
Abstract
Acute kidney injury (AKI) is a significant issue in public health, displaying a high occurrence rate and mortality rate. Ferroptosis, a form of programmed cell death (PCD), is characterized by iron accumulation and intensified lipid peroxidation. Recent studies have demonstrated the pivotal significance of ferroptosis in AKI caused by diverse stimuli, including ischemia-reperfusion injury (IRI), sepsis and toxins. Autophagy, a multistep process that targets damaged organelles and macromolecules for degradation and recycling, also plays an essential role in AKI. Previous research has demonstrated that autophagy deletion in proximal tubules could aggravate tubular injury and renal function loss, indicating the protective function of autophagy in AKI. Consequently, finding ways to stimulate autophagy has become a crucial therapeutic strategy. The recent discovery of the role of selective autophagy in influencing ferroptosis has identified new therapeutic targets for AKI and has highlighted the importance of understanding the cross-talk between autophagy and ferroptosis. This study aims to provide an overview of the signaling pathways involved in ferroptosis and autophagy, focusing on the mechanisms and functions of selective autophagy and autophagy-dependent ferroptosis. We hope to establish a foundation for future investigations into the interaction between autophagy and ferroptosis in AKI as well as other diseases.
1. Introduction
AKI is a prevalent and serious condition caused by a range of physiological and pathological factors, resulting in a rapid deterioration of renal function [Citation1,Citation2]. Recent reports indicate that the occurrence rate of AKI has remained elevated and is continuing to rise each year [Citation3]. Additionally, studies have shown that AKI raises the likelihood of chronic kidney disease in affected patients. Despite blood purification being a common treatment, there have been limited advancements in finding effective therapeutics to prevent the occurrence and progression of AKI [Citation4,Citation5].
The pathogenesis of AKI involves various factors, including oxidative stress, inflammation, hypoxia, pericyte injury, and microvascular dysfunction [Citation6]. These factors can be caused by surgery, nephrotoxic drugs, ischemia, and sepsis [Citation6–8]. In response to these stressors, autophagy serves as a cytoprotective mechanism by eliminating and recycling damaged molecules and organelles to promote cellular adaptation [Citation9,Citation10]. Autophagy exists in three forms: macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA), each differing in how cargo is transported to lysosomes. Macroautophagy (hereafter called autophagy) can be categorized as nonselective autophagy (degrades parts of cytoplasm and organelles at random) and selective autophagy (clears specific cargos such as proteins, pathogens, and damaged organelles) [Citation11,Citation12]. Previous studies have demonstrated that the absence of autophagy in proximal tubules results in more severe tubular injury and renal function loss [Citation13,Citation14]. Kaushal GP and Shah SV also valued effective pharmacological inducers of autophagy as a crucial treatment for AKI [Citation15]. With further studies on autophagy, some types of selective autophagy have been reported as drivers of ferroptosis under certain conditions [Citation16]. The precise regulation of selective autophagy could potentially serve as a novel target for AKI treatment.
Recently, numerous studies have reported that the pharmaceutical blockage of ferroptosis has a positive effect on reducing renal injury in different AKI models, suggesting a significant role for ferroptosis in AKI [Citation17,Citation18]. Ferroptosis can additionally trigger the release of intracellular immunogenic molecules and proinflammatory cytokines, thereby contributing to the advancement of tubular injury [Citation19–21]. Initially described as a distinct form of regulated cell death (RCD), ferroptosis was first observed in cancer cells with oncogenetic RAS mutations. It distinguishes itself from apoptosis, necrosis, and autophagy by its unique characteristics in morphology, biochemistry, and genetics [Citation16,Citation22]. However, recent research on selective autophagy, including ferritinophagy (elimination of ferritin), lipophagy (elimination of lipids), and chaperone-mediated autophagy, has revealed that autophagy is closely connected with ferroptotic cell death [Citation16,Citation23]. It has been discovered that certain types of autophagy actually promote ferroptotic cell death, and these autophagy-dependent processes have significant implications in various areas such as anticancer therapies, neurodegenerative diseases, liver fibrosis, ischemic stroke, and even AKI [Citation24–31]. These studies have shed light on the interplay between the autophagic machinery and ferroptosis, highlighting the transition of autophagy from a pro-survival to a lethal function. Consequently, it is imperative to review the connection between them as well as the distinct roles of different selective autophagy processes in AKI. While numerous studies have discussed the mechanisms of either ferroptosis or autophagy in relation to AKI, limited attention has been given to the simultaneous involvement of both autophagy and ferroptosis, the interplay of their molecular pathways, and the possibility of therapeutic strategies. This review delves into the pathways of ferroptosis and autophagy (specifically focusing on selective autophagy), offering a thorough insight into their molecular mechanisms, their crosstalk in AKI, and the promise of ferroptosis and autophagy pathways for treating AKI.
2. Ferroptosis
2.1. Overview of ferroptosis
Iron is essential for living organisms as it serves various important biological functions, including cellular oxygen transport, enzyme catalysis, and bioenergetic reactions [Citation32,Citation33]. Iron has the ability to accept and donate electrons, allowing it to participate in the Fenton reaction: Fe2+ + H2O2 → Fe3+ + HO• + HO−. The Fenton reaction produces HO• (hydroxyl radical), which can cause nonselective damage to most biomolecules such as lipids [Citation34,Citation35]. The presence of redox-active iron is tightly regulated under normal conditions. However, intracellular iron overload, particularly in its ferrous form, can lead to lipid peroxidation [Citation36]. This form of iron-dependent cell death, characterized by excessive lipid peroxidation, was named ferroptosis in 2012 [Citation22]. Further research on ferroptosis has identified several key regulators and mechanisms that could potentially serve as new targets for diseases associated with ferroptosis, such as AKI [Citation17,Citation18,Citation37]. To explore the connection between autophagy and ferroptosis in AKI, our focus will now be on understanding how autophagy affects the key regulators and central events of ferroptosis.
2.2. Mechanisms and key regulators of ferroptosis
Ferroptosis was recognized as a ROS-dependent cell death with two central biochemistry events, including iron overload and lipid peroxidation. In addition, several critical regulators of ferroptosis, especially system Xc− and GPX4, have been extensively researched as antioxidant systems. Next, we will provide a brief overview of these major regulators and their relationship with several types of selective autophagy ().
Figure 1. Ferritinophagy, mitophagy, and chaperone-mediated autophagy are related to ferroptosis in AKI. (1) Ferritinophagy increases intracellular iron (Fe2+) to promote ferroptosis via autophagic degradation of ferritin. (2) Mitophagy can eliminate damaged mitochondria, which is a major source of ROS generation and mainly protects tubular cells against stimuli. (3) Chaperone-mediated autophagy is involved in GPX4 degradation for ferroptosis.
TF: transferrin; TFR: transferrin receptor; STEAP3: six-transmembrane epithelial antigen of Prostate 3; ROS: reactive oxygen species; LC3: microtubule associated protein 1 light chain 3; SQSTM1: sequestosome 1; NCOA4: Nuclear Receptor Coactivator 4; SLC7A11: solute carrier family 7 member 11; SLC3A2: solute carrier family 3 member 2; GSH: glutathione; GPX4: glutathione peroxidase 4; CMA,chaperone-mediated autophagy; HSP90: heat shock protein 90; LAMP2A: lysosome-associated membrane protein type 2a; ACSL4: CoA synthetase long chain family member 4; LPCAT3: lysophosphatidylcholine acyltransferase 3; LOXs: lipoxygenases; PUFA: polyunsaturated fatty acids; PUFA-CoA: polyunsaturated fatty acyl CoA; PUFA-PL: phospholipid-bound polyunsaturated fatty acids; LOOH: lipid hydroperoxide; VDAC: voltage-dependent anion channel.
