
Abstract
The transient receptor potential canonical 6 (TRPC6) channel, a nonselective cation channel that allows the passage of Ca2+, plays an important role in renal diseases. TRPC6 is activated by Ca2+ influx, oxidative stress, and mechanical stress. Studies have shown that in addition to glomerular diseases, TRPC6 can contribute to renal tubular disorders, such as acute kidney injury, renal interstitial fibrosis, and renal cell carcinoma (RCC). However, the tubule-specific physiological functions of TRPC6 have not yet been elucidated. Its pathophysiological role in ischemia/reperfusion (I/R) injury is debatable. Thus, TRPC6 may have dual roles in I/R injury. TRPC6 induces renal fibrosis and immune cell infiltration in a unilateral ureteral obstruction (UUO) mouse model. Additionally, TRPC6 overexpression may modify G2 phase transition, thus altering the DNA damage checkpoint, which can cause genomic instability and RCC tumorigenesis and can control the proliferation of RCC cells. This review highlights the importance of TRPC6 in various conditions of the renal tubular system. To better understand certain renal disorders and ultimately identify new therapeutic targets to improve patient care, the pathophysiology of TRPC6 must be clarified.
Introduction
Ca2+ is critical as a sensor and effector for maintaining cellular homeostasis. An excessive increase in the Ca2+ level can result in various functional abnormalities that affect cell membrane permeability, enzyme activity and cell signal transduction. Transient receptor potential (TRP) channels are a type of integral membrane protein that plays an important part in modifying membrane potential or intracellular Ca2+ concentration. TRP channels can also be classified as canonical (TRPC), vanilloid (TRPV), or melastatin (TRPM), among others. TRPC channels are nonselective cation channels that are encoded by TRPC1-7 and serve as cellular sensors for a wide range of physical and environmental stimuli and contribute to the exquisite control of Ca2+ levels [Citation1]. Mutations in TRPCs that result in a loss or gain of function can cause hereditary illnesses and disorders in several physiological systems. Among the TRPC family members, the transient receptor potential canonical 6 (TRPC6) channel has attracted the most interest. TRPC6 contains six transmembrane proteins (S1-S6), with their NH2 and COOH termini facing the cytoplasm. There is a hypothetical pore region between S5 and S6, which constitutes a nonselective cation channel (). The domain structure of TRPC6 raises the possibility of extensive connections with other proteins and cytoplasmic components [Citation2].
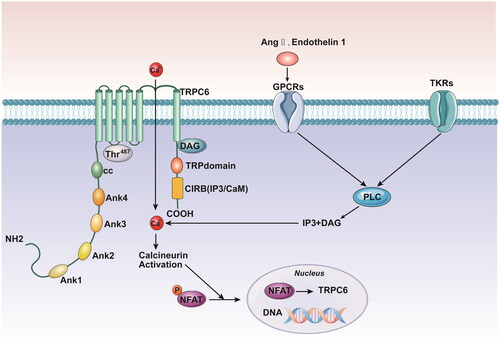
Figure 1. TKRs and GPCRs both induce PLC activation. PLC generates DAG and IP3, which in turn stimulates the TRPC6 activation, Ca2+ influx, calcineurin activation, dephosphorylation of NFAT, and translocation of NFAT to the nucleus. NFAT stimulates transcription of multiple genes including TRPC6, resulting in a positive feedback loop. TKRs: tyrosine kinase receptors; GPCRs: G protein coupled receptors; PLC: phospholipase C; DAG: diacylglycerol; IP3: inositol 1,4,5 trisphosphate; NFAT: nuclear factor of activated T cells.
TRPC6 is broadly distributed in kidney tissues, such as glomerular visceral, mesangial, tubular epithelial cells, and renal vasculature [Citation3]. The relationship between TRPC6 and glomerular selectivity and its involvement in renal proteinuria disorders have received much attention. TRPC6 has been implicated in the pathophysiology of kidney diseases, such as focal segmental glomerulosclerosis, diabetic nephropathy (DN), and immune-mediated kidney diseases [Citation4]. However, TRPC6 is also present in the renal tubular system. Owing to the rich energy metabolism in the kidney and the role of the reabsorption and concentration after glomerular filtration, tubular epithelial cells (TECs) act as sensors, effectors, and targets of renal ischemia/reperfusion (I/R) injury. Most filtered Ca2+ can be reabsorbed in renal tubules [Citation5]. Disrupting Ca2+ reabsorption can promote crystal precipitation, which can lead to nephrolithiasis, inflammation, fibrosis, and eventually, to chronic kidney disease [Citation6,Citation7]. Interestingly, in a unilateral ureteral obstruction (UUO) mouse model, TRPC6 was shown to contribute to renal fibrosis and immune cell infiltration [Citation8]. Additionally, studies have shown that renal clear cell carcinoma (RCC) originates from renal tubular epithelial cells and that TRPC6 overexpression may control the proliferation of RCC cells [Citation9]. The abovementioned TRPC6-related tubular diseases always have poor outcomes. Therefore, it is of great clinical importance to actively explore their pathological mechanisms and develop effective targeted therapeutic strategies. However, the tubule-specific physiological functions of TRPC6 have not yet been elucidated. This review focuses on recent advances in the understanding of the physiological and pathophysiological roles of TRPC6 in the regulation of the renal tubular system.
Molecular mechanisms of TRPC6 activation
TRPC6 can be activated by receptor-operated channel (ROC) stimulation
ROCs are cation channels that contribute to phospholipase C (PLC)-dependent Ca2+ cell entry following the stimulation of G protein-coupled receptors (GPCRs) or tyrosine kinase receptors (TKRs). GPCRs and TKRs induce PLC activation, which in turn stimulates Ca2+ influx. PLC generates the second messengers diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3) via the hydrolysis of phosphatidylinositol 4,5-bisphosphate (PIP2), and these messengers have different effects on TRPC6 activation. While DAG can activate receptor-operated Ca2+ channels to cause persistent influx of extracellular Ca2+, IP3 can activate cellular store-operated Ca2+ channels to release intracellular Ca2+ [Citation4,Citation10]. The activation of TRPC6 stimulates a positive feedback loop that promotes further expression of TRPC6 [Citation4]. Angiotensin II (Ang II) or endothelin 1 can bind to GPCRs and then induce continuous calcium influx. Excessive Ca2+ activates calcineurin, which results in the dephosphorylation of nuclear factor of activated T cells (NFAT) and its translocation to the nucleus. A positive feedback loop is created when NFAT induces the transcription of the trpc6 gene, which enhances TRPC6 expression and amplifies Ca2+ influx [Citation4,Citation11]. An increase in the cytosolic Ca2+ level can activate Ca2+/calmodulin-dependent kinase II (CaMKII), which in turn can further increase Ca2+ influx via TRPC6. Molecular research revealed that the distal domain of the C-terminal CaM/IP3R-binding domain (CIRB) and Thr487, which were found to be located in the intracellular loop, were two spatially distinct locations of the TRPC6 channel that are critical for CaMKII-mediated TRPC6 regulation [Citation12] (). However, a different line of evidence indicates that static Ca2+ levels also affect subsequent receptor-mediated activation of the TRPC6 channel via CaMKII [Citation13].
