Abstract
Considerable effort has been invested in developing methods for predicting the crystalline structure(s) of a given compound, ideally starting from no more than a structural formula of the molecule. Reliable computational predictions would be of great value in many areas of materials chemistry, from the design of materials with novel properties to the avoidance of an undesirable change of form in the late stages of development of an industrially important molecule. Methods used in crystal structure prediction are reviewed, with particular focus on the most common approach – global lattice energy minimization. Progress and current limitations are highlighted, with reference to examples from the literature and the results of blind tests organized to objectively monitor developments in the field.
1. Motivation for computational prediction of crystal structures
The ab initio prediction of crystal structures is a rapidly growing area of research and developments are being driven by both academic interest in the problem and the potential benefits to any industry that relies on delivering a product in crystalline form. The long-term aim of computational crystal structure prediction (CSP) is to propose the crystal structure(s) that will be observed for a given molecule, given no other information than the structural formula and conditions of crystallization.
The structure of a crystal is a necessary starting point for understanding and predicting its physico-chemical properties. Furthermore, the calculated properties of predicted crystal structures could be used as a screen for crystals with novel properties, such as a high non-linear optical response (which requires a non-centrosymmetric packing of the molecules). The calculations can be performed in advance of synthesizing the molecule, so they have the potential to streamline research by narrowing down experimental investigations to the most promising candidates. CSP is also of great interest to the pharmaceutical industry because of the prevalence of polymorphism (different crystal structures of a given compound) of pharmaceutical molecules. The occurrence of polymorphs is a potentially disastrous problem, should it occur without warning late in the processing cycle of a pharmaceutical molecule. The most notorious example of the consequences of unanticipated polymorphism is the temporary withdrawal from the market of the protease inhibitor Ritonavir (Citation 1 , Citation 2 ); the scientists dealing with the impact of the new polymorph concluded that it ‘is highly advisable to put enough resources to carry on exhaustive research to identify the most stable and all possible polymorphs’ (Citation 1 ). Computational methods of predicting crystal structures could play a large part in this research, both in identifying all possible polymorphic forms and judging their relative stabilities – an examination and evaluation of all possible crystal structures of a given compound is a step towards the understanding and, perhaps, control of polymorphism.
While industrial applications may be a major motivation for the development of CSP, surveys of the CSP literature up to 2004 ((Citation 3 , Citation 4 ); covering about 20 years since the first CSP studies were reported) showed that, up to that point, pharmaceuticals made up only 4% of systems studied, and pigments a further 3%. Hydrocarbons, on the other hand, represented 22% of molecules studied. The types of molecules that had been studied reflect a greater focus on method development, where relatively simple model systems are being examined to assess the assumptions and methodologies, than applications of CSP to industrially important molecules. However, over the past few years, the size and complexity of systems studied has increased significantly, with some impressive applications to structure prediction of larger, flexible pharmaceutical molecules (Citation 5 , Citation 6 ), as well as salts (Citation7–10), cocrystals (Citation11–13) and solvates (Citation14–17).
Over 20 years ago, Maddox (Citation 18 ) described our inability to predict a molecule's crystal structure as a ‘scandal’ and ‘one would have thought that, by now, it should be possible to equip a sufficiently large computer with a sufficiently large program, type in the formula of the chemical and obtain, as output, the atomic coordinates of the atoms in a unit cell’. Over the next few years, programs started appearing that attempted to do just this and, in 1994, Gavezzotti (Citation 19 ) addressed the fundamental question ‘Are crystal structures predictable?’ – while reviewing the difficulties and necessary advances in methodology, his answer wavered between ‘No’ and a qualified ‘Maybe’. Since then, the many approaches that are being developed have been assessed in four blind tests of CSP (Citation20–23), where the predictions were performed in advance of the real crystal structures being made available. There were some successes under these blind test conditions during the first three of these blind tests (held between 1999 and 2004; (Citation20–22)) but their results highlighted clearly a lack of reliability in the computational predictions. The latest blind test, held in 2007 (Citation 23 ) demonstrated significantly improved results, showing that great steps are being made towards an invaluable technology.
2. Computational methods
Much of our understanding of the principles of close packing in molecular crystals was developed by Kitaigorodskii (Citation 24 , Citation 25 ) in the middle of last century. Qualitatively, the projections of a molecule fit into the hollows of its neighbours to minimize free volume in the crystal and ‘the mutual orientation of molecules in a crystal is conditioned by the shortest distances between the atoms of adjacent molecules’ (Citation 24 ). The quantification of close packing started with the determination of volume increments for common functional groups (e.g. on average, a methyl group adds 23.5 Å3 to the volume per molecule of a crystal) and average intermolecular (or van der Waals) atomic radii in crystals. Using these parameters, the packing coefficient of the overwhelming majority of organic molecular crystals was found to fall in the range of 0.65–0.75 (Citation 26 ). The description of molecules in crystals by their atomic radii and analysis of their packing in terms of atomic contacts naturally leads to an atom–atom approach in the modelling of molecular crystals, where intermolecular interactions are described in terms of pairwise interactions between atoms. It is the suitability of the atomistic approach to computational methods that has led to a developing understanding of the energetics and, more recently, dynamics in molecular crystals.
Thanks to increasing computing power available at relatively low cost, some tasks have become possible that were thought to be unmanageable in the early days of crystal structure analysis. Particularly, while describing the many possibilities for close packing in molecular crystals, Kitaigorodskii (Citation 24 ) remarked that it ‘would not, of course, be feasible to carry out one-by-one examination of all possible packings of molecules for unit cells of different symmetries and different dimensions’. In a way, this task can now be performed on high performance, or even desktop, computers in studies aimed at predicting the crystal structure of a molecule. For most of the crystal structures in such studies, the ‘examination’ is normally limited to a computation of some scoring function (usually the lattice energy) while a selection of the most promising structures is often investigated in more detail.
The most common approach to the challenge of CSP has been to search for all possible crystal packings and calculate their lattice energies (Citation 3 ). The lowest energy structure (the global minimum) is assumed to be the most likely crystal structure and crystals with competitive energies are possible polymorphs. In this approach, kinetics of crystallization are ignored and the two major challenges are: (i) the search for all possible ways to pack the molecule in a crystal lattice (Citation 27 ) and (ii) accurately calculating the energies of these putative structures (Citation 28 ). Such calculations are becoming somewhat reliable for small molecules, at least in proposing the observed forms amongst a handful of possibilities (Citation29–31).
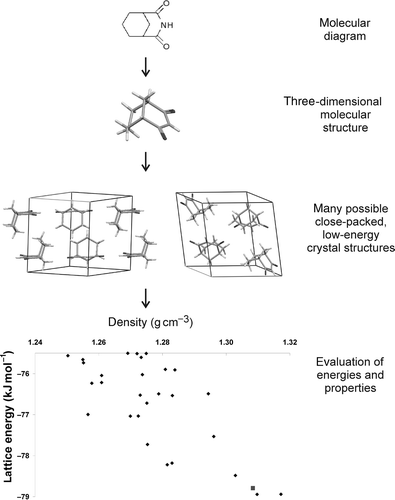
While there are many variations in methodology, the main steps in this approach are nearly always the same (): building a molecular model from the known chemical bonding in the molecule; generating all possible ways of packing the molecule in a reasonable crystal structure and evaluating energies and properties of these putative structures to decide which are most likely. Each of these steps presents its own challenges and requires careful attention; a poor choice or implementation of methodology at any stage will seriously reduce the chances of locating all likely polymorphs. Specifics of the approaches currently being applied to CSP are described in the following sections.
Figure 1. Diagrammatic summary of the lattice energy minimization approach to CSP. Structures and data reported in (Citation 31 ).
2.1. The molecular model
The starting point of energy or property calculations for a known crystal structure is the set of atomic coordinates, taken directly from that structure. Slight corrections might be made to account for errors in experimental determinations, such as normalization of bond lengths to hydrogen atoms in X-ray determined crystal structures (Citation 32 ). However, in a prediction study, we are interested in crystals with unknown structures – no atomic coordinates are known, so our first step must be to build a three-dimensional model of the molecule. This is our building block for generating potential crystal structures.
The most ambitious use of CSP is to perform the predictions in advance of even synthesizing the molecule. The computational results for a molecule might influence whether the time and expense is then invested in its synthesis. In such an application, the starting point for calculations could be as simple as the chemical diagram defining atom connectivity. Even where one crystal structure is known and CSP is part of a screen for other polymorphs, it is best practice to take a neutral starting point instead of the molecular geometry from the known crystal structure, so as not to bias calculations towards that known form. This molecular structure is normally the result of a geometry optimization of the isolated molecule. At least for small molecules, calculating the three-dimensional molecular structure is fairly routine – force fields can provide a starting point or quantum mechanics may be used for greater accuracy. The development of density functional theory (DFT) has reduced the cost of high quality electronic structure calculations and extended the size of molecule for which quantum mechanics is affordable.
There are two main considerations at this stage: the number of stable conformations and, for a given conformation, the effect of packing forces on the molecular geometry. The equilibrium geometry of the molecule in a crystal is a balance of the forces within the molecule with the forces between molecules. These two influences are often on different scales – the interactions between molecules are much weaker than the covalent bonds that hold molecules together. For this reason, the geometry of the isolated molecule is generally a good starting point for generating possible crystal structures. Large changes in bond lengths or angles generally require more energy than can be supplied by intermolecular forces; bond lengths in crystal and free-state conformations, determined experimentally or computationally, typically differ by hundredths of an Å, while differences in bond angles are usually smaller than a few degrees (Citation 24 , Citation33–36). The intramolecular parameters that are most significantly affected by the crystalline environment are torsion (dihedral) angles – usually rotations about single bonds, but also pyramidalization of atoms (e.g. the nitrogen atom in an amine group) or puckering of rings. Such changes in geometry are often possible at a small cost in conformational energy. The energy cost for such distortions, which is specific to the atoms involved and their exact bonding environment, can be calculated from a force field or quantum mechanics. The energy profile places limits on packing-induced distortions; conformational strain energy in crystals rarely exceeds 1 kcal mol−1 (∼4 kJ mol−1 (Citation 37 )). For rotation about a single bond, this often corresponds to a possible 20–30° rotation away from the equilibrium position. A search of the Cambridge Structural Database (CSD (Citation 38 )) for molecules that are similar to that of interest can also highlight the range of likely conformations.
Most of the development of CSP methodologies has focussed on small, fairly rigid molecules. Therefore, many software packages developed for the prediction of crystal structures constrain the molecule to being rigid, at least in the initial generation of trial structures (Citation 27 ). Even if the molecular structure is allowed to relax in a final lattice energy minimization, conformational energy barriers are rarely crossed at that stage and the molecule normally remains close to the initial chosen geometry. For this reason, if several stable conformations of the isolated molecule exist, then each distinct conformation must be treated separately – a geometry for each conformation is optimized using a force field or quantum mechanical method and predictions must then be performed for each conformer (Citation 5 , Citation 6 , Citation 29 ).
In many studies, the molecule is kept rigid throughout all steps of the CSP calculations, so we must recognize the influence of small changes in the assumed molecular structure on calculated structures and their energies. As an example, small differences in the assumed molecular geometry of paracetamol (acetaminophen) lead to large variations in the calculated energy difference between the two known polymorphs (Citation 39 ): 3.6 kJ mol−1 with an ab initio optimized molecular geometry, and ranging from 1.0 to 7.2 kJ mol−1 with molecular geometries taken from various structural determinations. CSP usually generates a multitude of possible crystal structures within a small energy range (sometimes 50+ in a 5 kJ mol−1 window above the global minimum (Citation 29 )), so changes in relative energies of a few kJ mol−1 can rearrange the relative stabilities of a large number of the predicted crystal structures. The lattice energy minimization approach relies on getting these relative energies correct, so the choice of molecular geometry is a crucial step in a successful prediction. In the case of paracetamol, the ab initio (HF/6-31G**) optimized molecular structure led to successful predictions of the two common polymorphs as the two lowest energy structures (Citation 40 ).
In many molecules, there are one or more ‘soft’ degrees of freedom and a continuum of conformations is accessible – perhaps 20° either side of the equilibrium for the rotation about a single bond. In such cases, the rigid molecule approach will not be able to describe all possible crystal structures. Such molecular flexibility affects both the generation of crystal structures and the refinement (energy minimization) of these trial structures – the full range of conformations must be accounted for in the search and the conformation must be allowed to adjust to its environment in each individual crystal structure. The simplest solution to this problem is the molecular mechanics approach – the intramolecular degrees of freedom are described by simple mathematical models, whose parameters are sometimes fitted to reproduce quantum mechanical energy calculations.
A classic example of a soft molecular distortion is the twist angle between the phenyl rings in biphenyl. In the isolated molecule, the angle between the rings is approximately 45° (Citation 41 ), the molecule is planar in the room temperature crystal and only slightly twisted in the low-temperature incommensurate phases (Citation 42 ). For this molecule, predicting structures with a fixed gas phase optimized molecular structure would certainly fail. However, a simple description of the intramolecular potential can adequately describe both the isolated molecule and crystal phase conformations (Citation43–45).
2.2. Crystal structure generation
Having decided on a starting molecular geometry, or a set of conformations, and possibly a force field to describe their flexibility, the next stage in the structure prediction is to explore the possible close packed arrays of this three-dimensional molecular model. In the early days of modelling molecular crystals, hypothetical alternatives to known crystal structures were sometimes derived from the packing of similar molecules (see Pertsin and Kitaigorodskii (Citation 26 ) and references therein). The development and implementation of algorithms for exploring all crystal packing possibilities of a molecule started in the mid-1980s with Dzyabchenko's (Citation 46 ) search for the low-energy crystal structures of benzene. Many more approaches have since been applied to the problem, starting in the early 1990s (e.g. (Citation47–49)). Here, an attempt is made to overview the methods that are currently in use.