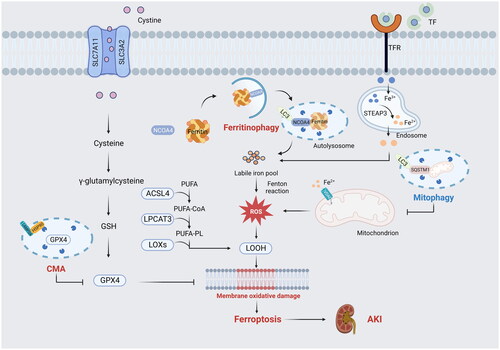
2.2.1. Iron overload
Iron plays an important role in the production of ROS through enzymatic or non-enzymatic reactions [Citation32]. Normally, in the serum, Fe3+ ions typically bind to transferrin (TF) and are then transported into cells via the membrane transferrin receptor 1 (TFR1). The metalloreductases reduce Fe3+ ions to Fe2+ ions, which are subsequently transported to the intracellular iron pool. Excess Fe2+ ions are stored in the iron-storage protein ferritin, which consists of ferritin light chain (FTL) and ferritin heavy chain 1 (FTH1) [Citation32,Citation38,Citation39]. The process of ferritin degradation in lysosomes, known as nuclear receptor coactivator 4 (NCOA4)-mediated ferritinophagy, plays a critical role in increasing free iron [Citation40]. When the regulation of iron metabolism is disrupted, the degradation of FTH1 resulting from the alteration of NCOA4 and FTR1 triggers the excessive buildup of Fe2+ ions. This leads to the generation of high levels of ROS and even ferroptotic cell death [Citation24,Citation40,Citation41]. Furthermore, iron has been identified as a promoter of lipoxygenases (LOXs), which mediate and catalyze the peroxidation of polyunsaturated fatty acids (PUFAs) [Citation36,Citation42].
2.2.2. Lipid peroxidation
As a free radical-driven reaction, lipid peroxidation drives the occurrence of ferroptosis [Citation22,Citation43]. Since phospholipid-bound polyunsaturated fatty acid (PUFA-PL) is well-known for its susceptibility to peroxidation catalyzed by LOXs and cytochrome P450 oxidoreductase (POR), increased production of PUFA-PL accelerates the deposition of lipid hydroperoxide [Citation36,Citation42]. During ferroptosis, CoA synthetase long chain family member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) are key mediators of PUFA-PL synthesis. ACSL4 catalyzes the biochemical reaction of CoA and PUFA to generate PUFA-CoA. LPCAT3 participates in the esterification of PUFA-CoA to promote the formation of PUFA-PL [Citation44,Citation45]. The extent of lipid peroxidation and subsequent ferroptosis depends on the accumulation of PUFAs; increased levels of PUFAs heighten the susceptibility to ferroptosis [Citation20]. Studies have shown that lipophagy mediated by Rab7a (a member of the RAS oncogene family) increases the deposition of free fatty acids (FFA) via triggering the degradation of lipid droplets, leading to more lipid peroxidation [Citation46]. Lipid droplet, synthesized in the endoplasmic reticulum (ER), is a lipid storage organelle that can be rapidly mobilized to release fatty acids [Citation46]. In addition, clockophagy is a recently discovered form of selective autophagy that targets the Aryl hydrocarbon receptor Nuclear Translocator Like Protein 1 (ARNTL) for degradation during ferroptosis induced by ferroptosis inducers, such as RSL3 and FIN56, in ferroptosis-sensitive cancer cell lines like HT1080 and HL-60 cells [Citation47–49]. Autophagic ARNTL degradation promotes ferroptotic cell death through blocking HIF1α (hypoxia inducible factor 1 subunit alpha)-dependent fatty acid uptake and lipid storage [Citation47,Citation49]. HIF1α, a key transcription factor involved in the response to hypoxia, has a pro-survival function in ferroptosis by increasing the levels of fatty acid binding protein 3 (FABP3) and fatty acid binding protein 7 (FABP7) to facilitate lipid storage [Citation16,Citation47,Citation49]. The reduction of HIF1α levels resulting from ARNTL degradation hinders lipid storage and ultimately leads to increased lipid peroxidation during ferroptosis [Citation49].
2.2.3. System Xc−
System Xc−, a cysteine/glutamate antiporter system on the cytomembrane, controls the import of cystine to produce cysteine. It consists of two subunits: the light chain subunit (SLC7A11, solute carrier family 7 member 11) and the heavy chain subunit (SLC3A2, solute carrier family 3 member 2), and they maintain the production of glutathione (GSH, an enzyme-catalyzed antioxidant) by increasing the accumulation of intercellular cysteine [Citation22]. The synthesis and activity of GPX4 depend on GSH; GSH depletion will lead to the deposition of lipid hydroperoxide, ultimately resulting in ferroptosis [Citation22,Citation23]. Beclin1, a key regulator of autophagosome formation, has also been identified as a positive factor in ferroptosis by directly blocking system Xc−. System Xc− inhibitors like erastin induced the phosphorylation of beclin1 to promote the interaction between beclin1 and SLC7A11, resulting in the decreased activity of GPX4 and ultimate ferroptosis [Citation6].
2.2.4. GPX4
GPXs are a family of antioxidant enzymes that are widely expressed in various tissues. However, in the kidney, only GPX1, GPX3, and GPX4 have been found to be expressed [Citation50]. GPX4 serves as a phospholipid hydroperoxidase, which facilitates GSH to counteract lipoxygenase and decrease phospholipid hydroperoxide production. The synthesis and activity of GPX4 are controlled by selenium and GSH. Many studies have reported that the suppression of GPX4 synthesis and activity leads to the accumulation of lipid peroxidation, ultimately resulting in ferroptosis [Citation22,Citation42]. It has been reported that activation of CMA participates in the degradation of GPX4, and inhibiting CMA could stabilize GPX4 to reduce ferroptotic cell death [Citation48,Citation49]. While the cellular metabolic activity of GPX4 typically encourages ferroptosis, the breakdown of other proteins can actually hinder this process [Citation50]. Specifically, the breakdown of cysteine-rich extracellular proteins like albumin by lysosomal cathepsin D, followed by the export of cystine from the lysosome to the cytosol through cystinosin, can boost GSH synthesis and prevent ferroptosis in situations where extracellular free cystine is scarce [Citation50].
2.3. Ferroptosis in AKI
The development of AKI is extremely intricate, and the proximal tubule is vulnerable to different types of stimuli because of its anatomical characteristics and functions. While several molecular mechanisms have been proposed to trigger or worsen AKI, renal damage caused by ROS is believed to be one of the main contributors [Citation51]. In a recent investigation employing mice with inducible GPX4 deficiency, the mice exhibited a fatal outcome characterized by the extensive demise of renal tubular cells and the onset of acute renal failure within a fortnight following the deletion of GPX4. The longevity of GPX4-deficient mice potentially increased by approximately 35% when lipid peroxides were eliminated from the organism, further signifying the crucial involvement of ferroptosis in AKI [Citation52].
Recent studies on different types of AKI models have presented further direct evidence that ferroptosis is responsible for tubular injury and decreased renal function induced by different factors.