TRPC6 can be activated by oxidative stress
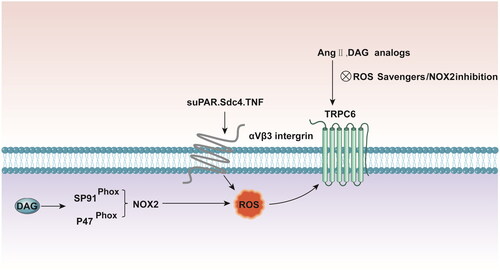
The second mechanism of TRPC6 activation involves oxidative stress. Accumulating evidence indicates that reactive oxygen species (ROS) play a role in the normal modulation of TRPC6 channels. Hydrogen peroxide can activate TRPC6 channels on the surface of podocytes and rapidly increase the cation current. In addition, hyperglycemia may promote NADPH oxidase 2 (NOX2)-dependent ROS generation and the activation of TRPC6, which contributes to diabetic nephropathy [Citation14–16]. ATP and Ang II, which participate in TRPC6 channel regulation, also increase the production of ROS by podocytes [Citation17,Citation18]. TRPC6 activation by Ang II and cell-permeable DAG analogs is suppressed in podocytes by ROS scavengers and by the pharmacological suppression of NOX2 [Citation14,Citation19]. Similarly, ROS quenchers can prevent ATP-induced TRPC6 activation [Citation20]. Additionally, TRPC6 coimmunoprecipitates with the catalytic subunit of NOX2, and the TRPC6-NOX2 interaction may require the scaffolding protein podocin because its knockdown eliminates the TRPC6-NOX2 protein-protein interaction [Citation19]. In podocytes, Ang II activates PLC via the Ang type 1 receptor (AT1R), which acts through the heterotrimeric Gq protein. This cascade produces DAG, which activates TRPC6 channels through a membrane-delimited mechanism. TRPC6 channels are sequestered in lipid rafts through interactions with podocin, which also interacts with NOX2. DAG stimulates the assembly of active NOX2 in lipid rafts, in part by mobilizing p47phox to the gp91phox catalytic subunit [Citation21,Citation22]. Interactions with podocin may also link TRPC6 to the underlying cytoskeleton via interactions with the CD2-associated protein (CD2AP) [Citation23]. A large body of evidence indicates that even before the estimated glomerular filtration rate (eGFR) is reduced, circulating soluble urokinase and plasminogen activator receptor (suPAR) levels strongly predict future deterioration in renal function [Citation24]. SuPAR binds directly to αVβ3 integrin, resulting in an increase in ROS generation, which is mediated in part by Rac1 and NOX2. An increase in the ROS levels triggers the activation of Src tyrosine kinases, which in turn can catalyze the phosphorylation of TRPC6 and modulate its function [Citation25]. Taken together, these findings indicate that the localized production of ROS is essential for TRPC6 activation ().
Figure 2. DAG stimulates assembly of active NOX2 at lipid rafts, in part by mobilizing p47phox to the gp91phox catalytic subunit. TRPC6 activation by Ang II and cell permeable DAG analogs is suppressed in podocytes by both ROS scavengers and pharmacologic suppression of NOX2. suPAR, Sdc4 and TNF bind directly to αVβ3-integrin resulting in increased ROS generation which upregulates podocyte TRPC6 channels. DAG: diacylglycerol; NOX2: NADPH oxidase 2; ROS: reactive oxygen species; suPAR: soluble urokinase plasminogen activator receptor; Sdc4: Soluble syndecan-4; TNF: tumor necrosis factor.
Recent research has demonstrated that reactive nitrogen species also influence TRPC channel function [Citation26–28]. Similar to ROS, reactive nitrogen species, such as nitric oxide, can influence TRPC signaling through site-specific changes in amino acid residues [Citation26]. Further research is necessary to determine whether this kind of redox reactions can affect the TRPC6 channel in kidney cells.
Multiple factors that regulate podocyte TRPC6 channels have been identified in the circulation. Soluble syndecan-4 (Sdc4) and tumor necrosis factor (TNF) can upregulate podocyte TRPC6 channels, which involves the activation of αVβ3 integrin [Citation29]. Exposure to suPAR but not to TNF leads to the loss of podocin. Many circulating factors may mediate interactions that ultimately destabilize podocytes, in part through TRPC6 dysregulation and, in most cases, through podocin loss [Citation30,Citation31].
TRPC6 can be activated by mechanical stress in podocytes
TRPC6 may be triggered by mechanisms other than ROC activation and oxidative stress. Recent research has suggested that TRPC6 is susceptible to mechanical stress. TRPC6 is activated in podocytes in response to a variety of mechanical stimuli, which appears to be different from the mechanism mediated by GPCRs. This mechanism persists in the presence of ROS quenchers and PLC inhibitors [Citation31,Citation32]. In podocytes, TRPC6 mediates stretch-induced cationic currents, which can be eliminated by TRPC6 knockdown or a highly selective TRPC6 inhibitor [Citation31,Citation32]. The mechanosensitivity is highly heterogeneous, varying from cell to cell and even within the same cell. It may depend on interacting proteins and the biophysics of the local membrane [Citation3]. Podocin modulates podocyte responses to mechanical and chemical stimuli in a complex manner [Citation33–35]. Huber et al. hypothesized that podocin was essential for TRPC6 mechanoactivation [Citation33]. One of the normal physiological roles of podocin is to suppress the mechanical activation of TRPC6 in podocytes [Citation33–35]. Given the general structure of the foot process and its underlying cytoskeleton, it is plausible that podocin retains TRPC6 channels within sterol-rich membrane domains and interacts with the cytoskeleton through CD2AP [Citation23]. Therefore, the ability of podocin to suppress the stretch activation of TRPC6 could involve multiple biophysical mechanisms. Cell surface mechanics are influenced by interactions with the underlying cytoskeleton, which can change the basal tension or curvature of the plasma membrane [Citation3].
The renal tubules are the primary sites of reabsorption of glomerular-filtered calcium, and they are very susceptible to oxidative stress. Thus, ROC stimulation and oxidative stress may activate TRPC6 in renal tubular diseases. Since TECs are similarly susceptible to mechanical stretch, mechanical stress within TECs may also activate TRPC6. As there have been only a few studies on the mechanism of TRPC6 activation in renal tubules, it is challenging to determine whether TRPC6 targets glomeruli or tubules. It is possible that TRPC6 acts on both the glomeruli and tubules.
Regulation of TRPC6 channels in renal I/R injury
TECs are the main sites of renal I/R injury because of their active metabolism [Citation36]. I/R injury, which is a major cause of acute kidney injury (AKI), is associated with renal dysfunction and tubular apoptosis. Abundant evidence has shown that Ca2+ overload, ROS production, mitochondrial dysfunction, and immune responses are crucial factors in tubular injury in I/R injury-induced AKI [Citation37–39]. TRPC6 has been reported to be involved in cell damage after I/R injury [Citation40,Citation41]. Moreover, TRPC6 has emerged as a functional element that controls calcium currents in immune cells, thereby regulating transendothelial migration, chemotaxis, phagocytosis, and cytokine release [Citation42]. Given the ubiquitous nature of TRPC6 channels and ROS production in the kidney, it is conceivable that the redox sensitivity of TRPC6 channels is involved in multiple physiological and pathological processes [Citation40]. However, the tubule-specific physiological functions of TRPC6 have not yet been determined. Its pathophysiological role in I/R injury is debatable. Thus, understanding the molecular mechanisms by which ROS alter TRPC6 channel activity and expression levels could aid in the development of specialized medications to treat kidney illnesses linked to TRPC6 and/or ROS.