2.2.1. Size of the search space
The generation of all plausible crystal structures means searching for all of the minima on the potential energy hypersurface, which is defined by the degrees of freedom that describe the geometry of the system. For crystal structures, these structural variables might be unit cell parameters or the positions of molecules in the unit cell. Such an energy surface could be sampled using a very simple approach of creating structures at points on a grid: M equally spaced values are chosen for each structural parameter. The M values for one variable must be combined with M values for each other variable so that a total of MN structures are generated, where N is the number of degrees of freedom. While the methods used in real studies are more advanced than such a grid-based approach, the complexity of the energy surface and the size of the search space clearly grow very rapidly with increasing dimensionality. The search for all possible crystal structures of a molecule is a challenge because of the large number of degrees of freedom. For a molecular crystal, there are six degrees of freedom in the unit cell dimensions (the three cell lengths a, b and c, and three angles α, β and γ) and, assuming rigid molecules, a further six degrees of freedom for each molecule in the unit cell – three positional and three orientational coordinates per molecule. When rotation about single bonds is possible, the packing can be strongly influenced by distortions of the molecular structure and some computational approaches relax the rigid molecule constraint. The conformational flexibility of such molecules can be treated by including a few variable torsion angles as searchable degrees of freedom, to be varied along with the unit cell parameters and molecular positions (see, e.g. (Citation 50 )).
Even with the dimensionality of the problem restricted by the rigid-body approximation, the search space is enormous; searching say 30 degrees of freedom (a unit cell containing four molecules) for all possible low-energy structures is an incredibly demanding task. Therefore, the problem is often simplified even further, thanks to the majority of organic molecules crystallizing in one of a small set of space groups. For example, 80% of homomolecular organic crystal structures have either one or a fraction of a molecule in the asymmetric unit (Z′ ≤ 1) in one of the six space groups: P21/c; P 1; P212121; P21; C2/c and Pbca (Citation 51 ). Therefore, most of the possible crystal structures for a molecule can be found by generating structures in a limited set of space groups with one molecule in the asymmetric unit; crystals with Z′ < 1 are automatically considered, as molecules with internal symmetry are free to find special positions in the crystal structures (e.g. the calculations can place a centrosymmetric molecule on a crystallographic centre of inversion). Generating structures within a given space group is a much more manageable task because restrictions are imposed on the relationship between molecules in the unit cell – only the position and orientation of the molecules in the asymmetric unit must be considered. The shape of the unit cell is also usually constrained by the space group. For example, the position and orientation of only one of the four molecules in a P21/c, Z′ = 1 unit cell must be varied independently and two of the angles defining the unit cell are fixed at 90°: 10 degrees of freedom must be searched instead of 30 for a general unit cell containing four rigid molecules.
The use of space group symmetry facilitates a search, but it means choosing a set of the most likely space groups in advance, so risks missing important structures in less common space groups – molecular crystal structures are known in all 230 space groups, although for many of these only a handful of structures have been reported. Furthermore, the contents of the asymmetric unit are fixed when using space group symmetry in a crystal structure search. While most molecules crystallize with Z′ ≤ 1, about 10% of homomolecular organic crystals have more than one molecule in the asymmetric unit (Z′ > 1 (Citation 52 )); 1 in 10 crystal structures would be missed in CSP unless searches are performed with several independent molecules in the asymmetric unit. Due to these risks, some approaches to generating crystal structures do not use symmetry to restrict the size of the search space. Instead, unconstrained unit cells are searched with a fixed total number of molecules in the unit cell, Z. Separate searches must then be performed for different Z. Such searches must use highly efficient algorithms (and powerful computers) for searching the many degrees of freedom. The space group symmetry of each predicted structure can then be identified at the end of the calculations; several programs are available to identify symmetry in crystals, for example PLATON (Citation 53 ) and the symmetry finder in Cerius2 (Citation 54 ).
2.2.2. Algorithms for structure generation
The difficulty of the search for all possible crystal structures will obviously increase with (a) less symmetry relating the molecules in the unit cell (Z′ > 1); and (b) increasing flexibility of the molecules. However, even with symmetry constraints, Z′ = 1, and the rigid molecule assumption, a complete search of the energy surface is a huge task and many approaches have been applied to the problem. A thorough review by Verwer and Leusen (Citation 27 ) in 1998 examined and contrasted the methods available at the time. The performance of many of the available methods has also been evaluated in the four blind tests of CSP (Citation20–23).
The most straightforward methods use a brute-force approach for searching all possible crystal structures – either systematically generating crystal structures on a grid of the multi-dimensional search space or generating a large number of crystals with random values for the parameters (cell lengths and angles, molecular positions and orientations). The CRYstal Structure CAlculation (CRYSCA) program, developed by Schmidt and co-workers (Citation 55 , Citation 56 ), is one example of such a program – trial structures are randomly generated and these are then energy minimized. The molecular packing analysis (MPA) program (Citation 57 ) performs a similar random search. Dzyabchenko's (Citation 50 , Citation 58 ) program (PMC) generates structures in a grid-based search and the program UPACK (Citation 14 , Citation 59 ) has capabilities for both grid searches and random generation of structures. UPACK was originally written to predict crystal structures for monosaccharides and has even been successfully applied to predicting structures of the hydrates of such flexible molecules (Citation 14 ) – very challenging searches. In both the grid and random search, many structures will have unrealistic densities; to avoid wasting computing time on energy-minimizing structures with large voids or overlapping molecules, the programs often consider only two of the cell lengths as independent variables, generating the third from an assumed cell volume.
For search problems with a fairly low dimensionality, the systematic grid search method seems slightly more effective than a random search (Citation 14 ). However, the size of a grid search can quickly become unmanageable with increasing degrees of freedom (e.g. when the asymmetric unit contains more than one molecule) and randomly generating structures might be the better approach for the higher dimensionality problems. However, for very complex problems, random searches can be unreliable at generating all low-energy structures, at least in a reasonable number of trials (van Eijck (Citation 14 ) found a limit of around 20 degrees of freedom with 5000 starting structures). Therefore, several variations on the random search have been developed to increase its efficiency, as well as adaptations that take advantage of parallel computing environments to significantly increase the number of trial structures that can be generated.
A recent adaptation of the random search replaces the random number generator with a low discrepancy sequence; such quasi-random sequences of numbers avoid the clusters and gaps of points that are often seen in random sampling, ensuring a more uniform (and therefore efficient) coverage of the energy surface. These sequences aim to provide the most uniform sampling possible at every point in the sequence. One benefit of such methods is that, unlike a grid search, the final number of trial structures does not have to be decided in advance of starting the search; this offers advantages for making the full use of available computing power. The procedures of both Della Valle and co-workers (Citation 60 ), and Karamertzanis and Pantelides (Citation 61 ) take this approach, using a Sobol’ (Citation 62 ) sequence of quasi-random numbers to generate their starting structures.
Another modification of the random search procedure is a Monte Carlo search where random moves on the potential energy surface are accepted or rejected based on a probability that is related to the change in energy associated with that move. One application of a Monte Carlo search applied to CSP is the simulated annealing algorithm (Citation 27 ) implemented in the Polymorph Predictor modules of the commercial programs Cerius2 (Citation 54 ) and Materials Studio (Citation 63 ). This algorithm evolved from the earlier work of Gdanitz and co-workers (Citation 48 , Citation 64 , Citation 65 ). At each step in this Monte Carlo search, random changes are made to the crystal's structural degrees of freedom, the change in energy (ΔE) is calculated and the step is accepted if its Boltzmann factor exp(−ΔE/kT) is greater than a random number in the range 0–1 (i.e. the Metropolis algorithm (Citation 66 )). The simulated annealing proceeds through a heating phase where the temperature is increased until any move is accepted, allowing all energy barriers on the potential energy surface to be crossed and the entire search space can be sampled. The simulation is then slowly cooled and, as the temperature decreases, fewer high energy steps are allowed and the search focusses in on the low-energy areas. Each accepted step through the heating and cooling cycle is retained as a trial structure, to be later refined by lattice energy minimization. Polymorph Predictor uses crystal symmetry in the searches, so the search is performed separately in each space group. Due to the stochastic nature of the process, several repeats of the simulated annealing must be performed to ensure a complete sampling of packing possibilities (a common mistake is to rely on the structures generated in a single heating and cooling cycle) – the convergence of the search can be assessed after each repeat of the temperature cycle. Unlike the pure random and grid searches, the set of trial structures resulting from simulated annealing depends on a model for the energy of the crystal; in the other methods, energy calculations are not necessary until the trial structures are refined during energy minimization. This energy dependence from the start allows the simulated annealing approach to preferentially sample areas of search space with relatively low energy, avoiding spending time on unfavourable, high-energy trial structures. This methodology has been successfully applied to some complex systems with many degrees of freedom, such as molecular salts (Citation 10 ) and structures with high Z′ (Citation 67 ).
A more recent Monte Carlo variation, the conformation-family Monte Carlo global optimization scheme, was originally developed to search the conformation space of proteins (Citation 68 ) and its application has been extended to molecular crystals in the program CRYSTALG (Citation 69 ). Conformation-family Monte Carlo differs from a classical Monte Carlo search by making steps between families, instead of between single structures. Here a family is a set of numerically different representations of a single structure; these are identical structures in different settings (in the same way that identical crystal structures can be described in space group settings P21/c and P21/n). Keeping such families of identical structures leads to a wider coverage of the search space and the algorithm is efficient enough to perform well without the use of space group symmetry. In other respects, the search is similar to the Polymorph Predictor simulated annealing, except that the sampling is performed at a constant temperature.
Polymorph Predictor and CRYSTALG achieve efficient sampling of all important (low energy) parts of the energy surface through special techniques of guiding the random steps between structures. A different approach is to distort the energy surface itself to create a more easily searched problem. Such a procedure, termed self-consistent basin-to-deformed-basin mapping, temporarily smoothes the potential energy surface to a surface with fewer minima. This deformed surface is easily searched and the small numbers of minima are traced back to the deepest minima on the original potential energy surface by removing the deformation in a stepwise fashion. The best structures from one step are carried forward and energy minimized on the new energy surface, with extra structures being generated by random perturbations of all parameters. This procedure has been implemented for crystal structure generation using either a diffusion equation or distance scaling method for the smoothing transformation (Citation 70 ). The methods proved to be very efficient, allowing the experimental crystal structures of benzene and S6 to be located with no use of space group symmetry – the searches were performed with up to eight independent molecules in the unit cell (51 degrees of freedom (Citation 70 )).
An alternative to the grid-based, random and Monte Carlo approaches is the very common global minimization methodology called a genetic algorithm, which is based on the ideas of biological evolution. The descriptors of the crystal structure (lattice dimensions and molecular positions) are stored in ‘genomes’, which undergo operations, such as crossover (mating), mutation and selection as a ‘population’ of trial crystals is evolved towards the best set of structures. The modified genetic algorithm for crystals and cluster structures (MGAC (Citation71–75)) program employs such a method to locate the lowest energy crystal structures. MGAC treats the molecular positions, orientations, flexible dihedral angles and unit cell angles as parameters. Cell lengths are then chosen to define the smallest asymmetric unit that encloses the molecules. The resulting crystal is energy minimized and the genetic algorithm operations (crossover, mutation and selection) are applied to these locally minimized structures.
An attractive feature of the genetic algorithm approach is the flexibility in the choice of scoring and penalty functions (which control selection). MGAC seeks to minimize the lattice energy, while another method, developed by Motherwell (Citation 76 ) and implemented in the RANCEL program, avoids the use of energy altogether. Instead, RANCEL uses a scoring function derived from structural information in the CSD (Citation 38 ). The crystal structures of molecules that are similar (in molecular formula and chemical functional groups) to the target molecule are retrieved from the CSD and intermolecular atom–atom distance frequency curves are derived for each pair of atom types. The genetic algorithm then searches for the crystal structures whose atom–atom distances fit these curves as well as possible. Penalty functions can be also defined to punish structures without certain structural features, such as common hydrogen-bonding motifs. The distance frequency curves seem to contain enough information about the three-dimensional packing around atoms that the correct crystal structure can sometimes be successfully predicted without recourse to any energy calculations (Citation 76 ). The method is particularly useful for modelling molecules containing atoms or functional groups that are considered difficult for traditional, energy-based modelling methods. In theory, such homology considerations could be combined with energy calculations in the selection of structures during a genetic algorithm search.
The MOLPAK software (Citation 49 ) describes crystal structures by a coordination geometry around a central molecule, instead of by their unit cell dimensions and space group. The most common coordination geometries (consisting of the 14 nearest neighbours to the central molecule) have been derived from the CSD and each of these is searched separately for the densest possible packings by systematically varying the orientation of the central molecule. At each orientation of the central molecule, a line of molecules is brought into close contact, lines are then packed together to form a two-dimensional grid, and these grids are then packed along the third dimension. The close packing between molecules is controlled by a repulsive interaction between atoms, and molecules are brought together until a threshold repulsive energy is reached. The densest crystals resulting from the search are then refined through energy minimization using a more complete energy model.