When organs rapidly recover from a sudden interruption of blood and oxygen supply, they become susceptible to injury related to oxidative stress and the inflammatory response, known as IRI. IRI is recognized as one of the primary causes of AKI [Citation53]. In IRI-induced AKI mice, the administration of ferrostatin-1 (Fer-1, a ferroptosis inhibitor that acts as a synthetic antioxidant) significantly mitigated tubular injury and renal function loss. The application of Fer-1 also shielded isolated renal tubule cells from hypoxic injury [Citation21,Citation54]. Our department reported that XJB-5-131, a novel generation of antioxidants, could mitigate renal tubule injury via inhibiting lipid peroxidation and subsequent ferroptosis [Citation54]. Moreover, Balzer MS, et al. indicated that ferroptosis was more pronounced in the long-term (30-min) IRI-induced AKI. Compared with the short-term (23 min) IRI-induced AKI, more severe renal function loss and subsequent fibrosis were demonstrated in the long-term (30 min) IRI-induced AKI [Citation55]. The administration of the ferroptosis inhibitor, liproxstatin (a radical-trapping antioxidant), was more effective in preventing long-term structural damage and fibrosis development [Citation55]. Nephrotoxic drugs, such as cisplatin and folic acid, are also known to be crucial factors that can trigger AKI. In folic acid-induced AKI, Fer-1 could also suppress ferroptotic cell death through improving the activity of GPX4 and decreasing iron deposition and lipid peroxidation accumulation [Citation56]. Similarly, ferroptotic cell death was observed in cisplatin-induced AKI, and Fer-1 enhanced renal function and reduced tissue damage by decreasing the deposition of lipid hydroperoxide [Citation57].
In 2019, Deng F, et al. reported an obvious change in the ferritinophagy biomarker NCOA4 in the tubules of patients with acute tubular necrosis. The researchers indicated that myo-inositol oxygenase (MIOX, an enzyme that catabolizes myo-inositol to d-glucuronate located in the proximal tubule) overexpression could promote NCOA4-mediated ferritinophagy and ultimate ferroptotic cell death in cisplatin-induced AKI [Citation30]. Subsequent studies have further supported the idea that ferritinophagy may be responsible for different AKI models. Excessive autophagic degradation of ferritin generates an increased accumulation of iron and lipid hydroperoxide, ultimately resulting in ferroptosis [Citation58–61]. Recent findings challenge earlier observations and indicate that certain types of selective autophagy, particularly ferritinophagy, contribute to ferroptosis in AKI. These findings indicate specific types of selective autophagy may not be cytoprotective, and highlight the potential of selective autophagy as a novel target for treating AKI. In the upcoming sections, we will explore different types of selective autophagy and their connection with ferroptosis, focusing on recent advancements in understanding the underlying molecular mechanisms.
3. The crosslinks between ferroptosis and autophagy in AKI
3.1. Overview of autophagy
Autophagy is a process that involves the transportation of cytoplasmic cargo to the lysosome for degradation and recycling [Citation9]. The process of autophagy is controlled by various autophagy-related genes (ATGs). The cargo in the cytosol is initially trapped inside the autophagosome, which requires the coordinated action of several ATG protein complexes, such as the Atg5-Atg12-Atg16L1 complex. Subsequently, the autophagosome is targeted and transported to the lysosome, leading to the fusion of the autophagosome and lysosome. Ultimately, the autophagosomal cargo is degraded by hydrolases in the autolysosome, and the resulting degradation products can be recycled to synthesize biomacromolecules [Citation62].
Nonselective autophagy randomly degrades various parts of the cytoplasm and organelles. In contrast, selective autophagy targets specific entities for degradation, including aggregated proteins, damaged organelles, and even invading pathogens [Citation11,Citation63]. Various forms of selective autophagy have been observed, including mitophagy (elimination of damaged mitochondria), lipophagy (elimination of lipids), ER-phagy (elimination of endoplasmic reticulum), xenophagy (elimination of pathogens and microorganisms), and ferritinophagy (elimination of ferritin) [Citation11]. Autophagy, as indicated by multiple recent studies, holds significant importance in both physiological processes and the advancement of diseases [Citation9]. Under normal physiological conditions, there exists a constitutive or basal level of autophagy that maintains cellular homeostasis by regulating the degradation of impaired proteins and organelles. In pathological conditions, a variety of cellular stressors, such as nutrient deprivation, hypoxia, oxidant injury, and other harmful insults, collectively trigger the activation of autophagy [Citation9,Citation10,Citation15,Citation64].
The pathogenesis of AKI also involves multiple stresses, including hypoxia, nutrient deprivation, energy depletion, oxidant injury, and ER stress, thereby triggering autophagy [Citation15,Citation64,Citation65]. Previous studies have shown that autophagy protects tubular cells from the deposition of detrimental molecules and damaged organelles to suppress the progression of tubular injury [Citation13,Citation14]. In 2011, Kimura, et al. reported that proximal tubule-specific Atg5-KO mice experienced more severe renal injury than WT mice when subjected to IRI [Citation13]. Further research on autophagy has revealed more specific mechanisms, especially selective autophagy, which have been found to promote ferroptotic cell death [Citation11,Citation63,Citation66]. Considering that the autophagic machinery can have lethal effects under certain conditions, it is imperative to investigate the specific functions of different selective autophagy in AKI.
3.2. Role of selective autophagy in ferroptosis in AKI
3.2.1. NCOA4- mediated ferritinophagy
Ferritinophagy is an intracellular process through which ferritin is degraded, releasing iron into the cytoplasm. This mechanism relies on the presence of NCOA4 as a selective cargo receptor, which interacts with FTH1 to enable the transportation of ferritin to the autophagosome [Citation40,Citation67]. NCOA4 is a 70-kDa protein that consists of two main isoforms, NCOA4α and NCOA4β, which mediate protein oligomerization. While previous studies have recognized NCOA4 as a coactivator of the androgen receptor and a regulator of DNA replication [Citation40,Citation68–70], it is now more commonly acknowledged as the core regulator in determining ferritinophagic flux. Mechanistically, the C-terminal structural domain of NCOA4 directly interacts with FTH1, leading to the binding of NCOA4 and ferritin. This interaction is necessary for the transport of ferritin from the cytoplasm to the autophagosome, enabling the degradation of ferritin in the lysosome and the release of iron [Citation71]. The autophagic degradation of ferritin is precisely controlled by the amount of intracellular iron [Citation40]. When cellular iron is abundant, the degradation of NCOA4 through the ubiquitin proteasome system decreases the levels of NCOA4 to suppress ferritinophagy. The ubiquitin ligase HECT and RCC1-like domain 2 (HERC2) is responsible for the ubiquitin-dependent turnover of NCOA4. Conversely, in situations of low cellular iron levels, the binding of NCOA4 and HERC2 is restricted to stabilize NCOA4 and enhance ferritinophagic flux [Citation31,Citation40,Citation71]. The differences between autophagy and ferritinophagy were summarized in a table ().
Table 1. Differences between autophagy and ferritinophagy.
Although ferritinophagy participates in the regulation of iron turnover to maintain intracellular iron balance, excessive ferritinophagic flux can lead to the increased deposition of free iron, which is associated with lipid peroxide deposition in ferroptosis [Citation72]. Several studies have demonstrated the significant impact of NCOA4-ferritinophagy on ferroptosis. Depletion of NCOA4 reduces intracellular free iron and lipid peroxide accumulation, resulting in the resistance of cells to ferroptosis [Citation40]. Yoshida et al. found that NCOA4 depletion in chronic obstructive pulmonary disease (COPD) alleviated iron overload and lipid peroxide deposition induced by GPX4 knockdown. Both genetic deletion and pharmacological inhibition of NCOA4 were reported to reduce ferroptotic cell death [Citation73]. Fang Y, et al. reported that a new ferroptosis inhibitor, synthesized compound 9a, disrupted the interaction between NCOA4 and FTH1 to suppress autophagic degradation of ferritin by directly binding to NCOA4. Compound 9a effectively mitigated the ischemic brain injury in vivo by protecting neuronal cells against ferroptosis [Citation28]. Conversely, exogenous overexpression of NCOA4 increases ferritinophagic flux and iron release from ferritin degradation, ultimately increasing sensitivity to ferroptosis [Citation72].