TRPC6 can aggravate kidney damage following renal I/R injury
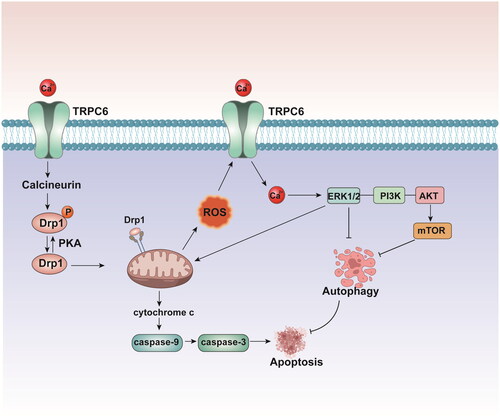
As the duration of ischemia increases, the kidney experiences more TEC loss into the lumen, more severe disruption of renal function, and more necrosis and apoptosis. ROS are generated under various pathological conditions, such as renal I/R injury and damage to multiple cellular organelles and processes. Overproduction of ROS causes oxidative stress and induces proximal tubular epithelial cells (PTCs) damage, which is the primary cause of renal I/R injury. TRPC6 functions as a downstream effector of ROS, and inhibiting ROS activity with N-acetyl-L-cysteine (NAC) eliminates H2O2-induced TRPC6 expression [Citation26,Citation43,Citation44]. Moreover, PTCs are particularly dependent on autophagy to maintain homeostasis and respond to oxidative stress. Intracellular Ca2+ is an important regulator of autophagy [Citation45]. TRPC6-mediated Ca2+ influx plays a pivotal role in suppressing cytoprotective autophagy, which is triggered by oxidative stress in PTCs. Hou et al. [Citation40]proposed that TRPC6-mediated Ca2+ influx regulated autophagic flux in PTCs undergoing oxidative stress through H2O2 exposure. Indeed, overexpression or silencing of TRPC6 can reduce or accelerate basal and oxidative stress-associated autophagy, respectively [Citation5]. TRPC6 is overexpressed during I/R injury, which leads to disrupted Ca2+ influx into TECs and in turn activates the ERK and PI3K/Akt/mTOR signaling pathways, leading to Akt phosphorylation, mTOR activation, and autophagy inhibition [Citation45]. These two pathways lead to apoptosis by inhibiting cytoprotective autophagy, which can remove damaged organelles in response to I/R injury. Moreover, activation of the ERK pathway induces excessive ROS production in mitochondria and exacerbates apoptosis [Citation45]. TRPC6-mediated Ca2+ entry suppresses cytoprotective autophagy and promotes TEC apoptosis, leading to the deterioration of renal function following I/R damage [Citation45]. A mechanistic analysis of an in vitro I/R model revealed that eliminating TRPC6-mediated Ca2+ entry could suppress the activity of the PI3K/Akt and ERK1/2 pathways, thus facilitating TEC survival. Inhibition of TRPC6 by genetic deletion or pharmacological blockade enhanced renoprotective autophagy by negatively modulating the PI3K/Akt/mTOR and Ras/Raf/ERK signaling pathways and attenuating H2O2-induced apoptosis in PTCs [Citation46] (). Furthermore, autophagy blockade prevents the protective effect of TRPC6 inhibition or knockout on H2O2-induced PTC apoptosis [Citation46]. Dynamin-related protein 1 (Drp1), an essential mediator of mitochondrial fission, is activated and accumulates in mitochondria during the early stages of TEC damage. Accumulating evidence has indicated that the levels of miRNAs change dramatically during the progression of AKI [Citation47,Citation48]. Drp1 inhibition by a siRNA has been shown to improve mitochondrial division and membrane permeability and induce apoptosis [Citation49]. Yang et al. [Citation49] reported that the overexpression of miR-26a inhibited TRPC6, which was accompanied by increased Drp1 protein levels, thus inhibiting the mitochondrial apoptosis pathway. Increasing miR-26a expression may be a potential therapeutic strategy against cell damage.
Figure 3. Overexpressed TRPC6 leads to damaged Ca2+ influx into TECs, which in turn,activates ERK and PI3K/Akt/mTOR signaling pathways, leading to Akt phosphorylation, mTOR activation, and autophagy inhibition and lead to cell apoptosis. Dephosphorylation of Drp1 by calcineurin promotes apoptosis by inducing mitochondrial fragmentation.TECs: tubular epithelial cells; Drp1: dynamin-related protein.
TRPC6 may be nephroprotective against renal I/R injury
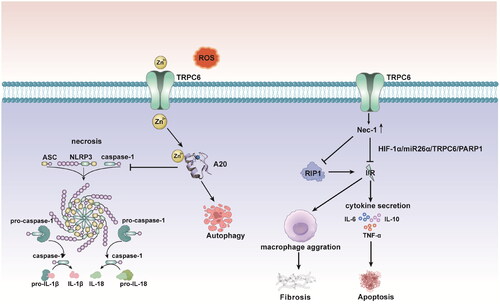
Cumulative research has provided evidence that TRPC6 may protect against renal I/R injury by alleviating the inflammatory response and inhibiting necroptosis [Citation41]. Necrostatin-1 (Nec-1), a receptor-interacting protein kinase-1 (RIP1) inhibitor, can effectively alleviate renal I/R injury [Citation50]. TRPC6 overexpression has been shown to dramatically boost the protective effect of Nec-1 against renal I/R injury, whereas TRPC6 knockdown can aggravate the oxidative stress response caused by I/R injury in human kidney-2 (HK-2) cells and increase the ratios of IL-1, IL-10, and TNF-α [Citation51]. Mechanistic investigations revealed that Nec-1 protected against renal I/R injury by suppressing necroptosis, oxidative stress, and inflammation. Nec-1 may exert its effects by mediating the HIF-1a/miR-26a/TRPC6/PARP1 signaling pathway [Citation51]. On the other hand, RIP1-dependent necroptosis has been shown to play a more important role than apoptosis in the pathophysiology of I/R injury. Shen et al. [Citation51] showed that the protein expression of RIP1 considerably increased in HK-2 cells following I/R insult, while siRNA-mediated knockdown of TRPC6 further promoted the I/R-mediated increase in RIP1 expression. However, 1-oleoyl-2-acetyl-sn-glycerol (OAG), a TRPC6 agonist, significantly reversed the necrotic effects of siRNA-mediated TRPC6 knockdown following I/R. Consistent with these results, the use of SKF96365, a TRPC inhibitor, and KN93, a specific inhibitor of Ca2+/CaMKII that regulates TRPC6 channel activity, was reported to further enhance the effects of siRNA-mediated TRPC6 knockdown on HK-2 cell necrosis after I/R [Citation41]. The generation of erythropoietin (EPO) is the ultimate response to I/R-induced anoxia in renal tissue. In a recent study, pretreatment with recombinant human erythropoietin (RhEPO) at 5,000 IU/kg was hypothesized to prevent I/R-induced AKI, which might be attributed in part to its ability to restore TRPC6 expression and collecting duct function [Citation3,Citation52,Citation53]. Further in vitro experiments on renal TECs supported this hypothesis, and TRPC6 was assigned a potential protective role under acute ischemic conditions [Citation36]. These findings suggest that TRPC6 may exert nephroprotective effects against AKI.
As a nonselective ion channel, TRPC6 is also markedly involved in transmembrane Zn2+ transport [Citation54]. Zn2+ is an essential element with antioxidant properties that may prevent renal cell apoptosis during I/R injury [Citation55]. After renal I/R-induced injury, the activities of major antioxidant enzymes, such as superoxide dismutase (SOD), catalase (CAT), and glutathione peroxidase (GPX), as well as the level of glutathione (GSH), are decreased, while Zn2+ supplementation stimulates the activities of these enzymes [Citation56]. It has recently been reported that TRPC6 can prevent necrosis and induce autophagy to alleviate I/R injury by accelerating Zn2+ influx [Citation36]. TRPC6 knockdown or overexpression in oxygen-glucose-deprived and reoxygenated (OGD-R) HK-2 cells could lead to a decrease or an increase in Zn2+ levels, respectively, and the latter occurred concomitantly with an increase in autophagic flux [Citation57]. Similarly, I/R injury leads to considerable increases in the serum levels of blood urea nitrogen (BUN) and creatinine, as well as to an increase in the fractional excretion of sodium (FENa), and these effects are reduced by ZnCl2 supplementation [Citation58]. Ogawa T and Mimura Y found that prior to I/R damage, zinc supplementation tended to improve the significant natriuresis and decreased eGFR [Citation59]. Recent research has examined the role of Zn2+ in activating the PI3K/Akt signaling pathway and the role of TRPC6 in inhibiting pyroptosis to attenuate TEC I/R injury [Citation57,Citation60]. TRPC6 suppression exacerbated renal injury and increased nucleotide-binding oligomerization domain-like receptor family pyrin domain-containing 3 (NLRP3), apoptosis-associated speck-like protein containing a caspase recruitment domain (ASC), caspase-1, IL-1, and IL-18 levels in an I/R injury murine model, and these effects could be alleviated by the administration of ZnCl2 [Citation57]. Pyroptosis is a form of programmed necrotic cell death that is induced by NLRP3-associated caspase-1 activation of gasdermin D, which forms pores in the plasma membrane, resulting in proinflammatory cell lysis [Citation61]. Interestingly, Zn2+ insufficiency is thought to mediate the activation of the NLRP3 inflammasome after ROS exposure and subsequently induce pyroptosis [Citation57]. Zn2+ influx and upregulation of the zinc finger protein A20, which inhibits the activation of NF-κB and plays a key role in NLRP3 activation, seem to control the cytoprotective and antipyroptotic effects of TRPC6 on I/R injury [Citation62] (). Furthermore, the expression of A20 was increased by ZnCl2 administration, and A20 knockdown impaired the antipyroptotic effect of ZnCl2, indicating that ZnCl2 acts as a protective factor through the A20 signaling pathway [Citation57].