One of the most recently developed methods employs molecular dynamics (MD) simulations to explore packing space. MD, where atoms follow Newton's equations of motion, would normally be inefficient at sampling all possible minima on the energy surface, because high-energy barriers between structures tend to confine the simulation to the neighbourhood of one crystal structure for long simulation times. The modified MD method, which has been named metadynamics (Citation 77 ), forces a quicker evolution of the structure by dynamically adding a biasing potential to the energy surface which guides the simulation away from all structures that have already been sampled. Because of the MD basis of the search, a metadynamics simulation samples the free energy surface. The free energy surface tends to be smoother than the lattice energy surface that is sampled by other temperature-free methods because low-energy barriers between related structures are overcome. Any minimum on the lattice energy surface whose potential well is shallower than about kBT will not be a stable structure at that temperature, so that a search for stable structures on the free energy surface will not locate these structures and leads to a smaller number of candidate structures than lattice energy-based methods. In the case of benzene, the result is that all known polymorphs are found as free energy minima, and no other unobserved structures remain (Citation 78 ). This contrasts with lattice energy searches that locate many possible crystal structures of benzene (Citation 79 ) and indicates that the many unobserved predicted structures lie in shallow lattice energy minima. These results are promising and show that MD simulations are an effective method of eliminating many of the unobservable structures that result from lattice energy searches. However, the effect of metadynamics on smoothing the energy surface is less effective for a system with strong hydrogen bonds and larger energy barriers between local minima on the lattice energy surface: for fluorouracil, the majority of crystal structures resulting from a lattice energy search are stable in a MD simulation at ambient conditions and only about 25% of the many lattice energy minima are eliminated by considering the free energy surface (Citation 80 ). Furthermore, the metadynamics simulations of fluorouracil only sampled crystal structures with the same hydrogen bond motif as the structure that was used as a starting point of the simulation. Overall, the metadynamics method could be useful for studying the stability of different structures and phase transitions between closely related structures, but does not seem to be an effective approach to producing all possible crystal structures of molecules that form strong intermolecular interactions.
All the above methods assume three-dimensional translational symmetry from the start, varying the unit cell dimensions and molecular positions simultaneously. A different class of methods starts by searching for stable clusters of molecules; translational symmetry is then applied to the most promising of these ‘nuclei’ to build trial crystal structures. The rationale for these methods is that certain strong interactions are crucial to the development of the crystal and that these interactions can easily be determined in small groups of molecules before worrying about how these will pack in the crystal lattice. One of the first CSP programs, Gavezzotti's (Citation 47 , Citation 81 ) PROMET, is based on this concept – small clusters are built from the molecular structure using the most common symmetry operators in organic crystals – the inversion centre, translation, 21 screw and glide. Translational symmetry is then imposed in one dimension, creating a string of molecules. If the string is stable, translational symmetry is imposed in a second direction to form layers, and the structure is finally expanded into a three-dimensional crystal. Many trial crystal structures can usually be generated from each cluster by systematically varying the three translation vectors (though one or more cell vectors are often fixed by one of the symmetry operators used to build the original cluster).
A similar approach is taken to generating crystals in Hofmann's (Citation 82 , Citation 83 ) FlexCryst program. However, unlike the atom–atom potentials that are used to judge the stability of clusters in PROMET, FlexCryst uses group-based interactions (e.g. phenyl ring–phenyl ring, methyl–phenyl or hydrogen bonds) in the initial search for clusters. Potentials that are trained by data mining (Citation 82 , Citation 84 ) are used to refine and judge the final crystals. A unique feature of FlexCryst is that the program discretizes configuration space – all interaction sites are moved to a point on a grid, allowing the scoring function to be tabulated and simply looked up for each interaction. This approach leads to significantly faster computation times than methods that require evaluation of potential functions for all atom–atom pairs. However, this comes at the expense of sacrificing accuracy in the final calculated structures.
In short, a host of methodologies has been thrown at the task of crystal structure generation – this review summarizes many, but certainly not all, approaches that have been applied to this problem. Few head-to-head comparisons of the programs have been made to assess their relative strengths. Even in the CSP blind tests (Citation20–23), whose results are discussed in a later section, each participant applied different computational resources to the project, so the search algorithms are not compared on a level playing field. As the blind tests continue, certain methods may emerge as the most reliable. For now, the choice of search algorithm depends on many factors: personal preference and experience; cost (in the case of commercial software); ease of implementation versus complexity of the problem; suitability to the computing setup of the research group; and the final aim of the calculations. An expensive and thorough search method would be preferred for polymorph screening applications, while faster approaches would be better suited in the context of testing hypotheses in crystal engineering and the design of targeted structures.
Progress is being made in developing more efficient approaches that allow more and more difficult problems to be addressed – multi-component crystals and highly flexible molecules. Furthermore, recent search algorithms are taking advantage of distributed and parallel computing, allowing energy minimization of 105–106 trial crystal structures (Citation 61 ). These very extensive searches should give a complete coverage of the search space, preventing any structure from being overlooked. Furthermore, such computing power allows more space groups (for search methods that rely on a pre-defined space group symmetry) or more values of Z (for search methods that treat all molecules in the unit cell independently) to be considered, so that crystal structures with less common symmetry need no longer be problematic.
2.2.3. Clustering of structures
The structure generation phase of CSP calculations typically creates 102–106 trial structures and these structures must usually be relaxed to minimize the energy or cost function before being analysed. Therefore, clustering algorithms are used at the final or intermediate stages to remove identical structures and save computing (and human) time. The clustering method is an important part of the process – it must be both quick and discriminating so that identical structures can be identified rapidly, but distinct structures are not erroneously discarded. The commonly used methods include comparison of: reduced cells (Citation 60 ); standardized unit cells, molecular positions and orientations (Citation 85 , Citation 86 ); the geometries of close atom–atom contacts (Citation 87 ); and distributions of inter-atomic separations (Citation 27 ).
2.3. Lattice energy calculations
There is a wide choice of methods for generating a set of trial crystal structures from the molecular model and most of these rely on energy calculations at some stage – to guide the crystal structure generation (e.g. during a Monte Carlo simulated annealing or genetic algorithm search), for refinement of the trial structures through energy minimization, and usually for the final ranking of the possible structures. After the final energy minimization, the results of such a search are often summarized as a plot of lattice energy against some property of the crystal structure (often the density, volume per molecule or packing coefficient), as at the bottom of . The set of structures, and their energies, is often referred to as the crystal energy landscape. Several types of landscape are possible, as illustrated in , using results taken from (Citation 31 ). In the simplest case, one crystal structure is thermodynamically favoured and separated from the rest of the possible structures by a large energy gap (). More often, many possible structures are located in a small energy window above the global minimum (). In all three of these searches shown in , one of the predicted structures (red squares in ) does correspond to the experimentally observed crystal structure. A cumulative ‘density of states’, counting the number of distinct crystal structures from the global minimum in lattice energy up to any ΔE (), highlights the differences in the results for these three molecules.
Figure 2. Possible outcomes of a CSP. Each point on the graph represents a distinct crystal structure. The large square data points represent the experimentally observed crystal structures. The same energy scale is used in all three plots. Results taken from the exp-6 + multipoles calculations in (Citation 31 ).
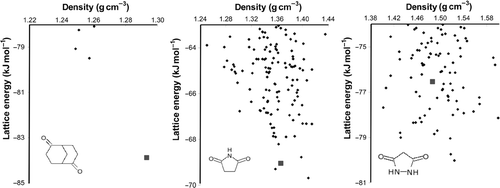
Figure 3. Cumulative number of computer-generated crystal structures within ΔE of the global minimum in lattice energy for three small organic molecules. Data taken from (Citation 31 ).
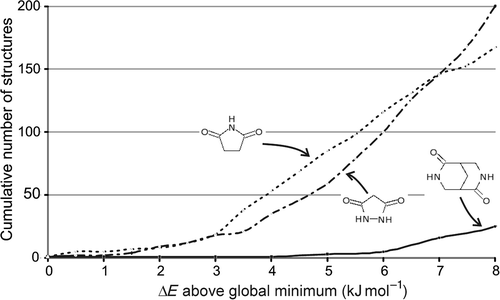
The first of these outcomes () is an ideal situation for prediction by lattice energy minimization. One structure is clearly the thermodynamically most stable, so that small errors and approximations in the model potential should be unimportant compared to the energy gap separating this structure from all other possibilities. Furthermore, kinetic influences during crystal nucleation and growth would have to be very strong to produce one of the much higher energy structures. Indeed, this lowest energy structure (red square in ) does correspond to the known crystal structure for this molecule. However, after studies of many model systems, we now recognize this type of situation as atypical; usually, many nearly equi-energetic possibilities are found in a structure search (e.g. 100+ different crystal structures within about 6 kJ mol−1 of the global minimum, dashed and dashed–dotted curves in ). This observation of multiple low-energy minima is a consistent problem in CSP – many more structures are found in a reasonable energy range than we would ever find as real polymorphs. As a consequence, the energy differences between possible structures are very small and minor changes to the way the energy is calculated could lead to significant reordering of the predicted structures. For one of these examples (), the four lowest energy predicted crystal structures are all within 1 kJ mol−1 and the known crystal structure does correspond to one of these. Energy differences on the order of 1 kJ mol−1 can come and go in lattice energy calculations, due to small changes in parameters, numerical considerations (e.g. tolerances used for convergence of the minimization) or details of the algorithm used to evaluate the lattice energies (e.g. summation cut-offs). Therefore, the energy differences between the observed structure and the two lower energy structures are smaller than the combination of errors and approximations in the energy calculations, and it is possible that the observed structure is in fact the thermodynamically most stable. However, in the third example (), there are many predicted structures with lower energies than the known crystal structure, by up to about 4 kJ mol−1 (a small amount of energy by many standards, but large in the context of CSP). This type of result makes one suspect that kinetic factors are at work; the observed crystal structure is thermodynamically metastable, but forms in preference to its competitors during nucleation and/or growth of the crystal. Such observations are important for us to develop an understanding of crystallization and polymorphism, but to be confident in the conclusions drawn from a lattice energy search, we must use reliable models to evaluate the relative energies.
There are many choices for how to calculate lattice energies of molecular organic crystals. The choice of model for a given molecule is always a balance between accuracy and simplicity – the ease of use and implementation, as well as computational expense. Typically, thousands of structures must be energy minimized during a prediction and the required accuracy in relative energies is usually very high. In fact, the goal of CSP has been the main motivation for the development of accurate models for intermolecular interactions in crystals; the most common models for energy calculations and advances in this area have been reviewed several times (see, e.g. (Citation 28 , Citation 88 , Citation 89 )).
2.3.1. Atomistic methods
The lattice energy of a crystal structure can be conceptually divided into the interactions between molecules and the conformational (intramolecular) energies of the molecules. When the molecular geometry is kept rigid throughout the calculations, the conformational energy is a constant for all of the predicted structures, so only the intermolecular energy must be evaluated. The total intermolecular energy in an assembly of molecules is usually approximated as a sum of pairwise potentials, UMN
The most commonly used approach in lattice energy studies of molecular crystals is the atom–atom potential method for the pairwise potentials, where the interaction between molecules M and N is expressed as a sum over atom–atom interactions:
Each pair of atoms is assigned a potential; the functional form of this atom–atom interaction potential is usually isotropic (i.e. spherically symmetric) and typically consists of van der Waals (i.e. repulsion and dispersion) and electrostatic terms. There are two commonly used forms for the repulsion and dispersion interactions: the n-6 or Lennard–Jones model
For the most common elements (C, N, O, H), there is an abundance of parameter sets, differing in the process used to derive values for the As, Bs and Cs. These parameters are usually derived empirically, to give the best possible fit to known structures or properties. Therefore, an important consideration in choosing a parameter set is the similarity of the molecule being studied to the molecules that were used in the parametrization. The fitting procedure usually involves varying the parameters simultaneously to reproduce the structures and properties (e.g. sublimation enthalpies, phonon frequencies) of known crystals – much of the early development of this empirical atom–atom approach and its application to the organic molecular solid state has been reviewed by Pertsin and Kitaigorodskii (Citation 26 ). Recently, the results of CSP searches have sometimes been incorporated in the parametrization process; the parameters are refined not only to reproduce structures and properties of known crystals, but also to ensure that the known crystal structure is as close as possible to the global minimum of a set of predicted structures for that molecule (Citation92–94).
While such empirically derived atom–atom models are very commonly used and do lead to successful prediction results, there are several limitations with using empirically parametrized models in crystal structure modelling. A large set of known crystal structures is needed to sample a sufficient range of possible types of interactions at various inter-atomic separations. There are often insufficient data to fit reliable parameters for less common atom types and functional groups. Furthermore, the model potential should represent the energy of a static, temperature-less configuration of molecules, so that a lattice energy minimized crystal structure approximates the true structure at T = 0 K. Temperature should only enter the calculations through dynamical simulations. However, the structural data used in empirical parametrization are often determined at a range of temperatures. Ideally, only low-temperature crystal structures are used. However, when too few low-temperature structures containing the atom types of interest exist, it is often necessary to utilize room temperature structures. Thus, calculations using empirical model potentials refer to an ill-defined temperature – a clear problem where the relative energies of polymorphs and the properties of crystal structures vary significantly with temperature.
Due to the problems with empirical atom–atom models, methods are being developed to systematically derive atom–atom model potentials for organic molecular crystals from quantum mechanical calculations on isolated molecules and small clusters (Citation95–104). Non-empirical models can be developed specifically for a given molecule or of a small family of molecules and quantum mechanical calculations can be used to probe many more interaction orientations and separations than are sampled in the known crystal structures used for empirical parametrization. Such non-empirically parametrized models usually lead to superior modelling of crystal structures (Citation 97 , Citation 101 ), properties (Citation 102 ) and more reliable lattice energy predictions of crystal structures than empirically parametrized model potentials (Citation 98 , Citation 100 , Citation 103 , Citation105–107). Furthermore, if the parameters are fitted to reproduce the pure intermolecular potential energy, then the resulting model potential is a valid starting point for dynamical simulations (discussed in Section 2.4), such as lattice dynamics or MD to investigate the temperature dependence of crystal structures, energies and properties.