Some studies have reported that ferritinophagy participates in regulating intracellular iron levels and ferroptosis in AKI models [Citation30,Citation58–61]. For example, Deng F, et al. described the function of NCOA4-mediated ferritinophagy in cisplatin-induced AKI, where inhibition of ferritinophagy flux, represented by increased expression of NCOA4 due to MIOX knockdown, resulted in reduced accumulation of free iron and less ferroptotic cell death [Citation30]. Recently, Zhou L and Deng F, et al. demonstrated that protein arginine methyltransferase 4 (PRMT4) interacted with NCOA4 to suppress ferritinophagy and subsequent ferroptosis during cisplatin-induced AKI [Citation74]. Another study demonstrated that cold-inducible RNA-binding protein (CRIBP, a regulator of inflammatory responses) upregulated ferroptosis to aggravate tubular injury and renal function loss in IRI-induced AKI, and the knockdown of CRIBP in vitro suppressed the development of ferroptosis. This was achieved through CRIBP activating ferritinophagy via interaction with the RNA-binding protein ELAVL1, facilitating ferritin degradation and iron deposition [Citation61]. ELAVL1 was reported as an activator of autophagy that can bind to the AU-rich elements within the F3 of the 3′-untranslated region of Beclin1 mRNA. ELAVL1 could also promote the activation of ferritinophagy to regulate ferroptosis in hepatic stellate cells [Citation26].
Similarly, a recent study showed that an overexpression of stimulator of interferon genes (STING) accelerated the progression of ferroptosis in renal IRI [Citation58]. Some studies reported that mitochondrial DNA leaked into the cytoplasm could activate the cGAS (cyclic GMP-AMP synthase)-STING pathway to trigger inflammation in cisplatin-induced and IRI-induced renal injury [Citation75,Citation76]. Jin L, et al. further demonstrated that STING could also directly interact with NCOA4, resulting in increased ferritin degradation and ultimate ferroptosis in tubular cells. Their study revealed that RSL3 (an inhibitor of GPX4) administration enhanced the interaction between STING and NCOA4 in vitro, and knockdown of NCOA4 significantly reduced lipid peroxidation levels in tubular cells subjected to RSL3 [Citation58]. Moreover, ferritinophagy was implicated in sepsis-induced AKI and patulin-induced AKI [Citation59,Citation60]. Tang Y, et al. reported that ferritinophagy participated in lipopolysaccharide (LPS)-induced renal damage. Isoliquiritigenin, a flavonoid from the Chinese traditional herb licorice, could decrease the expression of NOCA4 to reduce ferritinophagic flux and iron deposition, thereby promoting the recovery of GPX4 downregulation [Citation60]. Patulin, a toxic mycotoxin usually found in contaminated fruits like apples, could cause AKI by triggering ferroptosis. Hou Y, et al. showed that short-term high-dose intake of patulin could induce a marked change in the expression of NCOA4 and FTH1 in mice, indicating an increase in ferritinophagic flux [Citation59]. The role of ferritinophagy in different AKI models was summarized in a table ().
Table 2. The role of NCOA4-mediated ferritinophagy in various pathological conditions of AKI.
Although the relationship between NCOA4-mediated ferritinophagy and ferroptotic cell death has been extensively studied in various AKI models, additional research is needed to investigate the specific influence of directly regulating NCOA4 on ferroptotic cell death and subsequent renal injury.
3.2.2. Mitophagy
Mitophagy is defined as the selective degradation of damaged mitochondria via autophagic machinery, contributing to basal mitochondrial turnover and maintaining mitochondrial homeostasis [Citation77]. By clearing depolarized mitochondria, mitophagy promotes cell adaptation and protects them from ROS, which are primarily generated by damaged mitochondria [Citation78,Citation79].
PTEN-induced kinase 1 (PINK1) and parkin RBR E3 ubiquitin protein ligase (PRKN) are involved in the elimination of impaired mitochondria via triggering mitophagy. PINK1 is a mitochondrial serine/threonine kinase that contains a mitochondrial targeting sequence (MTS), while PRKN is a cytosolic ubiquitin enzyme 3 ligase [Citation80,Citation81]. Normally, PINK1 is translocated into the inner membrane by translocases. The MTS sequence of PINK1 is then removed, and PINK1 is ultimately degraded by peptidase and presenilin associated rhomboid like protein (PARL). However, when the damaged mitochondria lose membrane potential, the proteasomal degradation of PINK1 will be inhibited, leading to PINK1 stabilization and recruitment at the outer mitochondrial membrane (OMM) [Citation81,Citation82]. Since the switch of PRKN to active form requires PINK1, deposited PINK1 enables PRKN to be recruited to the OMM. PRKN, phosphorylated and activated by PINK1, promotes the polyubiquitination of OMM proteins, leading to the connection between damaged mitochondria and the autophagosome [Citation82]. It is well known that the PINK1-PRKN pathway mediates the activation of mitophagy, and there are different forms of mitophagy that contribute to the recycling of impaired mitochondria, including receptor-mediated mitophagy, ubiquitin-mediated mitophagy, and cardiolipin-mediated mitophagy [Citation81,Citation83].
Although there were a few studies for kidney disease that established the connection between mitophagy and ferroptosis in the past, sufficient evidence has demonstrated that mitophagy is responsible for promoting cellular adaptation and protecting the cell from various injury factors, such as ROS generated from damaged mitochondria [Citation82,Citation83]. Several studies have demonstrated the cytoprotective role of mitophagy in renal IRI [Citation84–86]. Tang C, et al. reported that the deficiency of Pink1 and Park2 led to more severe mitochondrial damage, ROS accumulation and inflammatory responses, emphasizing the significance of mitophagy in maintaining mitochondrial quality, alleviating tubule injury, and improving renal function during renal IRI [Citation84]. In cisplatin-induced AKI, the knockout of Pink1 and Park2 increased the production of ROS and aggravated tubule injury and renal function loss, demonstrating the cytoprotective function of mitophagy against cisplatin-induced renal injury [Citation87]. Similar results have been reported in sepsis-induced AKI and contrast-induced AKI, where mitophagy has shown a protective role against these stimuli [Citation88–90]. A recent study showed that the administration of melatonin, a circadian-regulating hormone, could mitigate the renal injury caused by sepsis through sirtuin 3 (SIRT3)-mediated mitophagy [Citation90]. Our department also showed the anti-inflammatory effect of melatonin in folic acid-induced AKI, suggesting the therapeutic potential of melatonin [Citation91]. Deng Z, et al. further demonstrated that melatonin supplementation could improve the activity of SIRT3, a histone deacetylase that regulates various biological mechanisms like autophagy, leading to increased mitophagic flux and the recovery of renal function [Citation90]. Vitamin D receptor (VDR) was identified as a crucial role in acute kidney damage caused by cisplatin. Paricalcitol, a VDR agonist commonly used as a pharmaceutical treatment for secondary hyperparathyroidism in CKD patients, could reduce the deposition of lipid peroxidation and malondialdehyde, ameliorate mitochondrial damage, and reverse the downregulation of GPX4, thereby suppressing ferroptosis [Citation92]. Further research reported that paricalcitol could also alleviate contrast-induced AKI by regulating mitophagy and decreasing mitochondrial damage [Citation93].