Figure 4. Nec-1can effectively alleviates renal I/R injury through mediation of the HIF-1a/miR-26a/TRPC6/PARP1 signaling pathway. TRPC6 overexpression boosts the protective effect of Nec-1 against renal I/R injury. TRPC6 siRNA deletion further enhances I/R-mediated elevation of RIP1 expression. Zn2+ influx and upregulation of the zinc inger protein A20 inhibiting the activation of NF-κB, which plays a key role in NLRP3 activation, and control the cytoprotective and antipyropoptic effects of TRPC6 in I/R injury. HIF: hypoxia-inducible factors; Nec-1: necrostatin-1; PARP1: poly(ADP-ribose)polymerase1; RIP1: receptor-interacting protein kinase-1-inhibitor; ASC: apoptosis-associated speck-like protein containing a caspase-recruitment domain.
In summary, TRPC6 is involved in Ca2+ and Zn2+ signaling during renal I/R injury. TRPC6 is a controversial player in TECs subjected to I/R injury. Upregulated levels of TRPC6 can increase Drp1 expression and ERK and PI3K/Akt/mTOR pathway activation, induce TEC apoptosis, and ameliorate I/R injury. Overexpression of TRPC6 can increase Nec-1 expression and Zn2+ influx, which inhibit TEC necrosis, oxidative stress, and inflammatory factor release, thus decreasing I/R injury. Low TRPC6 expression increases RIP1 expression, exacerbates oxidative stress and cytokine release, and worsens I/R damage. Interestingly, after I/R injury, TRPC6-/- mice, mice treated with TRPC6 inhibitors, and wild-type mice exhibited the same renal function and tubular damage. These findings demonstrate that TRPC6 inhibition has no effect on the short-term outcomes of AKI [Citation62,Citation63]. Further research is needed to better understand the role of TRPC6 in renal I/R damage.
TRPC6 overexpression may play a crucial role in renal interstitial fibrosis
Renal fibrosis is the principal process in chronic kidney disease (CKD), and it progresses to end-stage renal disease (ESRD). The initiation and development of renal fibrosis may involve complex but incompletely characterized interactions between injured tubules, pericytes, fibroblasts, endothelial cells, and inflammatory cells [Citation64]. Renal fibrosis is characterized by excessive accumulation and deposition of extracellular matrix components, which are mainly derived through epithelial-to-mesenchymal transition (EMT) and fibroblast activation [Citation65,Citation66]. TECs, which are the primary components of the kidney parenchyma, are vulnerable to various types of injury and act as a key source of myofibroblasts through EMT [Citation67]. TGF-β1 is a powerful inducer of EMT. It has recently been demonstrated that TRPC6 is an important component of the TGF-β1 signaling pathway. TGF-β1 upregulates the production of the TRPC6 protein via several pathways, such as the Smad3/ERK/NF-κB, Ras/Raf1/ERK, AKT/mTOR and ERK1/2 pathways, which promote EMT [Citation68–71]. In recent years, research on kidney fibrosis has revealed that TECs, rather than being targets or bystanders, are likely to be initiators of the interstitial fibrotic response to a range of injuries [Citation67,Citation72]. Although extensive studies on the role of TRPC6 in glomerulosclerosis have been conducted, the effect of TRPC6 on renal interstitial fibrosis has received less attention.
A common and extensively used experimental model for renal interstitial fibrosis is the UUO model. UUO results in marked alterations in renal hemodynamics and metabolism, followed by tubular injury and cell death via apoptosis or necrosis, in conjunction with the infiltration of macrophages and other inflammatory cells into the renal interstitium. This process has also been linked to phenotypic shifts in resident renal tubular cells, endothelial cells, and pericytes [Citation73]. In a UUO mouse model, TRPC6 promotes renal fibrosis and immune cell infiltration. TRPC6 has been implicated in the etiology of kidney fibrosis, and according to recent research, genetic blockade of this channel can reduce rat kidney fibroblast proliferation and myofibroblast differentiation in vitro [Citation8]. Histological and immunohistochemical analyses revealed less inflammatory cell infiltration (F4/80 and CD3) and fibrosis in the kidneys of Trpc6-/- mice with UUO than in those of WT mice with UUO(6). Compared to that in WT UUO kidneys, the expression of profibrotic genes in Trpc6-/- UUO kidneys was altered. Kong et al. used qRT–PCR to analyze the mRNA expression of the proinflammatory markers IL-1β, IL-6, and TNF-α. They discovered that the expression of these markers was considerably increased in response to UUO in the renal cortex of WT and Trpc6-/- kidneys [Citation8]. Furthermore, Trpc6 deficiency could affect immune cell function and, consequently, the expression of proinflammatory cytokines. The levels of all profibrotic markers were lower in the kidneys of Trcp6-/- UUO mice than in those of WT UUO mice, indicating that Trpc6 deficiency played a protective role in the UUO model. TRPC6 knockout and the selective TRPC6 inhibitor BI-749327 provided partial protection from renal fibrosis in a mouse UUO model [Citation74]. Taken together, these data are consistent with the hypothesis that an increase in TRPC6 expression plays a significant pathophysiological role in the development of progressive kidney disease.
EMT can be induced in fibrotic kidneys in a UUO model [Citation75,Citation76]. Grande and Lovisa et al. [Citation65,Citation77] reported that TECs underwent incomplete EMT. Partial EMT is associated with the basement membrane and promotes kidney fibrosis through three mechanisms. First, partial EMT induces cell cycle arrest, limiting future proliferation and repair; second, partial EMT results in the secretion of growth factors, such as TGF-β, and drives myofibroblast proliferation; and third, the pathogenic secretome promotes chronic kidney inflammation. The phenotypic changes in EMT were partially reversed in primary TECs derived from TRPC6-/- mice, and TRPC6 was upregulated in primary TECs from WT mice treated with TGF-β1. Additionally, compared with that in the control group, EMT was stimulated by HYP9, a TRPC6 agonist. These findings provide strong evidence that the antifibrotic effect of TRPC6 knockout is linked to EMT inhibition [Citation78]. Several studies have demonstrated that the activation of the Akt/mTOR and ERK1/2 pathways is critical in the process of EMT during fibrosis [Citation68,Citation69]. Consistent with previous reports, TRPC6–/– mice showed obvious downregulation of the Akt/mTOR and ERK1/2 pathways after UUO. In addition, compared with those in the control group, the Akt/mTOR and ERK1/2 pathways in the HYP9-stimulated group were upregulated, suggesting that TRPC6 deletion may limit EMT to alleviate kidney injury by downregulating the Akt/mTOR and ERK1/2 signaling pathways [Citation78]. Furthermore, klotho, a membrane and soluble protein, which is predominantly synthesized in the kidney, protects against renal fibrosis. UUO-induced interstitial fibrosis and tubular atrophy were significantly alleviated in klotho-overexpressing transgenic mice. The administration of soluble klotho significantly reduced obstruction-induced renal fibrosis in wild-type mice but not in Trpc6-knockout mice, indicating that klotho and TRPC6 inhibition act via the same pathway to prevent obstruction-induced renal fibrosis [Citation69,Citation79].
Although glomerular injury in DN is well established as the primary cause of kidney damage, tubular injury has recently been revealed to be also associated with DN progression and to be a major contributor to chronic kidney injury [Citation80]. Moreover, proliferation and apoptosis are crucial injurious events in DN-associated kidney injury, contributing to tubular atrophy and interstitial fibrosis [Citation81]. Accumulating evidence indicates that TRPC6 may also be involved in DN development. To determine the effect of increased TRPC6 expression on cell proliferation and apoptosis in high-glucose (HG)-treated TECs, TRPC6 siRNA and Ad-TRPC6 were used to silence and overexpress TRPC6, respectively. Emerging evidence suggests that TRPC6 contributes to inflammation in TECs under DN conditions [Citation11,Citation82]. Fu et al. [Citation82] reported that Ad-TRPC6 expression was associated with the release of IL-8 and IL-6 in HK-2 cells that were incubated with a high concentration of glucose. In addition, the TRPC6 siRNA inhibited inflammation and proliferation of HK-2 cells, and the knockout of TRPC6 reduced proteinuria and tubular injury in Akita mice [Citation82]. Under hyperglycemic conditions, NFAT is dephosphorylated and translocates to the nucleus in TECs. Once in the nucleus, NFAT targets downstream genes, such as TRPC6, which results in macrophage accumulation and inflammatory cytokine release, ultimately leading to tubulointerstitial inflammation and fibrosis [Citation11]. The NFAT signaling pathway was shown to be activated by the Ad-TRPC6 and deactivated by the TRPC6 siRNA. The effect of TRPC6 on HK-2 cells was eliminated by the NFAT signaling inhibitor FK-506. FK-506 blocks the activation of the JNK and p38 MAP kinases [Citation83,Citation84], which are involved in HK-2 cell proliferation and apoptosis [Citation85,Citation86]. Moreover, the inhibition of JNK phosphorylation and p38 expression can downregulate TRPC6 expression [Citation87,Citation88]. Consequently, p-JNK and p38 may play essential roles in TRPC6-mediated TEC injury following DN onset, and their exact roles should be explored in future studies.