Another attractive feature of non-empirically derived models is that sufficient relative orientations of atoms in molecules can be sampled to allow the orientational dependence of van der Waals interactions to be easily assessed and incorporated in the model. Anisotropy in atom–atom repulsion is known to be particularly accentuated in interactions involving certain atoms. For example, the polar flattening of halogen atoms (Citation 108 ) is sufficiently important to the crystal packing, thermal expansion and vibrational properties of chlorobenzenes that the isotropic atom–atom form cannot adequately describe these crystals (Citation 102 , Citation 109 ).
2.3.2. Electrostatic models
Besides the repulsion–dispersion model described above, almost all model potentials include a description of the electrostatic interactions between molecules. The simplest model assigns charges to each atom and the most appropriate schemes for partitioning the atomic charges choose these to reproduce the electrostatic potential (ESP) around the molecule (Citation 110 ). Along with an isotropic n-6 or exp-6 model, the inter-atomic interaction energy still depends only on the separation of the atoms.
While a set of atomic point charges can give a reasonable approximation to the molecular ESP, we know that the electron distribution around atoms in molecules is not spherically symmetric – the rearrangement of valence electrons upon bond formation results in localized lone pairs and π-electron density, which cannot be adequately described by the isotropic atom model. This has been recognized for some time and several improvements on the atomic charge model have been used in modelling molecular crystals. The simplest enhancement is to define off-nuclear charge sites, at bond centres and chemically sensible lone pair positions (Citation 111 , Citation 112 ), or at positions that are optimized to give the best fit to the molecular ESP (Citation 113 ). An even more elaborate representation of the molecular charge distribution consists of sets of multipoles (i.e. charge + dipole + quadrupole, etc.) centred on each atomic site. These multipoles can be derived directly from a calculated molecular wavefunction (Citation 114 , Citation 115 ) or fitted to the ESP, in the same way as charges are usually derived (Citation 99 , Citation 116 ). Atomic multipoles offer an important improvement in reproducing the molecular ESP – adding just dipoles and quadrupoles to the atomic charges typically halves root-mean-squared errors in the ESP around organic molecules (Citation 117 ). Unlike atomic charge models, such distributed multipole models have proven to be remarkably successful in predicting the hydrogen bond geometries in complexes of small molecules (Citation 118 , Citation 119 ). Distributed multipole models also show superior performance in reproducing the structures (Citation 32 , Citation 117 ), phonon frequencies (Citation 120 ) and mechanical properties (Citation 121 ) of molecular organic crystals. The use of anisotropic electrostatic and van der Waals models for modelling crystal structures is currently limited to a few specialized modelling packages, such as DMAREL (Citation 122 ) and its revised version, DMACRYS (Citation 123 ). The value and necessity of more accurate models than the isotropic atom–atom model should increase the popularity, as well as the availability of such calculations.
Extra terms in the model are sometimes included to account for the other contributions to the intermolecular interactions, such as polarization and higher order dispersion coefficients. Furthermore, flexible molecule models require additional terms to describe bond stretching, angle bending and torsions for rotations about bonds. The simplest approach is to include such intramolecular terms to the model potential via a molecular mechanics approach. At its simplest, the intramolecular potential might then take the form
These three terms model bond stretching, angle bending and bond torsions; bo and θo are equilibrium bond lengths and bond angles and the force constants K are often fitted to ab initio quantum mechanical calculations on isolated molecules. A major challenge in the development of energy calculations is the merging of accurate intermolecular models with highly accurate descriptions of the intramolecular energy. For example, the conformational dependence of atomic multipoles hampers their use for flexible molecules (Citation 117 ). One solution to the conformational dependence of the molecular electrostatic model is to recalculate the atom charges and multipoles as the molecular geometry adjusts to the crystal packing during lattice energy minimization (Citation 106 , Citation 124 ).
It is also clear from many studies that the molecular mechanics description of intramolecular energies is not sufficiently accurate for the final energy ranking of crystal structures in CSP studies of flexible molecules, where the conformation can vary between the possible crystal structures. One approach to achieve sufficiently accurate total (inter + intramolecular) energies for CSP of flexible molecules has been to couple atom–atom descriptions of intermolecular interactions with quantum mechanical electronic structure theory molecular energy calculations, either for the final energy evaluation of crystal structures that have been lattice energy minimized using molecular mechanics intramolecular energy models (Citation 5 , Citation 125 ) or iteratively throughout the lattice energy minimization procedure (Citation 124 ). While this approach has been successful, the calculations normally require at least one quantum mechanical molecular energy calculation per crystal structure. The computational cost of such calculations is therefore much greater than for rigid molecules, and the scaling of computing cost with molecular size is an issue for extending studies to larger, flexible molecules.
2.3.3. Performance of lattice energy minimization for CSP
The success of the global lattice energy minimization approach to CSP could be assessed by surveying published results to judge the rate of success and limitations of the general method. The formidable task of reviewing all published results of lattice energy minimization for CSP was performed in 2001 by Beyer et al. (Citation 3 ). While this survey does give a feel for the utility of global lattice energy minimization and a perspective on progress in the field up to about the year 2000, the variations in methodology and lack of details in many published studies prevent a real assessment of this approach to CSP. Another approach to assessing the approach is to perform CSP calculations on a large set of molecules using a consistent methodology. Such a study was performed on a set of 50 small, rigid organic molecules where each molecule was treated with exactly the same method (Citation 29 , Citation 31 ). The molecular structure for each of the 50 molecules was calculated using DFT calculations, trial crystal structures were generated using a simulated annealing approach and, because small molecules with limited flexibility were chosen for the study, the trial structures were then lattice energy minimized in the rigid-molecule approximation. Lattice energy calculations were performed using an empirically parametrized exp-6 repulsion–dispersion model potential and two electrostatic models were tested: ESP fitted atomic charges and atomic multipoles from a distributed multipole analysis of the calculated wavefunction. The crystal structures were first generated and lattice energy minimized with the simpler isotropic energy model and their energies were re-evaluated using the multipoles model to investigate the sensitivity of lattice energy ranking to the electrostatic model.
The first important results relate to the success of crystal structure generation, with the caveat that the chosen set of molecules was not a difficult test for the search algorithm because one of the criteria for the selection of molecules was that they crystallized in one of the nine most common space groups, with Z′ ≤ 1. Some of the 50 chosen molecules have known polymorphs and, in total, there were 62 known crystal structures that were considered as targets for the calculation. Fifty eight (94%) of the known crystal structures were easily located in the simulated annealing search, which was initially repeated four times in each space group. When found, the observed crystal structures are generally very well reproduced by the prediction, with unit cell parameters usually within 3–4% of those in the observed structure. We can conclude that this crystal structure search method works extremely well for these small rigid molecules. Of the remaining four known crystal structures that were not located in these initial searches, three were located after additional (up to 13) repeats of the simulated annealing temperature cycle. Only one of the 62 known crystal structures was not located and this failure was discovered to be a failure of the chosen energy model in reproducing that structure, rather than the search algorithm itself (Citation 29 ). These results demonstrate that the search problem is not a major obstacle to CSP, at least for crystal structures with Z′ = 1.
Given that the search method locates the known crystal structures, the overall success of the predictions depends on how well lattice energy minimization ranks the true crystal structures amongst the lists of computer generated possibilities. Two measures of the success of the lattice energy predictions were used to summarize the results for these 50 molecules: ΔE, the energy difference between the experimentally observed crystal structure and the lowest energy unobserved crystal structure; and N lower, the number of unobserved crystal structures with lower energy than the observed structure. The distributions of ΔE are shown in . The results were fairly encouraging, even using the simpler energy model that employed isotropic repulsion–dispersion and electrostatic atom–atom models. With this simple model, the experimentally observed crystal structures are found as either the global minimum energy structure or with ΔE < 0.5 kJ mol−1 in 36% of cases. The highest ΔE for any of the molecules was 7.3 kJ mol−1. These results were improved when the atomic partial charge model of electrostatic interactions was replaced by the more elaborate multipole expansions (charge, dipole, quadrupole, octupole and hexadecapole on each atom): the number of known crystal structures with ΔE < 0.5 kJ mol−1 was increased to 50% and the worst ΔE was reduced to 5.1 kJ mol−1 (). A third of the observed crystal structures for this set of molecules are ‘perfectly’ predicted, that is lower in energy than any unobserved predicted structure. In 69% of cases, five or fewer unobserved structures are assigned lower lattice energies than the real crystal structure with the exp-6 + multipoles model. With this energy model, a handful of the best structures from a global lattice energy search will normally contain the observed crystal structure.Footnote1
Figure 4. (a) Distributions of relative lattice energies of crystal structures of 50 small molecules from lattice energy minimization with exp-6 + atomic charge and exp-6 + atomic multipoles model potentials. (b) Distributions of relative lattice energies for hydrogen bonding and non-hydrogen bonding molecules, from the exp-6 + atomic multipoles calculations. Reprinted with permission from (31). Day, G.M.; Motherwell, W.D.S.; Jones, W. Beyond the Isotropic Atom Model in Crystal Structure Prediction of Rigid Molecules: Atomic Multipoles Versus Point Charges. Cryst. Growth Des. 2005, 5, 1023. Copyright 2005 American Chemical Society. Data taken from (Citation 29 , Citation 31 ).
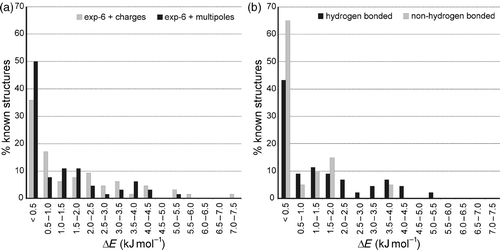
These results of this study demonstrate that a lattice energy search is a useful starting point for CSP and the improvement in prediction success that was attained by improving the energy model should motivate the further development of model potentials, as well as improvements in the thermodynamic model (e.g. by including dynamic/vibrational contributions to the free energy). Further analysis of how ΔE was influenced by molecular properties also suggested differences between hydrogen-bonding and non-hydrogen-bonding molecules (): non-hydrogen-bonding molecules are more often predicted at or near the global minimum in lattice energy than hydrogen-bonding molecules and almost all molecules whose known crystal structures are located with high ΔE are those with hydrogen-bonded functionality. The differences in results for hydrogen-bonding and non-hydrogen-bonding molecules probably result from a combination of factors. There may be inadequacies using the energy models applied in these studies in accurately modelling hydrogen bond energies and the relative stabilities of crystal structures containing different arrangements of hydrogen bonds. The polar environments in crystals of hydrogen-bonded molecules will polarize the molecular charge density, strengthening electrostatic interactions between molecules. However, the effect can vary greatly depending on the relative arrangement of polar functional groups. This polarization effect, which is responsible for cooperative effects in extended hydrogen bond networks, cannot be described using the pairwise additive atom–atom approach. Polarization models are now being developed to improve atom–atom models for polar, hydrogen-bonding molecules (Citation 126 , Citation 127 ). Apart from deficiencies in the energy model, the results in also suggest that hydrogen-bonding molecules are more likely to be kinetically trapped in metastable crystal structures than molecules with no hydrogen-bonding capability. This is physically reasonable from the point of view of the energy landscape controlling nucleation and crystal growth from solution. Strong directional interactions lead to high barriers between minima on the potential energy surface. Therefore, strong intermolecular interactions and the resulting molecular arrangements that are preferred in solution and the early stages of crystal nucleation are likely to persist in the final crystal structure, even if that arrangement of strong interactions does not lead to the thermodynamic global minimum, because the barriers to rearrangement into the lowest energy crystal structure are too large. Molecules that only interact via weak interactions, on the other hand, are relatively free to explore phase space in search of the deepest minimum on their smoother lattice energy surface.
2.3.3.1. Flexible molecules
Much of the development of methods for CSP has focussed on fairly simple, rigid molecules. However, the development of accurate methods of treating both inter- and intramolecular energy differences between crystal structures has extended the scope of global lattice energy minimization methods to molecules with a limited amount of conformational flexibility. No large-scale evaluations have been performed for more flexible molecules in the way that has been done for rigid molecules. However, a number of individual studies seem to indicate that current methods have similar success rates for moderately flexible molecules as seen above for rigid molecules. A few examples of flexible molecules for which current methods have been successful are (): glycol and glycerol, whose known crystal structures were predicted amongst the lowest energy calculated structures (Citation 106 ); aspirin, where the global lattice energy minimum corresponded to the stable polymorph (Citation 6 ) and another low-energy prediction was later discovered during an attempted co-crystallization (Citation 128 ); phenobarbital, where the global minimum in lattice energy corresponded to the thermodynamically stable polymorphic form (Citation 5 ); and piracetam, for which the structure of a new polymorph was predicted (Citation 129 ). Recently, several methods of refining the molecular geometry during lattice energy minimization were tested on a series of hydrophobic α-amino acids with varying degrees of conformational flexibility (alanine, valine, leucine and isoleucine) and enantiopure and racemic crystal structures all corresponded to either the global minimum or one of the 2–5 lowest energy structures from lattice energy searches, depending on the details of the method (Citation 130 ). The results of these investigations demonstrate promising progress in the size and flexibility of molecules for which CSP methods can be successfully applied.