Generally, mitophagy plays a pivotal role in maintaining mitochondrial homeostasis by suppressing the release of ROS from dysfunctional mitochondria [Citation94]. However, mitophagy could have complex effects on the development of ferroptosis, with some studies suggesting that it can both enhance and suppress ferroptosis [Citation95]. While excessive mitophagy can increase iron levels and promote lipid peroxidation and ferroptosis [Citation96,Citation97], mitophagy can also sequester iron in mitophagosomes during mild stress, reducing the availability of source materials for ROS [Citation98] Additionally, voltage-dependent anion channels (VDACs), located on MOM, are critical proteins involved in the exchange of iron between mitochondria and the cytosol. VDACs have been described as key players in the crosstalk between mitophagy and ferroptosis [Citation99,Citation100]. Erastin, an inducer of ferroptosis, targets System Xc− to decrease the activity and amount of GPX4 via suppressing the import of cysteine. Interestingly, erastin can also induce the opening of VDAC2/3, mitigating mitochondrial hyperpolarization and ROS formation to induce ferroptosis [Citation99,Citation101]. Inhibition of VDAC could further restrict the production and release of mitochondrial ATP, leading to inhibition of the mitochondrial tricarboxylic acid cycle and ultimately triggering an increase in ROS levels, which promotes ferroptotic cell death [Citation100]. Recently, Lin Q, et al. demonstrated that both Pink1-KO mice and Park2-KO mice exhibited more severe renal injury when exposed to cisplatin compared to WT mice. These knockout mice showed increased intracellular iron accumulation, aggravated lipid peroxidation, and lower expression of GPX4, indicating the protective role of mitophagy in renal injury via suppressing ferroptosis [Citation102]. However, there are a few studies that showed a detrimental effect of mitophagy in AKI [Citation103], so further research is imperative.
3.2.3. Lipophagy
In 2009, lipophagy was described as a form of selective autophagy that selectively degrades intracellular lipid droplets, indicating its crucial role in lipid metabolism. Lipophagy involves the autophagic digestion of lipid droplets, inducing the release of triglycerides, steroids, phospholipids, and free fatty acids into the cytoplasm [Citation104]. Lipid droplets contain small regulatory Rab GTPase molecular switch families, with Rab7a being identified as the key regulator for lipophagy in hepatocytes under specific conditions [Citation46,Citation105,Citation106]. Bai Y, et al. found that the knockdown of Rab7a inhibited RSL3-induced lipid droplets downregulation and MDA accumulation in HepG2 cells. This study demonstrated that Rab7a induces the recruitment of lipid droplets and facilitates their autophagic degradation, thereby promoting lipid peroxidation-mediated ferroptosis [Citation46]. Autophagic degradation of lipid droplets was also identified in kidney proximal tubular cells [Citation107]. Minami S, et al. demonstrated that lipophagy maintains energy homeostasis in the proximal tubule during prolonged starvation [Citation107]. Since lipid droplets are formed in renal tubules during AKI, lipophagy may also play a crucial role in AKI [Citation108]. Currently, there is limited research on lipophagy in AKI, we should value the functions of lipophagy and its relationship with ferroptosis in AKI.
3.3. Role of CMA in ferroptosis in AKI
As a cellular lysosome-mediated degradative mechanism, CMA transports selected protein substrates with a pentapeptide CMA-targeting motif into the lysosome, which is mediated by the binding of selected proteins with the chaperone heat shock-cognate protein of 70-KDa (HSC70) and their interaction with lysosome-associated membrane protein type 2a (LAMP-2A) [Citation49,Citation109]. CMA is essential in maintaining cellular protein quality control by regulating the degradation of damaged proteins [Citation49,Citation110].
Several studies have indicated that the activation of CMA contributes to the degradation of GPX4, and inhibiting CMA can stabilize GPX4, thereby reducing ferroptotic cell death [Citation49]. Wu Z, et al. examined the effect of HSC70 and LAMP-2A in ferroptosis and demonstrated that knockdown of LAMP-2A and HSC70 led to the restored GPX4 protein levels in HT-22 cells treated with erastin [Citation49]. Similarly, research on IRI-induced AKI showed that CMA participated in the degradation of GPX4, increasing the deposition of lipid hydroperoxide [Citation111]. Legumain, an asparaginyl endopeptidase, was found to potentially enhance chaperone-mediated autophagy of GPX4 by interacting with HSC70, heat shock protein 90 (HSP90), and GPX4 in tubular cells. The knockout of legumain and the administration of the pharmaceutical inhibitor RR-11a alleviated ferroptotic cell death, inflammation, tubular injury, and renal function loss, suggesting a novel therapeutic target for AKI [Citation111]. Recent reports have also identified other pharmacological inhibitors and activators of selective autophagy that can mitigate ferroptotic cell death in AKI ().
Table 3. Pharmacologic approaches targeting selective autophagy for AKI.
4. Conclusions and perspectives
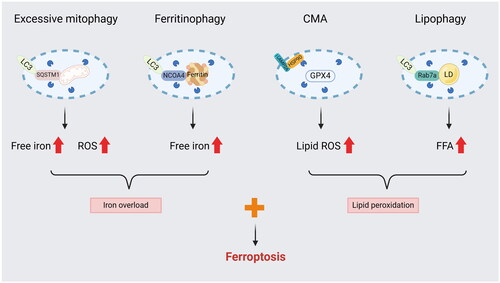
Recently, there has been a significant expansion in the studies of ferroptosis and autophagy in AKI, leading to a greater understanding of the mechanisms involved. Initially, studies separated ferroptosis from autophagy and ignored the connection between them. Now, more and more studies have demonstrated that autophagy and components of the autophagy machinery contribute greatly to ferroptotic cell death (). Although ferritinophagy and CMA have been linked to ferroptosis in certain AKI models, the functional role of lipophagy, clockophagy, and other selective autophagy in ferroptosis remains poorly understood.
Figure 2. Selective autophagy may serve as an enhancer of ferroptosis. (1) Excessive mitophagy could provide a source of iron and ROS from damaged mitochondrial degradation. (2) NCOA4-mediated ferritinophagy promotes iron accumulation by activating ferritin degradation. (3) CMA impairs the antioxidant system by promoting GPX4 degradation, leading to the deposition of lipid hydroperoxide. (4) Lipophagy increases the amount of FFA by inducing the breakdown of LD.
LC3: microtubule associated protein 1 light chain 3; SQSTM1: sequestosome 1; NCOA4: Nuclear Receptor Coactivator 4; HSP90: heat shock protein 90; LAMP2A: lysosome-associated membrane protein type 2a; LD: lipid droplet; Rab7a: a member of the RAS oncogene family; FFA: free fatty acid.
Prior research has primarily focused on the development of pharmacological inducers of autophagy that can selectively induce autophagy without impacting other metabolic pathways. Recent studies have suggested that different forms of selective autophagy may have distinct roles in AKI. Therefore, precise control over specific types of selective autophagy could be crucial. Given that various types of selective autophagy have been mechanistically linked to ferroptosis, targeting selective autophagy to reduce ferroptosis could offer a promising approach for treating AKI. Further exploration of the relationship between selective autophagy and ferroptosis is likely to uncover additional therapeutic targets for AKI.
In summary, according to current studies, the function of selective autophagy should be valued for its relation to ferroptosis in AKI. However, apart from ferritinophagy, mitophagy, and CMA, little is known about other types of selective autophagy in AKI, so further research is required to identify new targets for more effective and precise treatment.
irnf_a_2379601_sm5331.docx
Download MS Word (320.7 KB)irnf_a_2379601_sm5330.jpeg
Download JPEG Image (3.7 MB){kind=link}
irnf_a_2379601_sm5329.jpeg
Download JPEG Image (725.8 KB){kind=link}
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Mehta RL, Pascual MT, Soroko S, et al. Spectrum of acute renal failure in the intensive care unit: the PICARD experience. Kidney Int. 2004;66(4):1613–1621. doi: 10.1111/j.1523-1755.2004.00927.x.
- Hsu RK, McCulloch CE, Dudley RA, et al. Temporal changes in incidence of dialysis-requiring AKI. J Am Soc Nephrol. 2013;24(1):37–42. doi: 10.1681/ASN.2012080800.
- Liu KD, Goldstein SL, Vijayan A, et al. AKI! Now Initiative: recommendations for Awareness, Recognition, and Management of AKI. Clin J Am Soc Nephrol. 2020;15(12):1838–1847. doi: 10.2215/CJN.15611219.
- Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. 2012;81(5):442–448. doi: 10.1038/ki.2011.379.
- Venkatachalam MA, Weinberg JM, Kriz W, et al. Failed Tubule Recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol. 2015;26(8):1765–1776. doi: 10.1681/ASN.2015010006.