In a recent study, Jiang S et al. [Citation89] reported that TGF-β1 exposure induced fibroblast-myofibroblast transdifferentiation and upregulated the expression of TRPC6. TRPC6 expression is mediated by Yes-associated protein (YAP) translocation via p38 MAPK activation. Pharmacological inhibition or knockdown of TRPC6 suppresses fibroblast-myofibroblast transdifferentiation. These data demonstrated that TRPC6 played a vital role in TGF-β1-induced fibroblast–myoblast transdifferentiation of NRK-49F cells [Citation89].
TRPC6 drives tumorigenesis and the progression of RCC
RCC is the most frequent tumor arising from the cells lining the renal tubules [Citation90]. Different subtypes of RCC exist. Clear cell RCC (ccRCC) (70–80%) is the most common type of RCC, followed by papillary RCC (15%) [Citation91,Citation92]. Both originate from the proximal tubules. In RCC tissues, the TRPC6 channel was shown to be strongly expressed, and its expression level was linked to the RCC histological grade [Citation90]. TRPC6 overexpression may change G2 phase transition, thus altering the DNA damage checkpoint, which contributes to genomic instability and RCC tumorigenesis and regulates RCC cell proliferation [Citation90]. Conversely, the inhibition of TRPC6 mRNA expression is thought to prolong the G2/M phase transition in RCC cells, eventually providing sufficient time to ensure efficient DNA repair responses [Citation90]. Hepatocyte growth factor (HGF), which is a pleiotropic glycoprotein, reportedly increases the expression of TRPC6, which induces in vitro TEC growth, motility, scattering, and branching morphogenesis [Citation93]. In the case of hereditary papillary RCC, a gain-of-function mutation in the HGF-tyrosine kinase membrane receptor mesenchymal-epithelial transition(MET) factor generates uncontrolled cancer-promoting effects [Citation94]. In ccRCC, MET signaling also mediates vascular endothelial growth factor (VEGF) resistance [Citation92]. Lysine-deficient protein kinase 1 (WNK1), a major regulator of tubular electrolyte homeostasis, is highly expressed in distal convoluted tubules and connecting tubules [Citation95]. Furthermore, evidence has emerged regarding the key role of WNK1 in tumorigenesis [Citation96]. In ccRCC, increased WNK1 levels stimulate the phosphatidylinositol 4-kinase IIIα (PI4KIIIα) enzyme, which in turn triggers GPCR/PLC signaling and leads to the activation of TRPC6[5]. Kim et al. [Citation95]revealed that the WNK1-stimulated TRPC6/NFATc1 pathway functioned as a crucial component in the proliferation and migration of ccRCC cells. Overall, blocking the TRPC6/NFATc1 pathway markedly decreased the survival and proliferation of ccRCC cells. Moreover, hypoxia-inducible factor-1 (HIF-1) is expressed predominantly in TECs and plays a major role in both physiological and pathological processes, including tumorigenesis [Citation97,Citation98]. Glucose transporter 1 (GLUT1), which is a HIF-1 target gene, is essential for glucose uptake in most cells, including RCC cells [Citation97]. Under hypoxic conditions, TRPC6 regulates GLUT1 mRNA and protein levels via HIF-1, thereby affecting glucose uptake and consequent glucose metabolism in certain cancer cell types [Citation97].
In summary, TRPC6 is a critical factor in tumorigenesis and tumor progression in different RCC types [Citation95]. Inhibitors of MAPKs, which are the downstream effectors of WNK1, have been recommended for the treatment of RCC [Citation99]. Similarly, HIF-1, WNK1, TRPC6, GLUT1, and MET may be promising targets for the treatment of RCC [Citation92].
Conclusion
This review systematically presents the current knowledge and understanding of the physiological and pathophysiological roles of TRPC6 in renal tubules. TRPC6 is involved in I/R injury, renal interstitial fibrosis and RCC. These harmful conditions require much attention. This work provides a detailed introduction to the TRPC6 pathways in various scenarios. Notably, TRPC6 plays dual roles in I/R injury. TRPC6 also participates in the EMT process, which affects both tissue fibrosis and tumor growth. These findings have significant implications for future research on TRPC6. At present, few studies have been performed on TRPC6 in renal tubular disorders, and most of them are limited to basic research. This paper summarizes the role of TRPC6 in renal tubular diseases, which provides some guidance for future basic and clinical research. In clinical practice, obtaining blood or urine samples from patients and detecting the TRPC6 gene or mRNA would be very helpful.
The mechanisms of TRPC6 action in renal tubular diseases are diverse, and it is very meaningful to choose rational treatments according to the mechanism involved. As discussed above, Ang II and calcineurin are involved in the activation of TRPC6. Angiotensin-converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) may contribute to improving renal diseases by reducing the expression of TRPC6. CNIs can reduce TRPC6-induced calcium currents by directly inhibiting NFAT-induced TRPC6 transcription and dephosphorylation [Citation100]. Moreover, agonists of protein kinase G (PKG) can induce the phosphorylation of TRPC6 at threonine 69 (Thr69) and block the dephosphorylation of Thr69 by phosphodiesterase (PDE) family members, such as PDE5, which in turn increases the inhibition of TRPC6 [Citation11,Citation34]. ACEIs, ARBs, CNIs and PDE inhibitors may be effective therapeutic drugs because they inhibit both TRPC6 expression and activity. However, these drugs are mostly used for the treatment of TRPC6-mediated glomerular diseases, and their application in tubular diseases needs clinical validation. There is also very little information about the use of antioxidants in TRPC6-related kidney diseases. Overall, it is extremely challenging to translate medications from basic research to the clinic.
The overall effects of TRPC6 inactivation or knockout appear to be dependent on the disease model and may depend on the strain or species. Although we acknowledge the limitations of preclinical models, these model systems have significantly advanced our understanding of normal physiology and pathophysiological processes. Clinical data on TRPC6 are limited. Further investigations are needed to substantiate these findings and, ultimately, to obtain a deeper molecular knowledge of severe pathophysiological diseases. Increased knowledge of the TRPC6 function could lead to the development of targeted drugs. Following the introduction of additional targeted treatments, clinical care may improve.
Author contributions
All the above authors have contributed to this review. The main contents of this review were analyzed and written by Aiqin Sheng, assisted by Fei Liu, Qianhui Wang, Haidong Fu, and Jianhua Mao provided guidance and revision. All authors have read and agreed to the published version of the manuscript.
Disclosures statement
No potential conflict of interest was reported by the author(s).
Acknowledgment
The invaluable support of all staff of the department of nephrology in Children’s Hospital of Zhejiang University School of Medicine is gratefully acknowledged.
Additional information
Funding
References
- Inoue R, Kurahara LH, Hiraishi K. TRP channels in cardiac and intestinal fibrosis. Semin Cell Dev Biol. 2019;94:40–49. doi: 10.1016/j.semcdb.2018.11.002.
- Dryer SE, Reiser J. TRPC6 channels and their binding partners in podocytes: role in glomerular filtration and pathophysiology. Am J Physiol Renal Physiol. 2010;299(4):F689–F701. doi: 10.1152/ajprenal.00298.2010.
- Dryer SE, Roshanravan H, Kim EY. TRPC channels: regulation, dysregulation and contributions to chronic kidney disease. Biochim Biophys Acta Mol Basis Dis. 2019;1865(6):1041–1066. doi: 10.1016/j.bbadis.2019.04.001.
- Hall G, Wang L, Spurney RF. TRPC channels in proteinuric kidney diseases. Cells. 2019;9(1):44. doi: 10.3390/cells9010044.
- Englisch CN, Paulsen F, Tschernig T. TRPC channels in the physiology and pathophysiology of the renal tubular system: what do we know. Int J Mol Sci. 2022;24(1):181. doi: 10.3390/ijms24010181.