Figure 5. Flexible molecules studied by CSP methods.
2.3.4. Alternative methods for lattice energy calculations: solid-state DFT and semi-classical density sums
The atom–atom approach has greatly contributed to our understanding of the molecular solid state, but with the development of periodic (solid-state) DFT, quantum mechanical calculations on the periodic lattice are now feasible, though demanding on computational resources. Such calculations have the potential to solve many of the problems encountered with energy minimization using atom–atom models. All electron density and nuclear positions are optimized together, so that the molecular conformation can naturally adjust to the crystalline environment, without having to worry about the balance of inter- and intramolecular terms in an atom–atom force field. Furthermore, electrons can flow; so the electron distribution of a molecule does not need to be fixed from gas phase calculations. For example, the atomic charges (determined by Mulliken partitioning) in periodic DFT calculations on the polymorphs of glycine differ, on average, by 0.05 e from the corresponding atomic charges in the gas phase (Citation 131 ). This allowance for charge density reorganization enables energy calculations to include cooperative effects which might, for example, stabilize one hydrogen-bonding motif over another.
Naturally, periodic DFT calculations have been applied to the challenge of CSP. Rovira and Novoa (Citation 132 ) used periodic DFT calculations to re-explore the lowest energy acetic acid crystal structures that were generated with UPACK using an atom–atom force field. They demonstrated that DFT calculated energies do predict the low-pressure acetic acid crystal structure closer in energy to the global minimum than the force field used in the initial search. However, the computational expense for even this small molecule prevented all computer-generated crystal structures from being fully optimized (including cell constants) in the DFT calculations. While their results demonstrated promise for the use of periodic DFT in a final evaluation of energy differences between predicted structures, the calculations were shown to lack an adequate description of the long-range attractive dispersion contribution to intermolecular interaction energies, requiring a correction to be added to the pure DFT energy to yield reasonable agreement with experimental interaction energies.
More recently, Chisholm and co-workers (Citation 131 ) tested several flavours of DFT on the known and hypothetical polymorphs of glycine. DFT generally had problems reproducing the structures of the known polymorphs in this study and the errors were very sensitive to the choice of exchange-correlation functional – the local density approximation (LDA) leads to a serious overbinding of the crystal structures, while generalized gradient functionals (e.g. PW91) tended to underbind the crystals. Byrd and co-workers (Citation 133 ) made similar observations; PW91 calculations underestimated densities of a set of known molecular crystals by 10–15%. This underbinding is a result of the poor description of dispersion interactions in DFT calculations. Despite these problems in reproducing the densities of the known crystal structures, the energy ranking of the real polymorphs in the predicted lists was found to be about as good as predictions using an empirical atom–atom model potential. The LDA calculations get the correct order of stability of the polymorphs as well as assigning them lower energies than any of the other predicted structures. These results show promise for DFT calculations in CSP. However, glycine was chosen for this study as a system that is very well suited to solid-state DFT calculations – the molecule is zwitterionic in the crystal and the lattice energy is dominated by electrostatic contributions, which are reproduced very accurately by the DFT methods. Results would undoubtedly deteriorate for more weakly bound van der Waals crystals, where dispersion interactions are more influential.
To be generally useful for studying and predicting the crystal structures of organic molecules, DFT must overcome its poor treatment of the very important dispersion attraction between molecules. There has been significant effort in developing corrections to pure DFT to provide a better description of dispersion interactions. The most popular approach to date has been to augment the solid-state DFT energy calculation for a crystal with empirically derived C6/R6 terms (Citation134–137).
Neumann and Perrin (Citation 134 ) presented the first application of such dispersion-corrected DFT (DFT + D) methods to CSP of molecular organic crystals, using their method to lattice energy minimize the lowest energy predicted crystal structures that were generated using an atom–atom force field and simulated annealing method for six small molecules (acetylene, ethylene, ethane, methanol, acetic acid and urea). In this method, C6 coefficients were fitted to reproduce molecular C6 coefficients and this C6/R6 term is damped at short distances, with the parameters of the damping function empirically fitted to reproduce unit cell parameters of a set of low-temperature molecular organic crystal structures. The method was shown to reproduce the low-temperature experimentally observed crystal structures very accurately and finds the observed crystal structure as the lowest energy of the predictions for all but ethylene, where the observed structure corresponds to the second lowest energy predicted structure. The method has since then been further developed by fitting an atom–atom force field (including both non-bonded and bonding terms) specifically for each molecule to reproduce lattice energies and ESPs that are calculated using the DFT + D method (Citation 138 ). This ‘Tailor-made force field’ (TMFF) is used during the crystal structure generation procedure and a set of the resulting lowest energy crystal structures are re-optimized using DFT + D. The purpose of the TMFF is that it is sufficiently computationally efficient to be used during crystal structure generation (where the energies of a large number of structures are assessed) and, that it is sufficiently accurate to guarantee that a relatively small set of the TMFF-generated crystal structures will include the global minimum structure on the DFT + D lattice energy surface. The TMFF/DFT + D method for CSP has been tested in the 2007 blind test of CSP (Citation 23 ), whose results are discussed in Section 3.
A method falling somewhere between the atom–atom approach and all-electron quantum mechanics calculations is Gavezzotti's (Citation139–142) semi-classical density sums (SCDS) or ‘Pixel’ method. In SCDS calculations, a molecule is represented by a large number (∼104) of interaction sites representing the quantum mechanically calculated monomer electron density, instead of just the ∼101 nuclear positions used by atom–atom methods. Relatively, few parameters are used to calculate interaction energies between molecules – electron pixels interact to give the electrostatic, exchange-repulsion, dispersion and polarization energies (Citation 139 , Citation 140 ). Because each energy contribution is derived separately from the interactions of monomer charge densities using theoretically sound models, the Pixel method provides a better partitioning of the total lattice energy than empirical potentials. The individual contributions to the lattice energy can therefore be meaningfully analysed.
The method shows good promise – SCDS was tested for CSP by recalculating the energies of the putative crystal structures of a set of six small molecules (Citation 141 ) that were determined using atom–atom force field methods. The SCDS–Pixel calculations gives as good or better results than the atom–atom methods in the majority of cases, ranking the observed crystal structures of three of the six test molecules, naphthalene, naphthoquinone and pyridine, as the lowest energy of the computer-generated crystal structures.
The periodic electronic structure methods, such as DFT, as well as the SCDS–Pixel approaches are promising for recalculating the energies of lists of predicted crystal structures from atom–atom calculations. However, the computational expense of these approaches does limit their applicability in calculations beyond lattice energy minimization, such as dynamical methods discussed in the next section. To give a rough idea, single-point energy calculations on a typical crystal of a small (about 10 atoms) organic molecule using exp-6 + atomic charges, exp-6 + multipoles (including terms up to hexadecapole), SCDS–Pixel and DFT take on the order of 0.1 s, 1 s, 15 min and 1 h, respectively, on a modern workstation. Energy minimizations typically require 10–100 such energy calculations; meaningful MD requires, at minimum, tens or hundreds of thousands.
2.4. Beyond lattice energy calculations
Part of the motivation for developing CSP methods is to gain an understanding of, and to anticipate, polymorphism. Yet, the very existence of polymorphs tells us that the methods described so far cannot provide a complete understanding of, nor can they always predict, polymorphs. Even with a model potential that yields perfect energy differences between all possible crystal structures, lattice energy minimization can only be a first approximation and we must look beyond such calculations to fully understand polymorphism.
A pair of polymorphs can have one of two types of relationship – monotropic or enantiotropic. Monotropic polymorphs maintain their order of stability at all temperatures, while the free energies of enantiotropic polymorphs cross at some transition temperature. Both types of system tell us something about the limitations of predicting crystal structures from global minimization of the lattice energy of static crystal models. With enantiotropic polymorphs, each form is stable across a certain temperature range; below a transition temperature, the low-temperature form is the thermodynamically favoured polymorph, while above the transition temperature thermodynamics favours the other polymorph – the high temperature form. This behaviour tells us that temperature has an important effect on the relative stability of polymorphs. Lattice energy calculations are temperature-free, so they could never predict such phase transitions. The temperature of a lattice energy calculation is formally 0 K (and furthermore ignores zero-point vibrations of the molecules), so the relative lattice energies should be a representation of the low-temperature order of stability of the predicted crystal structures. In a monotropic pair of polymorphs, one polymorph is thermodynamically metastable at all temperatures below their melting point (i.e. the transition temperature between the polymorphs lies above their melting point). The fact that such systems are known (and are not uncommon) demonstrates that crystal structures other than that with the lowest possible free energy exist – kinetics can have a role in determining which structure is formed and can lead to a higher energy crystal structure if the most easily formed or fastest growing nuclei do not lead to the global minimum crystal structure.
Some progress has been made in improving the thermodynamic model to recover the temperature dependence of the relative energies (Section 2.4.1). However, the influence of kinetics on the final crystal structure is a much greater problem for molecular simulation. Ideally, we would model the crystallization process – from the nucleation event to growth of the bulk crystal, all in the presence of the solvent and any surfaces that could promote heterogeneous nucleation. Unfortunately, the time scales and size of the system that must be simulated are far beyond computational capabilities. However, much simpler kinetic considerations have formed a part of some CSP studies. One approach has been to test the usefulness of simple crystal growth calculations in highlighting kinetic preferences for certain crystal structures (Section 2.4.2), while others have used analyses of the many known crystal structures in the CSD to identify recurring structural features that might indicate a kinetic preference for their formation over energetically comparable alternatives (Section 2.4.3).
2.4.1. Dynamic contributions to the energy
Phase transitions between enantiotropic pairs of polymorphs are evidence that lattice energy modelling is not always sufficient to elucidate the relative stability of putative crystal structures. The thermodynamically favoured polymorph is the structure with the lowest free energy; under normal laboratory conditions of constant pressure, we consider the Gibbs free energy differences, ΔG, between structures:
Temperature, on the other hand, can have an important influence, even at ambient conditions. The difference in the heat capacities (and, thus, thermal contributions to U) between polymorphs is normally very small (Citation 26 ), so the internal energy differences (ΔU) between polymorphs are only weakly temperature dependent. However, the entropy is dominated by contributions from the low-energy lattice vibrations, which are strongly influenced by differences in crystal packing. These low-energy vibrations contribute most to the entropy of the crystal, so that entropy differences between polymorphs can be significant. Gavezzotti and Filippini (Citation 145 ) calculated vibrational contributions to entropy differences between 204 pairs of known polymorphs of organic molecules at T = 300 K. The study found that ΔS can approach 15 J mol−1 K−1 (TΔS ≈ 5 kJ mol−1 at room temperature), but is usually much smaller; ΔS < 6 J mol−1 K−1 (TΔS < 1.8 kJ mol−1 at room temperature) for about 80% of polymorphic pairs (). While the entropy differences do seem generally quite small, TΔS might often be greater than the lattice energy differences between predicted crystal structures (see Figures ).
Figure 6. Calculated differences in intermolecular vibrational entropy between known polymorphs. Data taken from (Citation 145 ).
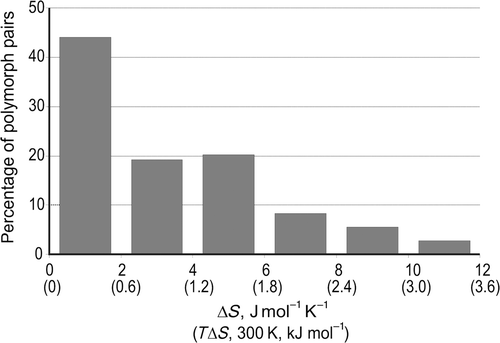
Despite their probable importance, the vibrational contributions to the relative free energies have rarely been considered in CSP studies. One reason for ignoring the vibrational energy is the computational expense – the calculations require an evaluation of the second derivatives (curvature) of the lattice energy surface with respect to atomic or molecular positions and such calculations would have to be performed for each predicted crystal structure. However, with modern computing power, calculating the second derivatives of the lattice energy is no longer a problem, even for the many structures typically generated in crystal structure searches. In fact, the lattice energy second derivatives should be calculated for each low-energy structure to ensure that a proper lattice energy minimum has been located by the energy minimization algorithm (Citation 29 , Citation 40 , Citation 60 , Citation 123 ) – energy minimizing routines sometimes locate saddle points on the lattice energy surface, especially when space group symmetry is imposed during minimization. Such saddle points do not correspond to observable crystal structures and can only be identified through analysis of the second derivatives of the lattice energy.
Another reason for ignoring vibrational energy contributions is that model potentials are often not considered to be accurate enough – if we are not confident in the accuracy of the small lattice energy differences, is it worth the computational expense of correcting for missing thermal effects? Finally, errors in the actual calculated frequencies are a concern. We expect the greatest differences between polymorphs to be in the lattice vibrations, the rigid-body motions of molecules with respect to each other, as opposed to angle bending and bond stretching within the molecules. Due to the weak intermolecular interactions involved, as well as the large masses, the frequencies of lattice vibrations normally fall in the narrow range from about 10 to 150 cm−1. Errors of say 10–20 cm−1, which seem typical when using simple model potentials (Citation 120 , Citation 146 ) are therefore relatively large and could lead to significant uncertainties in the computed vibrational partition function and hence the free energy. However, as the quality of model potentials is increasing, the accuracy of relative lattice energies and calculated frequencies is improving (Citation 31 , Citation 102 , Citation 120 ), and the neglect of vibrational energy contributions is now certainly as important as other errors in the calculations. As such, recent studies have examined the influence of vibrational energy contributions in CSP (Citation 21 , Citation 22 , Citation147–150) – some of these results are discussed in the following sections.