- Feng Q, Yu X, Qiao Y, et al. Ferroptosis and Acute Kidney Injury (AKI): molecular mechanisms and therapeutic potentials. Front Pharmacol. 2022;13:858676. doi: 10.3389/fphar.2022.858676.
- Thiele RH, Isbell JM, Rosner MH. AKI associated with cardiac surgery. Clin J Am Soc Nephrol. 2015;10(3):500–514. doi: 10.2215/CJN.07830814.
- Ow CPC, Trask-Marino A, Betrie AH, et al. Targeting oxidative stress in septic acute kidney injury: from theory to practice. J Clin Med. 2021;10(17):3798. doi: 10.3390/jcm10173798.
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;147(4):728–741. doi: 10.1016/j.cell.2011.10.026.
- Feng Y, He D, Yao Z, et al. The machinery of macroautophagy. Cell Res. 2014;24(1):24–41. doi: 10.1038/cr.2013.168.
- Vargas JNS, Hamasaki M, Kawabata T, et al. The mechanisms and roles of selective autophagy in mammals. Nat Rev Mol Cell Biol. 2023;24(3):167–185. doi: 10.1038/s41580-022-00542-2.
- Garza-Lombó C, Pappa A, Panayiotidis MI, et al. Redox homeostasis, oxidative stress and mitophagy. Mitochondrion. 2020;51:105–117. doi: 10.1016/j.mito.2020.01.002.
- Kimura T, Takabatake Y, Takahashi A, et al. Autophagy protects the proximal tubule from degeneration and acute ischemic injury. J Am Soc Nephrol. 2011;22(5):902–913. doi: 10.1681/ASN.2010070705.
- Jiang M, Liu K, Luo J, et al. Autophagy is a renoprotective mechanism during in vitro hypoxia and in vivo ischemia-reperfusion injury. Am J Pathol. 2010;176(3):1181–1192. doi: 10.2353/ajpath.2010.090594.
- Kaushal GP, Shah SV. Autophagy in acute kidney injury. Kidney Int. 2016;89(4):779–791. doi: 10.1016/j.kint.2015.11.021.
- Liu J, Kuang F, Kroemer G, et al. Autophagy-dependent ferroptosis: machinery and regulation. Cell Chem Biol. 2020;27(4):420–435. doi: 10.1016/j.chembiol.2020.02.005.
- Guo R, Duan J, Pan S, et al. The road from AKI to CKD: molecular mechanisms and therapeutic targets of ferroptosis. Cell Death Dis. 2023;14(7):426. doi: 10.1038/s41419-023-05969-9.
- Ni L, Yuan C, Wu X. Targeting ferroptosis in acute kidney injury. Cell Death Dis. 2022;13(2):182. doi: 10.1038/s41419-022-04628-9.
- Sun Y, Chen P, Zhai B, et al. The emerging role of ferroptosis in inflammation. Biomed Pharmacother. 2020;127:110108. doi: 10.1016/j.biopha.2020.110108.
- Tang D, Chen X, Kang R, et al. Ferroptosis: molecular mechanisms and health implications. Cell Res. 2021;31(2):107–125. doi: 10.1038/s41422-020-00441-1.
- Linkermann A, Skouta R, Himmerkus N, et al. Synchronized renal tubular cell death involves ferroptosis. Proc Natl Acad Sci USA. 2014;111(47):16836–16841. doi: 10.1073/pnas.1415518111.
- Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042.
- Tang D, Kang R, Berghe TV, et al. The molecular machinery of regulated cell death. Cell Res. 2019;29(5):347–364. doi: 10.1038/s41422-019-0164-5.
- Zhou B, Liu J, Kang R, et al. Ferroptosis is a type of autophagy-dependent cell death. Semin Cancer Biol. 2020;66:89–100. doi: 10.1016/j.semcancer.2019.03.002.
- Quiles Del Rey M, Mancias JD. NCOA4-mediated ferritinophagy: a potential link to neurodegeneration. Front Neurosci. 2019;13:238. doi: 10.3389/fnins.2019.00238.
- Zhang Z, Yao Z, Wang L, et al. Activation of ferritinophagy is required for the RNA-binding protein ELAVL1/HuR to regulate ferroptosis in hepatic stellate cells. Autophagy. 2018;14(12):2083–2103. doi: 10.1080/15548627.2018.1503146.
- Zheng Y, Zhao T, Wang J, et al. Curcumol alleviates liver fibrosis through inducing autophagy and ferroptosis in hepatic stellate cells. Faseb J. 2022;36(12):e22665.
- Fang Y, Chen X, Tan Q, et al. Inhibiting Ferroptosis through Disrupting the NCOA4-FTH1 Interaction: a new mechanism of action. ACS Cent Sci. 2021;7(6):980–989. doi: 10.1021/acscentsci.0c01592.
- Li C, Sun G, Chen B, et al. Nuclear receptor coactivator 4-mediated ferritinophagy contributes to cerebral ischemia-induced ferroptosis in ischemic stroke. Pharmacol Res. 2021;174:105933. doi: 10.1016/j.phrs.2021.105933.
- Deng F, Sharma I, Dai Y, et al. Myo-inositol oxygenase expression profile modulates pathogenic ferroptosis in the renal proximal tubule. J Clin Invest. 2019;129(11):5033–5049. doi: 10.1172/JCI129903.
- Yang Y, Cheng J, Lin Q, et al. Autophagy-dependent ferroptosis in kidney disease. Front Med. 2022;9:1071864. doi: 10.3389/fmed.2022.1071864.
- van Swelm RPL, Wetzels JFM, Swinkels DW. The multifaceted role of iron in renal health and disease. Nat Rev Nephrol. 2020;16(2):77–98. doi: 10.1038/s41581-019-0197-5.
- Soofi A, Li V, Beamish JA, et al. Renal-specific loss of ferroportin disrupts iron homeostasis and attenuates recovery from acute kidney injury. Am J Physiol Renal Physiol. 2024;326(2):F178–F188. doi: 10.1152/ajprenal.00184.2023.
- Stoyanovsky DA, Tyurina YY, Shrivastava I, et al. Iron catalysis of lipid peroxidation in ferroptosis: regulated enzymatic or random free radical reaction? Free Radic Biol Med. 2019;133:153–161. doi: 10.1016/j.freeradbiomed.2018.09.008.
- Lai CS, Piette LH. Spin-trapping studies of hydroxyl radical production involved in lipid peroxidation. Arch Biochem Biophys. 1978;190(1):27–38. doi: 10.1016/0003-9861(78)90250-3.
- Bayır H, Dixon SJ, Tyurina YY, et al. Ferroptotic mechanisms and therapeutic targeting of iron metabolism and lipid peroxidation in the kidney. Nat Rev Nephrol. 2023;19(5):315–336. doi: 10.1038/s41581-023-00689-x.
- Maremonti F, Meyer C, Linkermann A. Mechanisms and models of kidney tubular necrosis and nephron loss. J Am Soc Nephrol. 2022;33(3):472–486. doi: 10.1681/ASN.2021101293.
- McCullough K, Bolisetty S. Ferritins in Kidney Disease. Semin Nephrol. 2020;40(2):160–172. doi: 10.1016/j.semnephrol.2020.01.007.
- Shi H, Bencze KZ, Stemmler TL, et al. A cytosolic iron chaperone that delivers iron to ferritin. Science. 2008;320(5880):1207–1210. doi: 10.1126/science.1157643.
- Mancias JD, Wang X, Gygi SP, et al. Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature. 2014;509(7498):105–109. doi: 10.1038/nature13148.
- Ajoolabady A, Aslkhodapasandhokmabad H, Libby P, et al. Ferritinophagy and ferroptosis in the management of metabolic diseases. Trends Endocrinol Metab. 2021;32(7):444–462. doi: 10.1016/j.tem.2021.04.010.