- Awuah Boadi E, Shin S, Yeroushalmi S, et al. Modulation of tubular pH by acetazolamide in a Ca(2+) transport deficient mice facilitates calcium nephrolithiasis. Int J Mol Sci. 2021;22(6):3050. doi: 10.3390/ijms22063050.
- Shin S, Ibeh CL, Awuah Boadi E, et al. Hypercalciuria switches Ca(2+) signaling in proximal tubular cells, induces oxidative damage to promote calcium nephrolithiasis. Genes Dis. 2022;9(2):531–548. doi: 10.1016/j.gendis.2021.04.006.
- Kong W, Haschler TN, Nürnberg B, et al. Renal fibrosis, immune cell infiltration and changes of TRPC channel expression after unilateral ureteral obstruction in Trpc6–/– Mice. Cell Physiol Biochem. 2019;52(6):1484–1502. doi: 10.33594/000000103.
- Padala SA, Kallam A, Treasure Island (FL): StatPearls; 2024. .
- Markó L, Mannaa M, Haschler TN, et al. Renoprotection: focus on TRPV1, TRPV4, TRPC6 and TRPM2. Acta Physiol (Oxf). 2017;219(3):589–612. doi: 10.1111/apha.12828.
- Zhang S, Wang H, Liu Y, et al. Tacrolimus ameliorates tubulointerstitial inflammation in diabetic nephropathy via inhibiting the NFATc1/TRPC6 pathway. J Cell Mol Med. 2020;24(17):9810–9824. doi: 10.1111/jcmm.15562.
- Shi J, Geshi N, Takahashi S, et al. Molecular determinants for cardiovascular TRPC6 channel regulation by Ca2+/calmodulin-dependent kinase II. J Physiol. 2013;591(11):2851–2866. doi: 10.1113/jphysiol.2013.251249.
- Shi J, Mori E, Mori Y, et al. Multiple regulation by calcium of murine homologues of transient receptor potential proteins TRPC6 and TRPC7 expressed in HEK293 cells. J Physiol. 2004;561(Pt 2):415–432. doi: 10.1113/jphysiol.2004.075051.
- Anderson M, Roshanravan H, Khine J, et al. Angiotensin II activation of TRPC6 channels in rat podocytes requires generation of reactive oxygen species. J Cell Physiol. 2014;229(4):434–442. doi: 10.1002/jcp.24461.
- Ilatovskaya DV, Blass G, Palygin O, et al. A NOX4/TRPC6 pathway in podocyte calcium regulation and renal damage in diabetic kidney disease. J Am Soc Nephrol. 2018;29(7):1917–1927. doi: 10.1681/ASN.2018030280.
- Susztak K, Raff AC, Schiffer M, et al. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes. 2006;55(1):225–233. doi: 10.2337/diabetes.55.01.06.db05-0894.
- Kim EY, Anderson M, Dryer SE. Insulin increases surface expression of TRPC6 channels in podocytes: role of NADPH oxidases and reactive oxygen species. Am J Physiol Renal Physiol. 2012;302(3):F298–F307. doi: 10.1152/ajprenal.00423.2011.
- Kim EY, Anderson M, Dryer SE. Sustained activation of N-methyl-D-aspartate receptors in podoctyes leads to oxidative stress, mobilization of transient receptor potential canonical 6 channels, nuclear factor of activated T cells activation, and apoptotic cell death. Mol Pharmacol. 2012;82(4):728–737. doi: 10.1124/mol.112.079376.
- Kim EY, Anderson M, Wilson C, et al. NOX2 interacts with podocyte TRPC6 channels and contributes to their activation by diacylglycerol: essential role of podocin in formation of this complex. Am J Physiol Cell Physiol. 2013;305(9):C960–F971. doi: 10.1152/ajpcell.00191.2013.
- Roshanravan H, Dryer SE. ATP acting through P2Y receptors causes activation of podocyte TRPC6 channels: role of podocin and reactive oxygen species. Am J Physiol Renal Physiol. 2014;306(9):F1088–F1097. doi: 10.1152/ajprenal.00661.2013.
- Burnham DN, Uhlinger DJ, Lambeth JD. Diradylglycerol synergizes with an anionic amphiphile to activate superoxide generation and phosphorylation of p47phox in a cell-free system from human neutrophils. J Biol Chem. 1990;265(29):17550–17559. doi: 10.1016/S0021-9258(18)38200-0.
- Tyagi SR, Neckelmann N, Uhlinger DJ, et al. Cell-free translocation of recombinant p47-phox, a component of the neutrophil NADPH oxidase: effects of guanosine 5’-O-(3-thiotriphosphate), diacylglycerol, and an anionic amphiphile. Biochemistry. 1992;31(10):2765–2774. doi: 10.1021/bi00125a017.
- Schwarz K, Simons M, Reiser J, et al. Podocin, a raft-associated component of the glomerular slit diaphragm, interacts with CD2AP and nephrin. J Clin Invest. 2001;108(11):1621–1629. doi: 10.1172/JCI200112849.
- Hayek SS, Quyyumi AA, Reiser J. Soluble Urokinase Receptor and Chronic Kidney Disease. N Engl J Med. 2016;374(9):891. doi: 10.1056/NEJMc1515787.
- Kim EY, Hassanzadeh Khayyat N, Dryer SE. Mechanisms underlying modulation of podocyte TRPC6 channels by suPAR: role of NADPH oxidases and Src family tyrosine kinases. Biochim Biophys Acta Mol Basis Dis. 2018;1864(10):3527–3536. doi: 10.1016/j.bbadis.2018.08.007.
- Ma R, Chaudhari S, Li W. Canonical Transient Receptor Potential 6 Channel: a New Target of Reactive Oxygen Species in Renal Physiology and Pathology. Antioxid Redox Signal. 2016;25(13):732–748. doi: 10.1089/ars.2016.6661.
- Trebak M, Ginnan R, Singer HA, et al. Interplay between calcium and reactive oxygen/nitrogen species: an essential paradigm for vascular smooth muscle signaling. Antioxid Redox Signal. 2010;12(5):657–674. doi: 10.1089/ars.2009.2842.
- Yoshida T, Inoue R, Morii T, et al. Nitric oxide activates TRP channels by cysteine S-nitrosylation. Nat Chem Biol. 2006;2(11):596–607. doi: 10.1038/nchembio821.
- Kim EY, Roshanravan H, Dryer SE. Syndecan-4 ectodomain evokes mobilization of podocyte TRPC6 channels and their associated pathways: an essential role for integrin signaling. Biochim Biophys Acta. 2015;1853(10 Pt A):2610–2620. doi: 10.1016/j.bbamcr.2015.07.011.
- Davin JC. The glomerular permeability factors in idiopathic nephrotic syndrome. Pediatr Nephrol. 2016;31(2):207–215. doi: 10.1007/s00467-015-3082-x.
- Kim EY, Roshanravan H, Dryer SE. Changes in podocyte TRPC channels evoked by plasma and sera from patients with recurrent FSGS and by putative glomerular permeability factors. Biochim Biophys Acta Mol Basis Dis. 2017;1863(9):2342–2354. doi: 10.1016/j.bbadis.2017.06.010.
- Anderson M, Kim EY, Hagmann H, et al. Opposing effects of podocin on the gating of podocyte TRPC6 channels evoked by membrane stretch or diacylglycerol. Am J Physiol Cell Physiol. 2013;305(3):C276–C289. doi: 10.1152/ajpcell.00095.2013.
- Huber TB, Schermer B, Müller RU, et al. Podocin and MEC-2 bind cholesterol to regulate the activity of associated ion channels. Proc Natl Acad Sci USA. 2006;103(46):17079–17086. doi: 10.1073/pnas.0607465103.
- Kriz W, Lemley KV. Potential relevance of shear stress for slit diaphragm and podocyte function. Kidney Int. 2017;91(6):1283–1286. doi: 10.1016/j.kint.2017.02.032.
- Endlich N, Endlich K. The challenge and response of podocytes to glomerular hypertension. Semin Nephrol. 2012;32(4):327–341. doi: 10.1016/j.semnephrol.2012.06.004.