2.4.1.1. Lattice dynamics
The most straightforward approach to modelling the vibrations in crystals is through the methods of lattice dynamics. The theory, which is based on a normal mode analysis of the lattice energy surface, is well-developed for atomic and ionic crystals (Citation 151 ) and is readily extended to crystals of rigid molecules (Citation152–156). Much of the early application of lattice dynamics to molecular organic crystals was summarized by Pertsin and Kitaigorodskii (Citation 26 ) and the method is useful for investigating the pressure–temperature phase diagrams of simple molecular crystals (Citation 26 , Citation157–163).
As with most crystal properties, calculated vibrational frequencies are sensitive to the form and parametrization of the model potential. The motions of molecules in crystals are largely determined by the close contacts between neighbouring molecules, so the model for short range repulsive interactions is crucial; small changes to, say, the exp-6 parameters often result in variations of up to 10% (5–10 cm−1) in calculated frequencies (Citation 120 ). For molecules with polar functional groups, the frequencies of certain motions are also especially dependent on the electrostatic model. Atomic partial charge models often underestimate the energy required for hydrogen bond stretching and bending. A more detailed description of the intermolecular electrostatic interactions (such as atomic multipoles) stabilizes these modes, generally improving the agreement between experimental observations and the calculations. The electrostatic model seems to be even more important for describing vibrations that distort weaker interactions with polar groups or π-electron density in aromatic rings (Citation 120 ). In fact, for even moderately polar molecules, atomic charge models can be inadequate for modelling the vibrations (Citation 164 , Citation 165 ) and a more accurate description is crucial.
Here, we are interested in the contributions to the crystal free energy, which depend on the overall shape of the vibrational mode spectrum and contribute to the free energy through the sum of the frequencies, ω, in the zero-point energy:
2.4.1.2. The influence of vibrational energy contributions in CSP
Vibrational energy contributions to the relative energies of polymorphs have been considered in several studies of CSP (Citation 105 , Citation147–150), so we can examine their influence on the energies of the lists of predicted crystal structures.
The behaviour of hydrocarbon crystal structures is the most easily rationalized one. The crystals’ lattice energies are dominated by the repulsion and dispersion interactions, so that generally the denser structures are lowest in lattice energy, U latt (Citation 147 ). However, lower density structures allow more freedom for molecular motions and this greater freedom is reflected in lower frequency vibrations and greater vibrational entropy. Therefore, ΔS normally favours low density, higher lattice energy crystal structures. There is a resulting balance between the enthalpy and entropy, with increasing importance for TΔS as the temperature is raised; this is supported by the observation that the low-temperature polymorph in an enantiotropic pair is usually the higher density structure (Citation 166 ). Dunitz et al. (Citation 147 ) found that TΔS is often comparable to ΔU latt in the predicted structures of hydrocarbons and, because ΔS usually favours structures with higher U latt, the free energy differences between predicted crystal structures are even smaller than the lattice energy differences – the energy spacing between structures is compressed and the multiple minima problem (see Section 2.3) is compounded.
The entropy effect is less straightforward for molecules that interact through specific, directional interactions (such as the hydrogen bond). By optimizing such interactions, molecules can sacrifice some close packing, so that the structure with the best lattice energy is no longer necessarily one of those with highest density (e.g. see ). Furthermore, the vibrational frequencies are as sensitive to the arrangement of strong interactions as to the crystal density. Now the overall effect of vibrational entropy might not be to compress the predicted structures into a smaller energy range; for the hydrogen-bonding parabanic acid, the overall range in energies of the predicted crystal structures actually expands when the vibrational energy is added (the 18 lowest energy structures cover a range of 6.6 kJ mol−1 in lattice energy and 8.4 kJ mol−1 in free energy (Citation 150 )). For the moderately polar pyridine, the overall range in energy is about the same in lattice energy and free energy (Citation 149 ); shows how the ranking of predicted crystal structures of pyridine changes when vibrational contributions to the free energy are included. For both these rigid molecules, there is a large variation (up to about 2 kJ mol−1) in vibrational contributions to the relative energies and these contributions improve the energies of the real structures relative to the rest of the predictions.
Figure 7. The resulting re-ranking when vibrational energy contributions are added to the lattice energy for the predicted pyridine crystal structures. The positions of the two known polymorphs (Z′ = 4 Form I and Z′ = 1 Form II) are indicated. Data taken from (Citation 149 ).
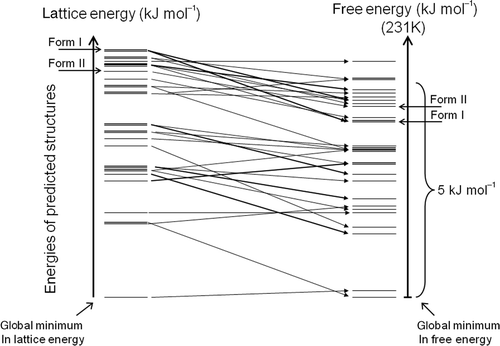
van Eijck (Citation 148 ) reported similar experiences with flexible molecules – the ranking of the real crystal structures of glycol and glycerol were both improved when the vibrational contributions to the free energy were taken into account. In this work, the individual contributions to the total energy were examined in detail, showing that differences in zero-point energy and entropy were equally important to the overall relative energies of predicted structures, each varying by up to about 3 kJ mol−1 amongst the lowest energy predicted crystal structures.
2.4.1.3. Molecular dynamics
While lattice dynamics seems to be a useful step, many molecular crystals are quite close to their melting point at ambient conditions. At these temperatures, there may be wide-amplitude dynamical motions, which could have significant anharmonic character, perhaps even leading to transitions between the possible crystal structures. The basis of lattice dynamics is harmonic and, while anharmonic corrections are possible, a more natural approach to studying such behaviour is MD, where the positions of molecules evolve via Newton's classical equations of motion and therefore naturally include anharmonic motions. While MD calculations are more computationally demanding than lattice dynamics, some MD studies of real (e.g. acetic acid (Citation 132 ), succinic anhydride (Citation 167 ), imidazole, 5-azauracil (Citation 168 )) and predicted (e.g. acetic acid (Citation 169 ), coumarin (Citation 170 ) and fluorouracil (Citation 80 )) molecular organic crystals have been performed.
Not only do MD simulations allow an evaluation of dynamic contributions to the energies of putative crystal structure, but the temperature allows conversion between structures separated by low-energy barriers, as discussed in Section 2.2.2 (Citation 169 ). The sheer number of nearly equi-energetic crystal structures that are found in lattice energy searches suggests a very bumpy potential energy surface. Visual inspection often suggests that many of these predicted structures are closely related and we can guess that some are separated by very small energy barriers (Citation 40 , Citation 149 , Citation 150 , Citation 169 ). As an extreme example, three very similar minima (differing by less than 0.01 Å in cell parameters and by only 0.25 kJ mol−1 in lattice energy) were found in a study of parabanic acid (Citation 150 ). The number of closely related lattice energy minima could be reality or an artefact of the atom–atom approach for describing intermolecular interactions. Whether they are real or artefacts of the method, the energy barriers between these structures are undoubtedly so small that thermal motion would average over the structures. While the lattice energy search does not give any information about the energy barriers between minima, a MD simulation can be used to ‘shake up’ the structures, providing enough thermal energy to allow small energy barriers to be crossed. Mooij and co-workers (Citation 169 ) took this approach in their study of acetic acid and, indeed, found that many of the structures interconvert during a short MD simulation at 200 K – many higher energy structures convert into (already found) lower energy structures, significantly reducing the final number of possible structures. The metadynamics approach to CSP (described in Section 2.2.2) includes this ‘smoothing out’ of the energy surface from the start of the calculations, sampling the free energy instead of the lattice energy surface when generating crystal structures.
2.4.2. Crystal properties and growth
While structure prediction is a considerable task, the structure of a crystal is really just a starting point for understanding or predicting its properties. A few of the many properties of interest are the solubility, colour, mechanical properties (e.g. compressibility or plasticity) and morphology. When CSP is used as part of a screen for unknown polymorphs, we might not only ask if polymorphism is likely, but also whether there would be an important variation in property X amongst possible polymorphs. Variations in properties could be advantageous if the polymorph with a desirable property could be reliably produced, or disastrous if a polymorph with unwanted properties cannot be suppressed. Of course, modelling the properties of putative structures could help in the structure prediction itself; perhaps the most relevant property in this respect is the crystal morphology, which provides insight into crystal growth.
2.4.2.1. Morphology prediction
The morphology of a crystal reflects the relative growth rates of its faces; if the relative growth rates in all possible directions are known, then the morphology can be predicted. There is a wide range of sophistication in predicting these relative growth rates and it would not be possible to provide a thorough review of the subject here. More thorough reviews can be found elsewhere (see, e.g. (Citation 171 ) and references therein). Instead, the most commonly used models are briefly described and some modern simulation methods are mentioned.
The simplest approach to morphology prediction uses a purely geometrical model – the Bravais–Friedel–Donnay–Harker (BFDH; (Citation 172 )) law, which states that the growth rate of each face is inversely proportional to the interplanar distance between layers parallel to that face. Because it only uses the geometry of the crystal, the BFDH model performs best when the strengths of interactions in the crystal are isotropic; most molecular crystals, with their hydrogen bonds and other directional interactions, do not obey this demand.
Hartman and Perdok (Citation 173 ) took the first steps in considering the anisotropy in strengths of interactions. Invoking the idea of strong bonds between growth units (typically, the individual molecules), they defined three types of crystal faces – flat (F), stepped (S) and kinked (K), with strong bonds in two directions (F), one direction (S) and no directions (K). In general, dominant crystal faces are F-faces. The strongest interactions between growth units are usually collected together in what is called a crystal graph and periodic chains of these interactions are called periodic bond chains (PBCs); stable F-faces are identified by searching for non-parallel, intersecting PBCs or ‘connected nets’. Only the growth rates of these F-faces need normally be considered for the crystal morphology. The most popular approach to predicting the relative growth rates is the attachment energy model – the attachment energy, E att, is defined as the energy per molecule released when a growth slice is attached to the crystal face (Citation 173 , Citation 174 ). A crystal face's growth rate always increases as the attachment energy increases, although the exact relationship depends on many factors. Variations with supersaturation, temperature, solvent effect and growth mechanism are ignored in the simple attachment energy model and the growth rates of all crystal faces are assumed to be linearly proportional to their attachment energies. Therefore, prediction starts with a search for F-faces, attachment energies are calculated for each of these faces and these define the morphology.
Such attachment energy calculations have been proposed as a way of distinguishing between nearly equi-energetic structures from lattice energy searches (Citation 40 ). As a very approximate consideration of kinetic differences between structures, the comparison of relative growth rates of different faces of a crystal is extended to the relative growth rates of faces on different, competing crystals. Initial studies have shown some promise for the attachment energy model in distinguishing real from unobserved crystal structures – predicted relative growth rates (as judged by the attachment energies to the morphologically dominant faces (Citation 40 , Citation 149 , Citation 150 , Citation 175 ) or the total integrated volume of the predicted morphology (Citation 102 , Citation 175 ) of the known structures of paracetamol, pyridine and parabanic acid are amongst the highest in all of the low-energy predicted crystal structures.
Important steps forward are being made towards more realistic models of crystal growth, such as Monte Carlo simulations which can incorporate growth mechanism, temperature and supersaturation (Citation 171 , Citation 176 , Citation 177 ); as methodologies of modelling growth of crystal faces become more advanced, they will surely play an increasingly important role in the ab initio predictive process from molecules to crystals to materials properties.
2.4.3. Prediction from known structures
An alternative to calculating physical properties to distinguish between the low-energy predicted crystal structures of a molecule is to make use of comparisons to the wealth of information in the CSD to highlight the most promising structures. Although crystal structures themselves provide no information on the kinetics of nucleation and growth, they are the end result of the crystallization process and so common patterns or interactions in the database may reflect thermodynamic as well as kinetic preferences. There are now over half a million crystal structures in the CSD and these must encode useful information for CSP. The database of known structures should be able to play an important role in the prediction of crystal structures, just as homology modelling is advancing predictive power in protein structure modelling (see Dunitz and Scheraga's (Citation 178 ) discussion of the similarities between protein and CSP).
First, we should distinguish between objective use of the CSD and subjective attempts to assess computer-generated structures using crystallographic experience. One might expect that, to the eye of an experienced chemical crystallographer, some calculated crystal structures might look less good than others. If so, it might be possible to select the true crystal structure(s) from a list of possibilities by visual inspection. A recent study asked a large group of attendees at the 2005 congress of the IUCr to visually assess sets of predicted structures of two small molecules and select the structure that they considered to be most likely. The surprising result was that, for both molecules, the observed crystal structure received the least votes (Citation 179 ). Clearly, for these molecules the unobserved low-energy predictions look just as plausible as the observed structures. Definite conclusions cannot be made from such a small test. However, it seems that a subjective assessment of computer-generated crystal structures is an unreliable method of assessing which of the low-energy possibilities will be observed; as long as a good quality model has been used during lattice energy minimization, all low-energy crystal structures will look reasonable. Therefore, assessment of predicted structures based on what has been seen before should rely on rigorous use of the CSD rather than simple intuition.
Information from the CSD has been used both in the initial generation of crystal structures and in the final choice of the most likely crystal structure of a molecule. Motherwell's (Citation 76 ) genetic algorithm search method, which scores structures by comparison to distributions of inter-atomic separations, has already been described. Hofmann (Citation 83 , Citation 84 ) used data mining of the CSD to obtain atom–atom potentials that can discriminate between real crystal structures and other plausible, but unobserved (wrong) structures. No a priori decision is made about the form of these trained potentials, but comparison to empirically derived models show that the data mining process does result in physically meaningful models. These trained potentials are used in Hofmann's (Citation 82 , Citation 83 ) FlexCryst program for CSP.