- Chen X, Li J, Kang R, et al. Ferroptosis: machinery and regulation. Autophagy. 2021;17(9):2054–2081. doi: 10.1080/15548627.2020.1810918.
- Yang WS, Kim KJ, Gaschler MM, et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci USA. 2016;113(34):E4966–75.
- Kagan VE, Mao G, Qu F, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol. 2017;13(1):81–90. doi: 10.1038/nchembio.2238.
- Dixon SJ, Winter GE, Musavi LS, et al. Human haploid cell genetics reveals roles for lipid metabolism genes in nonapoptotic cell death. ACS Chem Biol. 2015;10(7):1604–1609. doi: 10.1021/acschembio.5b00245.
- Bai Y, Meng L, Han L, et al. Lipid storage and lipophagy regulates ferroptosis. Biochem Biophys Res Commun. 2019;508(4):997–1003. doi: 10.1016/j.bbrc.2018.12.039.
- Yang M, Chen P, Liu J, et al. Clockophagy is a novel selective autophagy process favoring ferroptosis. Sci Adv. 2019;5(7):eaaw2238. doi: 10.1126/sciadv.aaw2238.
- Partch CL, Green CB, Takahashi JS. Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 2014;24(2):90–99. doi: 10.1016/j.tcb.2013.07.002.
- Liu J, Yang M, Kang R, et al. Autophagic degradation of the circadian clock regulator promotes ferroptosis. Autophagy. 2019;15(11):2033–2035. doi: 10.1080/15548627.2019.1659623.
- Brigelius-Flohé R, Maiorino M. Glutathione peroxidases. Biochim Biophys Acta. 2013;1830(5):3289–3303. doi: 10.1016/j.bbagen.2012.11.020.
- Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018;19(6):365–381. doi: 10.1038/s41580-018-0001-6.
- Wu Z, Geng Y, Lu X, et al. Chaperone-mediated autophagy is involved in the execution of ferroptosis. Proc Natl Acad Sci USA. 2019;116(8):2996–3005. doi: 10.1073/pnas.1819728116.
- Dixon SJ, Olzmann JA. The cell biology of ferroptosis. Nat Rev Mol Cell Biol. 2024;25(6):424–442. Feb 16. doi: 10.1038/s41580-024-00703-5.
- Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int. 2006;70(3):432–443. doi: 10.1038/sj.ki.5001565.
- Friedmann Angeli JP, Schneider M, Proneth B, et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180–1191. doi: 10.1038/ncb3064.
- Sharfuddin AA, Molitoris BA. Pathophysiology of ischemic acute kidney injury. Nat Rev Nephrol. 2011;7(4):189–200. doi: 10.1038/nrneph.2011.16.
- Zhao Z, Wu J, Xu H, et al. XJB-5-131 inhibited ferroptosis in tubular epithelial cells after ischemia-reperfusion injury. Cell Death Dis. 2020;11(8):629. doi: 10.1038/s41419-020-02871-6.
- Balzer MS, Doke T, Yang YW, et al. Single-cell analysis highlights differences in druggable pathways underlying adaptive or fibrotic kidney regeneration. Nat Commun. 2022;13(1):4018. doi: 10.1038/s41467-022-31772-9.
- Martin-Sanchez D, Ruiz-Andres O, Poveda J, et al. Ferroptosis, but Not Necroptosis, Is Important in Nephrotoxic Folic Acid-Induced AKI. J Am Soc Nephrol. 2017;28(1):218–229. doi: 10.1681/ASN.2015121376.
- Zhou L, Yu P, Wang TT, et al. Polydatin Attenuates Cisplatin-Induced Acute Kidney Injury by Inhibiting Ferroptosis. Oxid Med Cell Longev. 2022;2022:9947191–9947114. doi: 10.1155/2022/9947191.
- Jin L, Yu B, Wang H, et al. STING promotes ferroptosis through NCOA4-dependent ferritinophagy in acute kidney injury. Free Radic Biol Med. 2023;208:348–360. doi: 10.1016/j.freeradbiomed.2023.08.025.
- Hou Y, Wang S, Jiang L, et al. Patulin induces acute kidney injury in mice through autophagy-ferroptosis pathway. J Agric Food Chem. 2022;70(20):6213–6223. doi: 10.1021/acs.jafc.1c08349.
- Tang Y, Luo H, Xiao Q, et al. Isoliquiritigenin attenuates septic acute kidney injury by regulating ferritinophagy-mediated ferroptosis. Ren Fail. 2021;43(1):1551–1560. doi: 10.1080/0886022X.2021.2003208.
- Sui M, Xu D, Zhao W, et al. CIRBP promotes ferroptosis by interacting with ELAVL1 and activating ferritinophagy during renal ischaemia-reperfusion injury. J Cell Mol Med. 2021;25(13):6203–6216. doi: 10.1111/jcmm.16567.
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21(22):2861–2873. doi: 10.1101/gad.1599207.
- Debnath J, Gammoh N, Ryan KM. Autophagy and autophagy-related pathways in cancer. Nat Rev Mol Cell Biol. 2023;24(8):560–575. doi: 10.1038/s41580-023-00585-z.
- Xiang Y, Fu Y, Wu W, et al. Autophagy in acute kidney injury and maladaptive kidney repair. Burns Trauma. 2023;11:tkac059.
- Cui J, Bai X, Chen X. Autophagy and acute kidney injury. Adv Exp Med Biol. 2020;1207:469–480. doi: 10.1007/978-981-15-4272-5_34.
- Song X, Zhu S, Chen P, et al. AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system X-activity. Curr Biol. 2018;28(15):2388–2399.e5. doi: 10.1016/j.cub.2018.05.094.
- Galy B, Conrad M, Muckenthaler M. Mechanisms controlling cellular and systemic iron homeostasis. Nat Rev Mol Cell Biol. 2023;25(2):133–155. doi: 10.1038/s41580-023-00648-1.
- Santana-Codina N, Mancias JD. The role of NCOA4-mediated ferritinophagy in health and disease. Pharmaceuticals. 2018;11(4):114. doi: 10.3390/ph11040114.
- Santana-Codina N, Gikandi A, Mancias JD. The role of NCOA4-mediated ferritinophagy in ferroptosis. Adv Exp Med Biol. 2021;1301:41–57. doi: 10.1007/978-3-030-62026-4_4.
- Monaco C, Visconti R, Barone MV, et al. The RFG oligomerization domain mediates kinase activation and re-localization of the RET/PTC3 oncoprotein to the plasma membrane. Oncogene. 2001;20(5):599–608. doi: 10.1038/sj.onc.1204127.
- Mancias JD, Pontano Vaites L, Nissim S, et al. Ferritinophagy via NCOA4 is required for erythropoiesis and is regulated by iron dependent HERC2-mediated proteolysis. Elife. 2015;4:e10308. doi: 10.7554/eLife.10308.
- Hou W, Xie Y, Song X, et al. Autophagy promotes ferroptosis by degradation of ferritin. Autophagy. 2016;12(8):1425–1428. doi: 10.1080/15548627.2016.1187366.
- Yoshida M, Minagawa S, Araya J, et al. Involvement of cigarette smoke-induced epithelial cell ferroptosis in COPD pathogenesis. Nat Commun. 2019;10(1):3145. doi: 10.1038/s41467-019-10991-7.
- Zhou L, Deng Z, Wang Y, et al. PRMT4 interacts with NCOA4 to inhibit ferritinophagy in cisplatin-induced acute kidney injury. Faseb J. 2024;38(7):e23584.
- Maekawa H, Inoue T, Ouchi H, et al. Mitochondrial damage causes inflammation via cGAS-STING signaling in acute kidney injury. Cell Rep. 2019;29(5):1261–1273.e6. doi: 10.1016/j.celrep.2019.09.050.