- Pu Y, Zhao H, Shen B, et al. TRPC6 ameliorates renal ischemic reperfusion injury by inducing Zn(2+) influx and activating autophagy to resist necrosis. Ann Transl Med. 2022;10(5):249–249. doi: 10.21037/atm-21-5837.
- Schrier RW, Arnold PE, Van Putten VJ, et al. Cellular calcium in ischemic acute renal failure: role of calcium entry blockers. Kidney Int. 1987;32(3):313–321. doi: 10.1038/ki.1987.211.
- Meng XM, Ren GL, Gao L, et al. NADPH oxidase 4 promotes cisplatin-induced acute kidney injury via ROS-mediated programmed cell death and inflammation. Lab Invest. 2018;98(1):63–78. doi: 10.1038/labinvest.2017.120.
- Liao W, Wang Z, Fu Z, et al. p62/SQSTM1 protects against cisplatin-induced oxidative stress in kidneys by mediating the cross talk between autophagy and the Keap1-Nrf2 signalling pathway. Free Radic Res. 2019;53(7):800–814. doi: 10.1080/10715762.2019.1635251.
- Hou X, Huang M, Zeng X, et al. The Role of TRPC6 in Renal Ischemia/Reperfusion and Cellular Hypoxia/Reoxygenation Injuries. Front Mol Biosci. 2021;8:698975. doi: 10.3389/fmolb.2021.698975.
- Shen B, He Y, Zhou S, et al. TRPC6 May Protect Renal Ischemia-Reperfusion Injury Through Inhibiting Necroptosis of Renal Tubular Epithelial Cells. Med Sci Monit. 2016;22:633–641. doi: 10.12659/msm.897353.
- Ramirez GA, Coletto LA, Sciorati C, et al. Ion Channels and Transporters in Inflammation: special Focus on TRP Channels and TRPC6. Cells. 2018;7(7):70. doi: 10.3390/cells7070070.
- Ding Y, Winters A, Ding M, et al. Reactive oxygen species-mediated TRPC6 protein activation in vascular myocytes, a mechanism for vasoconstrictor-regulated vascular tone. J Biol Chem. 2011;286(36):31799–31809. doi: 10.1074/jbc.M111.248344.
- Zhao B, Yang H, Zhang R, et al. The role of TRPC6 in oxidative stress-induced podocyte ischemic injury. Biochem Biophys Res Commun. 2015;461(2):413–420. doi: 10.1016/j.bbrc.2015.04.054.
- Patergnani S, Danese A, Bouhamida E, et al. Various Aspects of Calcium Signaling in the Regulation of Apoptosis, Autophagy, Cell Proliferation, and Cancer. Int J Mol Sci. 2020;21(21):8323. doi: 10.3390/ijms21218323.
- Hou X, Xiao H, Zhang Y, et al. Transient receptor potential channel 6 knockdown prevents apoptosis of renal tubular epithelial cells upon oxidative stress via autophagy activation. Cell Death Dis. 2018;9(10):1015. doi: 10.1038/s41419-018-1052-5.
- Ren GL, Zhu J, Li J, et al. Noncoding RNAs in acute kidney injury. J Cell Physiol. 2019;234(3):2266–2276. doi: 10.1002/jcp.27203.
- Brandenburger T, Salgado Somoza A, Devaux Y, et al. Noncoding RNAs in acute kidney injury. Kidney Int. 2018;94(5):870–881. doi: 10.1016/j.kint.2018.06.033.
- Yang Y, Li ZL, Wang FM, et al. MicroRNA26a inhibits cisplatin-induced renal tubular epithelial cells apoptosis through suppressing the expression of transient receptor potential channel 6 mediated dynamin-related protein 1. Cell Biochem Funct. 2020;38(4):384–391. doi: 10.1002/cbf.3474.
- Linkermann A, Bräsen JH, Himmerkus N, et al. Rip1 (receptor-interacting protein kinase 1) mediates necroptosis and contributes to renal ischemia/reperfusion injury. Kidney Int. 2012;81(8):751–761. doi: 10.1038/ki.2011.450.
- Shen B, Mei M, Pu Y, et al. Necrostatin-1 Attenuates Renal Ischemia and Reperfusion Injury via Meditation of HIF-1α/mir-26a/TRPC6/PARP1 Signaling. Mol Ther Nucleic Acids. 2019;17:701–713. doi: 10.1016/j.omtn.2019.06.025.
- Shen S, Jin Y, Li W, et al. Recombinant human erythropoietin pretreatment attenuates acute renal tubular injury against ischemia-reperfusion by restoring transient receptor potential channel-6 expression and function in collecting ducts. Crit Care Med. 2014;42(10):e663–e672. doi: 10.1097/CCM.0000000000000542.
- Liu X, Zhang T, Xia W, et al. Recombinant human erythropoietin pretreatment alleviates renal glomerular injury induced by cardiopulmonary bypass by reducing transient receptor potential channel 6-nuclear factor of activated T-cells pathway activation. J Thorac Cardiovasc Surg. 2013;146(3):681–687. doi: 10.1016/j.jtcvs.2013.02.076.
- Gibon J, Tu P, Bohic S, et al. The over-expression of TRPC6 channels in HEK-293 cells favours the intracellular accumulation of zinc. Biochim Biophys Acta. 2011;1808(12):2807–2818. doi: 10.1016/j.bbamem.2011.08.013.
- Gammoh NZ, Rink L. Zinc in Infection and Inflammation. Nutrients. 2017;9(6):624. doi: 10.3390/nu9060624.
- Hadj Abdallah N, Baulies A, Bouhlel A, et al. Zinc mitigates renal ischemia-reperfusion injury in rats by modulating oxidative stress, endoplasmic reticulum stress, and autophagy. J Cell Physiol. 2018;233(11):8677–8690. doi: 10.1002/jcp.26747.
- Shen B, Mei M, Ai S, et al. TRPC6 inhibits renal tubular epithelial cell pyroptosis through regulating zinc influx and alleviates renal ischemia-reperfusion injury. Faseb J. 2022;36(10):e22527. doi: 10.1096/fj.202200109RR.
- Moslemi F, Talebi A, Nematbakhsh M. Protective Effect of Zinc Supplementation on Renal Ischemia/Reperfusion Injury in Rat: gender-related Difference. Int J Prev Med. 2019;10:68. doi: 10.4103/ijpvm.IJPVM_279_17.
- Ogawa T, Mimura Y. Antioxidant effect of zinc on acute renal failure induced by ischemia-reperfusion injury in rats. Am J Nephrol. 1999;19(5):609–614. doi: 10.1159/000013529.
- Rao K, Sethi K, Ischia J, et al. Protective effect of zinc preconditioning against renal ischemia reperfusion injury is dose dependent. PLoS One. 2017;12(7):e0180028. doi: 10.1371/journal.pone.0180028.
- Shi J, Gao W, Shao F. Pyroptosis: gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci. 2017;42(4):245–254. doi: 10.1016/j.tibs.2016.10.004.
- Chen X, Liu G, Yuan Y, et al. NEK7 interacts with NLRP3 to modulate the pyroptosis in inflammatory bowel disease via NF-κB signaling. Cell Death Dis. 2019;10(12):906. doi: 10.1038/s41419-019-2157-1.
- Zheng Z, Tsvetkov D, Bartolomaeus T, et al. Role of TRPC6 in kidney damage after acute ischemic kidney injury. Sci Rep. 2022;12(1):3038. doi: 10.1038/s41598-022-06703-9.
- Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7(12):684–696. doi: 10.1038/nrneph.2011.149.
- Lovisa S, LeBleu VS, Tampe B, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. 2015;21(9):998–1009. doi: 10.1038/nm.3902.
- Lovisa S, Zeisberg M, Kalluri R. Partial epithelial-to-mesenchymal transition and other new mechanisms of kidney fibrosis. Trends Endocrinol Metab. 2016;27(10):681–695. doi: 10.1016/j.tem.2016.06.004.
- Gewin LS. Renal fibrosis: primacy of the proximal tubule. Matrix Biol. 2018;68-69:248–262. doi: 10.1016/j.matbio.2018.02.006.
- Wang Z, Qu L, Deng B, et al. STYK1 promotes epithelial-mesenchymal transition and tumor metastasis in human hepatocellular carcinoma through MEK/ERK and PI3K/AKT signaling. Sci Rep. 2016;6(1):33205. doi: 10.1038/srep33205.