Sarma and Desiraju (Citation 180 ) suggest re-ranking the results of a lattice energy search by consideration of the crystal structures of similar molecules – chosen to have the same chemical functionality as the target molecule. In applying their method to one of the molecules in the 2001 CSP blind test (molecule IV, see ), they started with lists of the lowest energy crystal structures from Polymorph Predictor simulated annealing calculations using simple atom–atom model potentials. The known crystal structures of a set of 10 similar molecules were then unbuilt, the molecular structure exchanged for that of the blind test molecule and the resulting crystal structures were lattice energy minimized. The 10 resulting hypothetical crystal structures for the molecule were then compared to the Polymorph Predictor lists and those with good rankings in the lists were considered as the most probable crystal structures. One major choice for this particular molecule was whether it would crystallize with hydrogen bond dimers or chains. Of Sarma and Desiraju's (Citation 180 ) hypothetical structures with good lattice energy ranking, all but one formed hydrogen bond chains, which they proposed as the most likely hydrogen bond motif for molecule IV. They chose as their first prediction one of their hypothetical structures that was quite well-ranked on lattice energy (third using one of the force fields they used) and was derived from a highly similar molecule (with similarity based on molecular volume and surface area). This chosen crystal structure did, in fact, correspond to the observed crystal structure for molecule IV. Overall, the approach seems useful, but only when reliable intermolecular interactions are present and the crystal structures of enough similar molecules can be found in the CSD. This approach has been further tested, with mixed success (Citation 181 , Citation 182 ). Such comparisons will benefit from a rapidly expanding database of crystal structures.
Table 1. Diagrams and reference numbers of the target molecules in the CSP blind tests (Citation20–23).
Supplementing the physical approach (energy-based prediction methodologies) by comparisons to the CSD can offer short- and long-term benefits to crystal structure modelling. In the short term, the trends and patterns in the CSD have the potential to highlight the most likely candidates from the typically crowded list of lattice energy ranked putative crystal structures. In the longer term, these observations could guide developments of the physical approach. Take Sarma and Desiraju's (Citation 180 ) example of blind test molecule IV: their hypothetical structures derived from the CSD point to a preference for the hydrogen bond chain motif. Lattice energy predictions, on the other hand, consistently produced many possible crystal structures based on hydrogen bond dimers with as favourable, and often better, lattice energies (Citation 21 ). The fact that CSD-derived hypothetical structures show a preference for hydrogen bond chains suggests that the database holds some information that was not present in the lattice energy calculations. It is the nature of this information that is the question. Do the CSD-derived structures simply tell us that our model potentials are inadequate, that dynamical contributions to the energy are important, or that there is a kinetic preference for the formation of hydrogen bond chains during nucleation or crystal growth that defines the pattern of interactions in the final crystal structure? To discover the nature of the information that the CSD has provided, and so further our understanding of crystallization, the computational methods must be developed – improved energy models and dynamic modelling of the crystal, its nucleation and growth. In the short term, the CSD can be used for empirical guidance; in the long term, it should direct and motivate the development of simulation methodologies.
3. The blind tests of CSP
The ‘blind tests’ of CSP have been organized every 2–3 years since 1999 and hosted by the Cambridge Crystallographic Data Centre as an objective evaluation of current methods in CSP. The results of the four blind tests (Citation20–23) completed to date are summarized here.
Many of the active researchers in CSP were invited to test their methodologies in the blind tests; the number of participating research groups grew from 11 in 1999 to 17 in 2001, and 18 in 2004, and dropped slightly to 14 in 2007. In these international collaborations, the molecular diagrams of a set of target molecules () were circulated to the participants, while the recently determined crystal structures were withheld by an independent referee. Predicted crystal structures had to be submitted within 6 months of announcing the targets and each participant was allowed three predicted crystal structures for each molecule. Three categories of molecule have been used in each of the blind tests: small and rigid with only the elements C, N, O and H (category 1); rigid with more challenging elements or functional groups (category 2); and moderately flexible (category 3). In the first two blind tests (CSP1999 and CSP2001), the crystals were guaranteed to be in a ‘common’ space group with Z′ ≤ 1, while no restriction was placed on space group in the third round (CSP2004), except that Z′ ≤ 2. The rules were adjusted again in CSP2007 (Citation 23 ) so that the molecules in categories 1–3 were guaranteed to adopt a crystal structure with Z′ ≤ 1 (but with no restriction on space group). Instead, a new category was introduced specifically in CSP2007 to test predictions for crystal structures with more than one molecule in the asymmetric unit: in this year, a 1:1 cocrystal was issued as the challenge (target XV, ).
In addition to the three ‘official’ predictions that were allowed for each molecule, extended lists of predicted structures (about the 100 best structures from each participant) were analysed in CSP2004 and the authors reported enough information in the CSP2001 publication (Citation 21 ) to determine the success rate of the crystal structure generation step in each participant's method (). For the rigid molecules in categories 1 and 2, the structure generation was usually successful: 70–80% of participants generated the correct structure somewhere in their list for molecules IV, V, VIII and XII. The success of the crystal structure generation methods for molecules IX and XIII in category 2 was slightly lower and the problems with these molecules probably relate to shortcomings in modelling the intermolecular interactions involving halogen atoms. The problem was worst for the iodine-containing molecule IX, for which a spherical representation of the iodine atoms is particularly inadequate (Citation 22 ).
Table 2. Summary of results of the first four blind tests of CSP (Citation20–23). Statistics on the success of predictions are presented for each molecule and the molecules from the four blind tests are grouped by category.
Generating the observed crystal structure of molecule XI was problematic because the observed structure of this molecule contains two molecules in the asymmetric unit. Due to time constraints (this molecule was introduced half-way through the course of CSP2004 (Citation 22 )) or limitations of certain methodologies, the methods used by only eight of the 18 participants who attempted predictions could have generated crystal structures with the symmetry of the observed crystal (P21/c, Z′ = 2). Only four of these eight did have the correct crystal structure somewhere in their list. With the six extra degrees of freedom that must be sampled for the second molecule in the asymmetric unit, the dimensionality of the search space is clearly a problem for the crystal structure generation step in CSP. Considering that over 10% of homomolecular organic crystals have more than one molecule in the asymmetric unit (Citation 52 ), this is a serious problem and the robustness of the search methods must be improved. CSP for crystal structures with more than one molecule in the asymmetric unit is also important if the methods are to be applied to multicomponent crystal structures: salts, cocrystals and solvates. The success rate of crystal structure generation for the one cocrystal (XV) that has been included in a blind test was, again, low: only five of 12 participants who attempted CSP on this target generated the known structure. For this cocrystal, the difficulty of generating structures with two molecules in the asymmetric unit was compounded by having to consider two conformations of the acid.
For the flexible category three molecules in CSP2001 (VI) and CSP2004 (X), the success rate for generating the observed crystal structure somewhere in the extended list of predicted structures was below 50%. This failure to even generate the observed crystal structure guaranteed the failure of the prediction calculations for the majority of participants. A marked improvement was observed in CSP2007, when nine of 12 participants who attempted predictions on the flexible molecule XIV did generate the observed crystal structure. This encouraging improvement in search methods for the flexible molecule category probably reflects improvements in the search methodologies for flexible molecules. However, the calculations performed during the blind tests also revealed that the flexibility of the molecular structure of XIV was less important than for the molecules representing this category in the previous blind tests (Citation 23 ). This observation emphasizes the important point that it is not possible to guarantee the same level of difficulty from one blind test to another, because the difficulties involved in predicting crystal structures of a given molecule are often only apparent once the calculations have been performed. Therefore, while the blind tests are important, they cannot be considered a perfect measure of progress in the field of CSP.
The overall results of the first three tests demonstrated that prediction is possible under blind test conditions – while the overall success rate was low, eight of the 12 crystal structures were successfully predicted amongst the 3 allowed predictions by at least one participant. There were a few successes for the ‘easiest’ category of molecule, but no method was consistently successful (Citation20–22). The challenging functional groups in the category 2 set of rigid molecules (II, V and IX) lowered the success rate slightly, while the worst news is reserved for those interested in conformationally flexible molecules (III, VI and X). Only one successful prediction was made of a flexible molecule's crystal structure in these three blind tests.
The results of the fourth blind test demonstrate encouraging improved success rates (Citation 23 ). In total, the 14 participating research groups achieved 13 successful predictions spread across the four targets, 10 of which were submitted as the participant's first choice. These correct predictions included three for the flexible molecule and two for the cocrystal. Furthermore, the successes were not restricted to a small number of groups: seven of the 14 participants correctly predicted at least one of the structures, four of whom had multiple successes in CSP2007. One important observation is that five of the participating groups located the known crystal structures of all four CSP2007 targets somewhere in their lists of predictions. The methods employed by these groups were variations on random sampling of the structural variables (four groups) and Monte Carlo simulated annealing (one group) and their consistency of success is an indication that those methods are reliable for structure generation.
The other main observation from the CSP2007 blind test was that the method used by one participating group successfully predicted all four crystal structures, each as the number 1 (lowest energy) structure (Citation 183 ). These calculations used the empirically corrected DFT + D method parametrized by Neumann and Perrin (Citation 134 ) to lattice energy minimize and rank the lowest energy crystal structures for each blind test target. While the computational cost of their calculations was much greater than the other participants, the results are nonetheless impressive and demonstrate that the crystal structures of the relatively small molecules that have been studied in the blind tests are predictable by global lattice energy minimization methods. The focus of the most successful groups in CSP2007 has been on improving the description of the lattice energy surface, usually without including other contributions to the free energies of the crystal structures or any consideration of the crystal growth conditions. In these cases, it seems that the molecules find the global energy minimum structure during crystallization and that the global lattice energy minimum for each target corresponds to the free energy minimum.
The results of all four blind tests are discussed in detail in the four main publications (Citation20–23), and these studies provide a wealth of useful comparative information of the various methods employed for CSP. The blind tests should be continued as a means of objectively monitoring the progress that is made in developing methods for CSP and the fifth blind test is currently underway. Reflecting the progress that has been made in methods for CSP, the targets of the fifth blind test include a molecular salt, a hydrate of a flexible molecule and a larger, much more flexible molecule than has previously been set as a target ().
Figure 8. The targets of the fifth blind test of CSP.
4. Concluding remarks and the future
In two and a half decades of increasing interest and effort, there have been considerable advances in methodology for the prediction of crystal structures from molecular information alone. The requirements for this goal bring together many challenges for molecular modelling: structure searching and global minimization methodologies; data mining of the CSD; development of intermolecular model potentials, force fields for molecular flexibility and first principles electronic structure mechanical methods; and development of dynamical simulations.
An arsenal of methods has been developed for generating all of the possibilities for the crystal packing of a molecule. At least for small molecules, this will surely soon be a solved problem – some methods already seem consistently successful (Citation 22 , Citation 23 ). As these search methods have been coupled to the significant progress in modelling the static energies of the crystal structures of rigid molecules, we are nearing a true representation of the energy landscape for crystals of small molecules. At least, we are piecing together a highly accurate and comprehensive static picture, where the relative depths of the valleys in the surface, and their corresponding structure, are known. A limiting factor now is our static representation of crystals, ignoring the vibrations of molecules about their equilibrium positions and the corresponding contributions to the energy and entropy. In the few cases where such dynamic considerations have been included in the thermodynamic model for CSP, generally improved results have been reported (i.e. known crystal structures are more reliably found at, or closer to, the global free energy minimum).
There has been recent progress in methods that can be applied to CSP of flexible molecules. Judging from the blind test results and the limited number of published studies, structure generation for flexible molecules seems to be less reliable than for rigid molecules and is hampered by the need to adequately sample all crystal packing possibilities of all relevant conformations. The need to simultaneously model intra- and intermolecular energies to the requisite accuracy for CSP (where relative energies are sometimes measured in fractions of a kJ mol−1) is a further challenge, but promising results have been reported using accurate atom–atom intermolecular models combined with quantum mechanical molecular energy calculations.
4.1. Requirements for progress in CSP and future prospects
4.1.1. Structure searching
To become a generally useful tool in crystal engineering and industry, it is necessary to be able to generate all possible crystal structures for rigid and flexible molecules, even in the less common space groups and with multiple independent molecules – salts, cocrystals, and solvates are of obvious interest and even pure (neat) crystals have Z′ > 1 often enough to be a worry. The algorithms for generating structures are very advanced, so considering these complex systems is now largely a matter of computing power – the parallelization of programs for crystal structure generation and the use of distributed computing now allows complete sets of structures to be compiled when an accurate enough energy model is available. (The last point is crucially important for flexible molecules; poor force fields can undermine powerful search methods.)
4.1.2. The thermodynamic model
The continued development of energy models is necessary for refinement of our thermodynamic model. Several promising approaches are being pursued: non-empirically derived, molecule-specific anisotropic atom–atom model potentials and SCDS–Pixel or quantum mechanical solid-state electronic structure calculations as alternatives to the atom–atom approach.
To make progress with flexible molecules, the increasingly accurate intermolecular models are being combined with better models for molecular flexibility than are currently provided by molecular mechanics methods. The use of quantum mechanically calculated conformational energies along with anisotropic atom–atom models of intermolecular interactions has yielded promising results. A similar route has been envisioned by Gavezzotti (Citation 184 ) for the coupling of SCDS–Pixel and quantum mechanically calculated conformational energies. One major challenge here is the scalability of the calculations to larger molecules, where molecular quantum mechanical calculations of a sufficiently high level for reliable conformational energies dominate the overall computing time needed for CSP.