- Doke T, Mukherjee S, Mukhi D, et al. NAD + precursor supplementation prevents mtRNA/RIG-I-dependent inflammation during kidney injury. Nat Metab. 2023;5(3):414–430. doi: 10.1038/s42255-023-00761-7.
- Onishi M, Yamano K, Sato M, et al. Molecular mechanisms and physiological functions of mitophagy. Embo J. 2021;40(3):e104705.
- Okamoto K. Organellophagy: eliminating cellular building blocks via selective autophagy. J Cell Biol. 2014;205(4):435–445. doi: 10.1083/jcb.201402054.
- Bravo-San Pedro JM, Kroemer G, Galluzzi L. Autophagy and mitophagy in cardiovascular disease. Circ Res. 2017;120(11):1812–1824. doi: 10.1161/CIRCRESAHA.117.311082.
- Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004;304(5674):1158–1160. doi: 10.1126/science.1096284.
- Wang Y, Cai J, Tang C, et al. Mitophagy in acute kidney injury and kidney repair. Cells. 2020;9(2):338. doi: 10.3390/cells9020338.
- Shiba-Fukushima K, Inoshita T, Hattori N, et al. PINK1-mediated phosphorylation of Parkin boosts Parkin activity in Drosophila. PLoS Genet. 2014;10(6):e1004391. doi: 10.1371/journal.pgen.1004391.
- Iorio R, Celenza G, Petricca S. Mitophagy: molecular mechanisms, new concepts on parkin activation and the emerging role of AMPK/ULK1 axis. Cells. 2021;11(1):30. doi: 10.3390/cells11010030.
- Tang C, Han H, Yan M, et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia-reperfusion injury. Autophagy. 2018;14(5):880–897. doi: 10.1080/15548627.2017.1405880.
- Tang C, Han H, Liu Z, et al. Activation of BNIP3-mediated mitophagy protects against renal ischemia-reperfusion injury. Cell Death Dis. 2019; Sep 1210(9):677. doi: 10.1038/s41419-019-1899-0.
- Livingston MJ, Wang J, Zhou J, et al. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy. 2019;15(12):2142–2162. doi: 10.1080/15548627.2019.1615822.
- Wang Y, Tang C, Cai J, et al. PINK1/Parkin-mediated mitophagy is activated in cisplatin nephrotoxicity to protect against kidney injury. Cell Death Dis. 2018;9(11):1113. doi: 10.1038/s41419-018-1152-2.
- Wang Y, Zhu J, Liu Z, et al. The PINK1/PARK2/optineurin pathway of mitophagy is activated for protection in septic acute kidney injury. Redox Biol. 2021;38:101767. doi: 10.1016/j.redox.2020.101767.
- Lin Q, Li S, Jiang N, et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019;26:101254. doi: 10.1016/j.redox.2019.101254.
- Deng Z, He M, Hu H, et al. Melatonin attenuates sepsis-induced acute kidney injury by promoting mitophagy through SIRT3-mediated TFAM deacetylation. Autophagy. 2024;20(1):151–165. doi: 10.1080/15548627.2023.2252265.
- Zhu F, Chong Lee Shin OL, Xu H, et al. Melatonin promoted renal regeneration in folic acid-induced acute kidney injury via inhibiting nucleocytoplasmic translocation of HMGB1 in tubular epithelial cells. Am J Transl Res. 2017;9(4):1694–1707.
- Hu Z, Zhang H, Yi B, et al. VDR activation attenuate cisplatin induced AKI by inhibiting ferroptosis. Cell Death Dis. 2020;11(1):73. doi: 10.1038/s41419-020-2256-z.
- Bae E, Kim JH, Jung MH, et al. Paricalcitol attenuates contrast-induced acute kidney injury by regulating mitophagy and senescence. Oxid Med Cell Longev. 2020;2020:7627934–7627913. doi: 10.1155/2020/7627934.
- Ashrafi G, Schwarz TL. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013;20(1):31–42. doi: 10.1038/cdd.2012.81.
- Li J, Jia YC, Ding YX, et al. The crosstalk between ferroptosis and mitochondrial dynamic regulatory networks. Int J Biol Sci. 2023;19(9):2756–2771. doi: 10.7150/ijbs.83348.
- Granata S, Votrico V, Spadaccino F, et al. Oxidative stress and ischemia/reperfusion injury in kidney transplantation: focus on ferroptosis, mitophagy and new antioxidants. Antioxidants. 2022;11(4):769. doi: 10.3390/antiox11040769.
- Rademaker G, Boumahd Y, Peiffer R, et al. Myoferlin targeting triggers mitophagy and primes ferroptosis in pancreatic cancer cells. Redox Biol. 2022;53:102324. doi: 10.1016/j.redox.2022.102324.
- Yu F, Zhang Q, Liu H, et al. Dynamic O-GlcNAcylation coordinates ferritinophagy and mitophagy to activate ferroptosis. Cell Discov. 2022;8(1):40. doi: 10.1038/s41421-022-00390-6.
- Yagoda N, von Rechenberg M, Zaganjor E, et al. RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature. 2007;447(7146):864–868. doi: 10.1038/nature05859.
- Su L, Zhang J, Gomez H, et al. Mitochondria ROS and mitophagy in acute kidney injury. Autophagy. 2023;19(2):401–414. doi: 10.1080/15548627.2022.2084862.
- Fang D, Maldonado EN. VDAC regulation: a mitochondrial target to stop cell proliferation. Adv Cancer Res. 2018;138:41–69. doi: 10.1016/bs.acr.2018.02.002.
- Lin Q, Li S, Jin H, et al. Mitophagy alleviates cisplatin-induced renal tubular epithelial cell ferroptosis through ROS/HO-1/GPX4 axis. Int J Biol Sci. 2023;19(4):1192–1210. doi: 10.7150/ijbs.80775.
- Liu D, Liu Y, Zheng X, et al. c-MYC-induced long noncoding RNA MEG3 aggravates kidney ischemia-reperfusion injury through activating mitophagy by upregulation of RTKN to trigger the Wnt/β-catenin pathway. Cell Death Dis. 2021;12(2):191. doi: 10.1038/s41419-021-03466-5.
- Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–1135. doi: 10.1038/nature07976.
- Kiss RS, Nilsson T. Rab proteins implicated in lipid storage and mobilization. J Biomed Res. 2014;28(3):169–177. doi: 10.7555/JBR.28.20140029.
- Schroeder B, Schulze RJ, Weller SG, et al. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology. 2015;61(6):1896–1907. doi: 10.1002/hep.27667.
- Minami S, Yamamoto T, Takabatake Y, et al. Lipophagy maintains energy homeostasis in the kidney proximal tubule during prolonged starvation. Autophagy. 2017;13(10):1629–1647. doi: 10.1080/15548627.2017.1341464.
- Li H, Dixon EE, Wu H, et al. Comprehensive single-cell transcriptional profiling defines shared and unique epithelial injury responses during kidney fibrosis. Cell Metab. 2022;34(12):1977–1998.e9. doi: 10.1016/j.cmet.2022.09.026.
- Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012;22(8):407–417. doi: 10.1016/j.tcb.2012.05.006.
- Cuervo AM, Wong E. Chaperone-mediated autophagy: roles in disease and aging. Cell Res. 2014;24(1):92–104. doi: 10.1038/cr.2013.153.
- Chen C, Wang D, Yu Y, et al. Legumain promotes tubular ferroptosis by facilitating chaperone-mediated autophagy of GPX4 in AKI. Cell Death Dis. 2021;12(1):65. doi: 10.1038/s41419-020-03362-4.
- Xu L, Cai J, Li C, et al. 4-Octyl itaconate attenuates LPS-induced acute kidney injury by activating Nrf2 and inhibiting STAT3 signaling. Mol Med. 2023;29(1):58. doi: 10.1186/s10020-023-00631-8.