence of the successful establishment of our model. Inflammatory factors play a vital role in causing organ function damage in LPS-induced sepsis. Previous studies have shown that stimulating LPS for 24 h leads to a significant increase in the expression of IL-1β, IL-6, and TNF-α in both the kidney and serum [Citation9,Citation27]. In our study, we observed a significant increase in mRNA and serum levels of IL-1β, IL-6, TNF-α, and IL-18 after 12 h of LPS stimulation. Notably, the H-151 + LPS group exhibited a significant inhibition of these inflammatory factors compared to the LPS group. Septic-associated acute kidney injury is characterized by the apoptosis of renal tubular cells. To evaluate the severity of renal apoptosis, TUNEL staining is commonly employed. BCL-2 and BAX are crucial proteins involved in the apoptosis pathway [Citation28]. By utilizing TUNEL staining and Western blotting, we observed a significantly higher proportion of apoptotic positive cells in the LPS group compared to the saline group. Furthermore, the ratio of BAX/BCL2 was also significantly higher in the LPS group. However, treatment with H-151 demonstrated a down-regulation in this increasing trend, indicating its anti-apoptotic properties through the inhibition of the STING–TBK1 pathway. Therefore, our experiments provide evidence that H-151 can alleviate inflammation and apoptosis in septic kidney injury by inhibiting the activation of the STING–TBK1 pathway.
However, this study has certain limitations. We only utilized a STING inhibitor to demonstrate the potential improvement of SA-AKI outcome by inhibiting the STING–TBK1 pathway. We did not employ STING knockout mice or conduct in vitro experiments to further validate the role of the STING–TBK1 pathway in SA-AKI. Moreover, the downstream mechanism of the STING–TBK1 pathway in SA-AKI remains a topic for future investigation.
5. Conclusions
In conclusion, this study demonstrates that the activation of the STING–TBK1 pathway exacerbates renal dysfunction, inflammatory response, and apoptosis in cases of septic kidney injury. Therefore, targeting the inhibition of the STING–TBK1 pathway could potentially serve as a novel therapeutic approach for the treatment of septic kidney injury.
Author contributions
Lei Xia, Yan-cun Liu, and Song-tao Shou designed the study. Lei Xia, Jie-yu Liu, and Jia-hui Jiang conducted the experiments. Yu-xin Dong, Qi-hui Liu and Tian-yi Zhang analyzed the data. Lei Xia, Jie-yu Liu wrote the manuscript. Yan-fen Chai and Song-tao Shou revised the paper and provided funding. All authors read and approved the final manuscript.
Ethical approval
All members included in this research, or their families, provided informed consent, and the Ethics Committee members at the General Hospital of Tianjin Medical University were sent a copy of the experimental procedures performed in this study for approval. The Animal Ethics Committee in the General Hospital of Tianjin Medical University granted its approval to all animal research experiments, based on the National Institute of Health (NIH) regulations (Ethical permit number: IRB2021-DW-52).
Consent form
All members included in this research, or their families, provided informed consent, and the Ethics Committee members at the General Hospital of Tianjin Medical University were sent a copy of the experimental procedures performed in this study for approval.
Acknowledgements
We appreciate the assistance provided by the FigDraw platform in terms of image resources.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Any data involved in this study can be requested from the corresponding author.
Additional information
Funding
References
- Singer M, Deutschman CS, Seymour CW, et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA. 2016;315(8):1–12. doi: 10.1001/jama.2016.0287.
- van Engelen TSR, Wiersinga WJ, Scicluna BP, et al. Biomarkers in sepsis. Crit Care Clin. 2018;34(1):139–152. doi: 10.1016/j.ccc.2017.08.010.
- Peerapornratana S, Manrique-Caballero CL, Gómez H, et al. Acute kidney injury from sepsis: current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 2019;96(5):1083–1099. doi: 10.1016/j.kint.2019.05.026.
- Gómez H, Kellum JA. Sepsis-induced acute kidney injury. Curr Opin Crit Care. 2016;22(6):546–553. doi: 10.1097/MCC.0000000000000356.
- Schrier RW, Wang W. Acute renal failure and sepsis. N Engl J Med. 2004;351(2):159–169. doi: 10.1056/NEJMra032401.
- Poston JT, Koyner JL. Sepsis associated acute kidney injury. BMJ. 2019;364:k4891. doi: 10.1136/bmj.k4891.
- Ishikawa H, Barber GN. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature. 2008;455(7213):674–678. doi: 10.1038/nature07317.
- Tanaka Y, Chen ZJ. STING specifies IRF3 phosphorylation by TBK1 in the cytosolic DNA signaling pathway. Sci Signal. 2012;5(214):ra20. doi: 10.1126/scisignal.2002521.
- Li N, Zhou H, Wu H, et al. STING–IRF3 contributes to lipopolysaccharide-induced cardiac dysfunction, inflammation, apoptosis and pyroptosis by activating NLRP3. Redox Biol. 2019;24:101215. doi: 10.1016/j.redox.2019.101215.