- Qu H, Liu L, Liu Z, et al. Blocking TBK1 alleviated radiation-induced pulmonary fibrosis and epithelial-mesenchymal transition through Akt-Erk inactivation. Exp Mol Med. 2019;51(4):1–17. doi: 10.1038/s12276-019-0240-4.
- Yu L, Lin Q, Liao H, et al. TGF-β1 induces podocyte injury through Smad3-ERK-NF-κB pathway and Fyn-dependent TRPC6 phosphorylation. Cell Physiol Biochem. 2010;26(6):869–878. doi: 10.1159/000323996.
- Ge P, Wei L, Zhang M, et al. TRPC1/3/6 inhibition attenuates the TGF-β1-induced epithelial-mesenchymal transition in gastric cancer via the Ras/Raf1/ERK signaling pathway. Cell Biol Int. 2018;42(8):975–984. doi: 10.1002/cbin.10963.
- Liu BC, Tang TT, Lv LL, et al. Renal tubule injury: a driving force toward chronic kidney disease. Kidney Int. 2018;93(3):568–579. doi: 10.1016/j.kint.2017.09.033.
- Chevalier RL, Forbes MS, Thornhill BA. Ureteral obstruction as a model of renal interstitial fibrosis and obstructive nephropathy. Kidney Int. 2009;75(11):1145–1152. doi: 10.1038/ki.2009.86.
- Lin BL, Matera D, Doerner JF, et al. In vivo selective inhibition of TRPC6 by antagonist BI 749327 ameliorates fibrosis and dysfunction in cardiac and renal disease. Proc Natl Acad Sci U S A. 2019;116(20):10156–10161. doi: 10.1073/pnas.1815354116.
- Iwano M, Plieth D, Danoff TM, et al. Evidence that fibroblasts derive from epithelium during tissue fibrosis. J Clin Invest. 2002;110(3):341–350. doi: 10.1172/JCI15518.
- Klahr S, Morrissey J. Obstructive nephropathy and renal fibrosis: the role of bone morphogenic protein-7 and hepatocyte growth factor. Kidney Int Suppl. 2003;64(87): 105–112. doi: 10.1046/j.1523-1755.64.s87.16.x.
- Grande MT, Sánchez-Laorden B, López-Blau C, et al. Snail1-induced partial epithelial-to-mesenchymal transition drives renal fibrosis in mice and can be targeted to reverse established disease. Nat Med. 2015;21(9):989–997. doi: 10.1038/nm.3901.
- Zhang Y, Yin N, Sun A, et al. Transient receptor potential channel 6 knockout ameliorates kidney fibrosis by inhibition of epithelial-mesenchymal transition. Front Cell Dev Biol. 2020;8:602703. doi: 10.3389/fcell.2020.602703.
- Wu YL, Xie J, An SW, et al. Inhibition of TRPC6 channels ameliorates renal fibrosis and contributes to renal protection by soluble klotho. Kidney Int. 2017;91(4):830–841. doi: 10.1016/j.kint.2016.09.039.
- Okamura DM, Pasichnyk K, Lopez-Guisa JM, et al. Galectin-3 preserves renal tubules and modulates extracellular matrix remodeling in progressive fibrosis. Am J Physiol Renal Physiol. 2011;300(1):F245–F253. doi: 10.1152/ajprenal.00326.2010.
- Habib SL. Diabetes and renal tubular cell apoptosis. World J Diabetes. 2013;4(2):27–30. doi: 10.4239/wjd.v4.i2.27.
- Fu Y, Wang C, Zhang D, et al. Increased TRPC6 expression is associated with tubular epithelial cell proliferation and inflammation in diabetic nephropathy. Mol Immunol. 2018;94:75–81. doi: 10.1016/j.molimm.2017.12.014.
- Matsuda S, Shibasaki F, Takehana K, et al. Two distinct action mechanisms of immunophilin-ligand complexes for the blockade of T-cell activation. EMBO Rep. 2000;1(5):428–434. doi: 10.1093/embo-reports/kvd090.
- Sullivan JA, Adams AB, Burlingham WJ. The emerging role of TH17 cells in organ transplantation. Transplantation. 2014;97(5):483–489. doi: 10.1097/TP.0000000000000000.
- Lu H, Zhang XY, Wang YQ, et al. Andrographolide sodium bisulfate-induced apoptosis and autophagy in human proximal tubular endothelial cells is a ROS-mediated pathway. Environ Toxicol Pharmacol. 2014. 37(2): 718–28. doi: 10.1016/j.etap.2014.01.019.
- Park JW, Cho JW, Joo SY, et al. Paricalcitol prevents cisplatin-induced renal injury by suppressing apoptosis and proliferation. Eur J Pharmacol. 2012;683(1–3):301–309. doi: 10.1016/j.ejphar.2012.03.019.
- Davis J, Burr AR, Davis GF, et al. A TRPC6-dependent pathway for myofibroblast transdifferentiation and wound healing in vivo. Dev Cell. 2012;23(4):705–715. doi: 10.1016/j.devcel.2012.08.017.
- Meng Y, Li WZ, Shi YW, et al. Danshensu protects against ischemia/reperfusion injury and inhibits the apoptosis of H9c2 cells by reducing the calcium overload through the p-JNK-NF-κB-TRPC6 pathway. Int J Mol Med. 2016;37(1):258–266. doi: 10.3892/ijmm.2015.2419.
- Jiang S, Gu L, Hu Y, et al. Inhibition of TRPC6 suppressed TGFβ-induced fibroblast-myofibroblast transdifferentiation in renal interstitial NRK-49F cells. Exp Cell Res. 2022;421(1):113374. doi: 10.1016/j.yexcr.2022.113374.
- Song J, Wang Y, Li X, et al. Critical role of TRPC6 channels in the development of human renal cell carcinoma. Mol Biol Rep. 2013;40(8):5115–5122. doi: 10.1007/s11033-013-2613-4.
- Paner GP, Chumbalkar V, Montironi R, et al. Updates in Grading of Renal Cell Carcinomas Beyond Clear Cell Renal Cell Carcinoma and Papillary Renal Cell Carcinoma. Adv Anat Pathol. 2022;29(3):117–130. doi: 10.1097/PAP.0000000000000341.
- Nandagopal L, Sonpavde GP, Agarwal N. Investigational MET inhibitors to treat Renal cell carcinoma. Expert Opin Investig Drugs. 2019;28(10):851–860. doi: 10.1080/13543784.2019.1673366.
- Rampino T, Gregorini M, Guidetti C, et al. KCNA1 and TRPC6 ion channels and NHE1 exchanger operate the biological outcome of HGF/scatter factor in renal tubular cells. Growth Factors. 2007;25(6):382–391. doi: 10.1080/08977190801892184.
- Finley DS, Pantuck AJ, Belldegrun AS. Tumor biology and prognostic factors in renal cell carcinoma. Oncologist. 2011;16 (Suppl 2):4–13. doi: 10.1634/theoncologist.2011-S2-04.
- Kim JH, Hwang KH, Eom M, et al. WNK1 promotes renal tumor progression by activating TRPC6-NFAT pathway. Faseb J. 2019;33(7):8588–8599. doi: 10.1096/fj.201802019RR.
- Moniz S, Jordan P. Emerging roles for WNK kinases in cancer. Cell Mol Life Sci. 2010;67(8):1265–1276. doi: 10.1007/s00018-010-0261-6.
- Li S, Wang J, Wei Y, et al. Crucial role of TRPC6 in maintaining the stability of HIF-1α in glioma cells under hypoxia. J Cell Sci. 2015;128(17):3317–3329. doi: 10.1242/jcs.173161.
- Choudhry H, Harris AL. Advances in hypoxia-inducible factor biology. Cell Metab. 2018;27(2):281–298. doi: 10.1016/j.cmet.2017.10.005.
- Chauhan A, Semwal DK, Mishra SP, et al. Combination of mTOR and MAPK inhibitors-A potential way to treat renal cell carcinoma. Med Sci. 2016;4(4):16. doi: 10.3390/medsci4040016.
- Hall G, Rowell J, Farinelli F, et al. Phosphodiesterase 5 inhibition ameliorates angiontensin II-induced podocyte dysmotility via the protein kinase G-mediated downregulation of TRPC6 activity. Am J Physiol Renal Physiol. 2014;306(12):F1442–F1450. doi: 10.1152/ajprenal.00212.2013.