- Maekawa H, Inoue T, Ouchi H, et al. Mitochondrial damage causes inflammation via cGAS–STING signaling in acute kidney injury. Cell Rep. 2019;29(5):1261–1273.e6. doi: 10.1016/j.celrep.2019.09.050.
- Decout A, Katz JD, Venkatraman S, et al. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat Rev Immunol. 2021;21(9):548–569. doi: 10.1038/s41577-021-00524-z.
- Haag SM, Gulen MF, Reymond L, et al. Targeting STING with covalent small-molecule inhibitors. Nature. 2018;559(7713):269–273. doi: 10.1038/s41586-018-0287-8.
- Gong W, Lu L, Zhou Y, et al. The novel STING antagonist H151 ameliorates cisplatin-induced acute kidney injury and mitochondrial dysfunction. Am J Physiol Renal Physiol. 2021;320(4):F608–F616. doi: 10.1152/ajprenal.00554.2020.
- Li J, Lu Y, Lin G. Blocking cGAS/STING signaling protects against sepsis-associated acute liver injury. Int Immunopharmacol. 2022;113(Pt A):109276. doi: 10.1016/j.intimp.2022.109276.
- Matsumoto M, Tanaka T, Yamamoto T, et al. Hypoperfusion of peritubular capillaries induces chronic hypoxia before progression of tubulointerstitial injury in a progressive model of rat glomerulonephritis. J Am Soc Nephrol. 2004;15(6):1574–1581. doi: 10.1097/01.asn.0000128047.13396.48.
- Bonventre JV, Yang L. Cellular pathophysiology of ischemic acute kidney injury. J Clin Invest. 2011;121(11):4210–4221. doi: 10.1172/JCI45161.
- Zarbock A, Gomez H, Kellum JA. Sepsis-induced acute kidney injury revisited: pathophysiology, prevention and future therapies. Curr Opin Crit Care. 2014;20(6):588–595. doi: 10.1097/MCC.0000000000000153.
- Wei Q, Dong G, Chen JK, et al. Bax and Bak have critical roles in ischemic acute kidney injury in global and proximal tubule-specific knockout mouse models. Kidney Int. 2013;84(1):138–148. doi: 10.1038/ki.2013.68.
- Schoggins JW, MacDuff DA, Imanaka N, et al. Pan-viral specificity of IFN-induced genes reveals new roles for cGAS in innate immunity. Nature. 2014;505(7485):691–695. doi: 10.1038/nature12862.
- Liu YP, Zeng L, Tian A, et al. Endoplasmic reticulum stress regulates the innate immunity critical transcription factor IRF3. J Immunol. 2012;189(9):4630–4639. doi: 10.4049/jimmunol.1102737.
- Bai J, Cervantes C, Liu J, et al. DsbA-L prevents obesity-induced inflammation and insulin resistance by suppressing the mtDNA release-activated cGAS–cGAMP–STING pathway. Proc Natl Acad Sci U S A. 2017;114(46):12196–12201. doi: 10.1073/pnas.1708744114.
- Hu Q, Ren H, Li G, et al. STING-mediated intestinal barrier dysfunction contributes to lethal sepsis. EBioMedicine. 2019;41:497–508. doi: 10.1016/j.ebiom.2019.02.055.
- Shen H, Jin L, Zheng Q, et al. Synergistically targeting synovium STING pathway for rheumatoid arthritis treatment. Bioact Mater. 2023;24:37–53. doi: 10.1016/j.bioactmat.2022.12.001.
- Berger G, Marloye M, Lawler SE. Pharmacological modulation of the STING pathway for cancer immunotherapy. Trends Mol Med. 2019;25(5):412–427. doi: 10.1016/j.molmed.2019.02.007.
- Hu S, Gao Y, Gao R, et al. The selective STING inhibitor H-151 preserves myocardial function and ameliorates cardiac fibrosis in murine myocardial infarction. Int Immunopharmacol. 2022;107:108658. doi: 10.1016/j.intimp.2022.108658.
- Pan Y, You Y, Sun L, et al. The STING antagonist H-151 ameliorates psoriasis via suppression of STING/NF-κB-mediated inflammation. Br J Pharmacol. 2021;178(24):4907–4922. doi: 10.1111/bph.15673.
- Chen Y, Wei W, Fu J, et al. Forsythiaside A ameliorates sepsis-induced acute kidney injury via anti-inflammation and antiapoptotic effects by regulating endoplasmic reticulum stress. BMC Complement Med Ther. 2023;23(1):35. doi: 10.1186/s12906-023-03855-7.
- Bruckheimer EM, Cho SH, Sarkiss M, et al. The Bcl-2 gene family and apoptosis. Adv Biochem Eng Biotechnol. 1998;62:75–105. doi: 10.1007/BFb0102306.