Inadequacies in the treatment of the long-range dispersion energy mean that periodic DFT for molecular organic crystals currently relies on empirically derived corrections. The development of improved solid-state quantum mechanical methods is a very active area of development and increasingly available computing power at relatively low cost makes such calculations feasible for CSP.
Dynamical simulations of molecules in crystals are a requirement for real thermodynamics and an understanding of the role of temperature and entropy, as well as low-energy transitions between structures. While most studies still focus on static lattice energies rather than free energies, methods of including the effects of dynamics in crystals should develop an increasingly prominent role in modern CSP methodologies.
4.1.3. Kinetics
As we have known all along, the thermodynamic model is only a starting point – a reliable method of CSP will need to reflect all factors that influence the crystallization of a molecule. We do need to be modelling the kinetics of crystal growth and nucleation. Simulations of crystal growth are advancing in sophistication, while the nucleation of molecular crystals is still a great, largely unexplored, challenge. Very simple considerations of crystal growth can give an initial indication of the relative growth rates of competing crystal structures and, until (if ever) nucleation and growth kinetics can be fully understood by molecular simulation, data mining of the CSD could help guide the final selection of the most likely structures from the crowded lists of low-energy crystal structures.
The goal of CSP has required a significant level of development and careful validation of methods. While there is still much needed in terms of development of methods, the methods have reached a good level of reliability, at least for reasonably small molecules, to be a useful complementary method to experimental polymorph screening and possibly to help guide crystal engineering experiments for the design and discovery of new molecular materials.
Acknowledgements
There are many to thank for their contributions to the development of the field of CSP. Personally, I thank Professor William Jones, Dr W.D. Sam Motherwell and Dr Neil Feeder for many fruitful discussions and Professor Sally L. Price for introducing me to this fascinating and challenging research problem, as well as for passing on some of her vast experience in modelling intermolecular forces. I also thank the Royal Society for funding my own research via a University Research Fellowship and the Cambridge Crystallographic Data Centre for its hosting of the CSP blind tests.
Notes
Note
1. Rice and Sorescu (Citation 30 ) presented a similar study of CSP for a large set of molecules with somewhat different results. Of the experimentally observed crystal structures 64% (111 of 174) were located as the lowest energy structure in the list of computer-generated structures. The very high success rate in this study could be due to an incomplete sampling of packing space (only 85% of observed structures were located at all) and the fact that rigid molecular models were taken directly from the experimentally determined crystal structures. This choice of molecular model, rather than an optimized molecular geometry, will bias the lattice energy ranking towards the observed crystal structures.
Related Research Data
References
- Chemburkar , SR , Bauer , J , Deming , K , Spiwek , H , Patel , K , Morris , J , Henry , R , Spanton , S , Dziki , W , Porter , W , Quick , J , Bauer , P , Donaubauer , J , Narayanan , BA , Soldani , M , Riley , D and McFarland , K . 2000 . Dealing with the Impact of Ritonavir Polymorphs on the Late Stages of Bulk Drug Process Development . Org. Process Res. Dev. , 4 : 413 – 417 .
- Bauer , J , Spanton , S , Henry , R , Quick , J , Dziki , W , Porter , W and Morris , J . 2001 . Ritonavir: An Extraordinary Example of Conformational Polymorphism . Pharm. Res. , 18 : 859 – 866 .
- Beyer , T , Lewis , T and Price , SL . 2001 . Which Organic Crystal Structures are Predictable by Lattice Energy Minimisation? . CrystEngComm. , 3 : 178 – 212 .
- Price , SL . 2004 . The Computational Prediction of Pharmaceutical Crystal Structures and Polymorphism . Adv. Drug Deliv. Rev. , 56 : 301 – 319 .
- Day , GM , Motherwell , WDS and Jones , W . 2007 . A Strategy for Predicting the Crystal Structures of Flexible Molecules: The Polymorphism of Phenobarbital . Phys. Chem. Chem. Phys. , 9 : 1693 – 1704 .
- Ouvrard , C and Price , SL . 2004 . Toward Crystal Structure Prediction for Conformationally Flexible Molecules: The Headaches Illustrated by Aspirin . Cryst. Growth Des. , 4 : 1119 – 1127 .
- Antoniadis , CD , D’Oria , E , Karamertzanis , PG , Tocher , DA , Florence , AJ , Price , SL and Jones , AG . 2010 . A Computationally Inspired Investigation of the Solid Forms of (R)-1-phenylethylammonium-(S)-2-phenylbutyrate . Chirality , 22 : 447 – 455 .
- D’Oria , E , Karamertzanis , PG and Price , SL . 2010 . Spontaneous Resolution of Enantiomers by Crystallization: Insights from Computed Crystal Energy Landscapes . Cryst. Growth Des. , 10 : 1749 – 1756 .
- Gourlay , MD , Kendrick , J and Leusen , FJJ . 2007 . Rationalization of Racemate Resolution: Predicting Spontaneous Resolution through Crystal Structure Prediction . Crys. Growth Des. , 7 : 56 – 63 .
- Leusen , FJJ . 2003 . Crystal Structure Prediction of Diastereomeric Salts: A Step toward Rationalization of Racemate Resolution . Cryst. Growth Des. , 3 : 189 – 192 .
- Görbitz , CH , Dalhus , B and Day , GM . 2010 . Pseudoracemic Amino Acid Complexes: Blind Predictions for Flexible Two-component Crystals . Phys. Chem. Chem. Phys. , 12 : 8466 – 8477 .
- Karamertzanis , PG , Kazantsev , AV , Issa , N , Welch , GWA , Adjiman , CS , Pantelides , CC and Price , SL . 2009 . Can the Formation of Pharmaceutical Cocrystals be Computationally Predicted? II. Crystal Structure Prediction . J. Chem. Theory Comput. , 5 : 1432 – 1448 .
- Cruz Cabeza , AJ , Day , GM , Motherwell , WDS and Jones , W . 2006 . Prediction and Observation of Isostructurality Induced by Solvent Incorporation in Multicomponent Crystals . J. Am. Chem. Soc. , 128 : 14466 – 14467 .
- van Eijck , BP and Kroon , J . 2000 . Structure Predictions Allowing more than One Molecule in the Asymmetric Unit . Acta Crystallogr., Sect. B , B56 : 535 – 542 .
- Hulme , AT and Price , SL . 2007 . Toward the Prediction of Organic Hydrate Crystal Structures . J. Chem. Theory Comput. , 3 : 1597 – 1608 .
- Cruz-Cabeza , AJ , Day , GM and Jones , W . 2008 . Towards Prediction of Stoichiometry in Crystalline Multicomponent Complexes . Chem. Eur. J. , 14 : 8830 – 8836 .
- Cruz-Cabeza , AJ , Karki , S , Fabian , L , Friscic , T , Day , GM and Jones , W . 2010 . Predicting Stoichiometry and Structure of Solvates . Chem. Commun. , 46 : 2224 – 2226 .
- Maddox , J . 1988 . Crystals from First Principles . Nature , 335 : 201
- Gavezzotti , A . 1994 . Are Crystal Structures Predictable? . Acc. Chem. Res. , 27 : 309 – 314 .
- Lommerse , JPM , Motherwell , WDS , Ammon , HL , Dunitz , JD , Gavezzotti , A , Hofmann , DWM , Leusen , FJJ , Mooij , WTM , Price , SL , Schweizer , B , Schmidt , MU , van Eijck , BP , Verwer , P and Williams , DE . 2000 . A Test of Crystal Structure Prediction of Small Organic Molecules . Acta Crystallogr., Sect. B , B56 : 697 – 714 .
- Motherwell , WDS , Ammon , HL , Dunitz , JD , Dzyabchenko , A , Erk , P , Gavezzotti , A , Hofmann , DWM , Leusen , FJJ , Lommerse , JPM , Mooij , WTM , Price , SL , Scheraga , H , Schweizer , B , Schmidt , MU , van Eijck , BP , Verwer , P and Williams , DE . 2002 . Crystal Structure Prediction of Small Organic Molecules: A Second Blind Test . Acta Crystallogr., Sect. B , B58 : 647 – 661 .
- Day , GM , Motherwell , WDS , Ammon , HL , Boerrigter , SXM , Della Valle , RG , Venuti , E , Dzyabchenko , A , Dunitz , JD , Schweizer , B , van Eijck , BP , Erk , P , Facelli , JC , Bazterra , VE , Ferraro , MB , Hofmann , DWM , Leusen , FJJ , Liang , C , Pantelides , CC , Karamertzanis , PG , Price , SL , Lewis , TC , Nowell , H , Torrisi , A , Scheraga , HA , Arnautova , YA , Schmidt , MU and Verwer , P . 2005 . A Third Blind Test of Crystal Structure Prediction . Acta Crystallogr., Sect. B , B61 : 511 – 527 .
- Day , GM , Cooper , TG , Cruz-Cabeza , AJ , Hejczyk , KE , Ammon , HL , Boerrigter , SXM , Tan , JS , Valle , RGD , Venuti , E , Jose , J , Gadre , SR , Desiraju , GR , Thakur , TS , van Eijck , BP , Facelli , JC , Bazterra , VE , Ferraro , MB , Hofmann , DWM , Neumann , MA , Leusen , FJJ , Kendrick , J , Price , SL , Misquitta , AJ , Karamertzanis , PG , Welch , GWA , Scheraga , HA , Arnautova , YA , Schmidt , MU , Streek , Jvd , Wolf , AK and Schweizer , B . 2009 . Significant Progress in Predicting the Crystal Structures of Small Organic Molecules – A Report on the Fourth Blind Test . Acta Crystallogr., Sect B , B65 : 107 – 125 .
- Kitaigorodskii , AI . 1973 . Molecular Crystals and Molecules , Vol. 29 , New York and London : Academic Press .
- Kitaigorodskii , AI . 1961 . Organic Chemical Crystallography , New York : Consultants Bureau .
- Pertsin , AJ and Kitaigorodskii , AI . 1987 . The Atom-Atom Potential Method: Applications to Organic Molecular Solids , Vol. 43 , Berlin : Springer-Verlag .
- Verwer , P and Leusen , FJJ . 1998 . Computer Simulation to Predict Possible Crystal Polymorphs . Rev. Comput. Chem. , 12 : 327 – 365 .
- Price , SL . 2004 . Quantifying Intermolecular Interactions and their Use in Computational Crystal Structure Prediction . CrystEngComm , 6 : 344 – 353 .
- Day , GM , Chisholm , J , Shan , N , Motherwell , WDS and Jones , W . 2004 . An Assessment of Lattice Energy Minimisation for the Prediction of Molecular Organic Crystals Structures . Cryst. Growth Des. , 4 : 1327 – 1340 .
- Rice , BM and Sorescu , DC . 2004 . Assessing a Generalized CHNO Intermolecular Potential through Ab Initio Crystal Structure Prediction . J. Phys. Chem. B , 108 : 17730 – 17739 .
- Day , GM , Motherwell , WDS and Jones , W . 2005 . Beyond the Isotropic Atom Model in Crystal Structure Prediction of Rigid Molecules: Atomic Multipoles Versus Point Charges . Cryst. Growth Des. , 5 : 1023 – 1033 .
- Coombes , DS , Price , SL , Willock , DJ and Leslie , M . 1996 . Role of Electrostatic Interactions in Determining the Crystal Structures of Polar Organic Molecules. A Distributed Multipole Study . J. Phys. Chem. , 100 : 7352 – 7360 .
- Warshel , A , Huler , E , Rabinovich , D and Shakked , Z . 1974 . Examination of Intramolecular Potential Surfaces of Flexible Conjugated Molecules by Calculation of Crystal Structures. Equilibrium Geometries of Chalcones and Diphenyloctatetraene in the Crystal and Gaseous State . J. Mol. Struct. , 23 : 175 – 191 .
- Colapietro , M , Domenicano , A , Portalone , G , Schultz , G and Hargittai , I . 1987 . Molecular Structure of p-Diaminobenzene in the Gaseous Phase and in the Crystal . J. Phys. Chem. , 91 : 1728 – 1737 .
- Harris , NJ and Lammertsma , K . 1997 . Ab Initio Density Functional Computations of Conformations and Bond Dissociation Energies for Hexahydro-1,3,5-trinitro-1,3,5-triazine . J. Am. Chem. Soc. , 119 : 6583 – 6589 .
- Riehn , C , Degen , A , Weichert , A , Bolte , M , Egert , E , Brutschy , B , Tarakeshwar , P and Kim , KS . 2000 . The Molecular Structure of para-Cyclohexylaniline. Comparison of Results Obtained by X-ray Diffraction with Gas Phase Laser Experiments and Ab Initio Calculations . J. Phys. Chem. A , 104 : 11593 – 11600 .
- Allen , FH , Harris , SE and Taylor , R . 1996 . Comparison of Conformer Distributions in the Crystalline State with Conformational Energies Calculated by Ab Initio Techniques . J. Comput. Aided Mol. Des. , 10 : 247 – 254 .
- Allen , FH . 2002 . The Cambridge Structural Database: A Quarter of a Million Crystal Structures and Rising . Acta Crystallogr., Sect. B , B58 : 380 – 388 .
- Beyer , T and Price , SL . 2000 . The Errors in Lattice Energy Minimisation Studies: Sensitivity to Experimental Variations in the Molecular Structure of Paracetamol . CrystEngComm , 2 : 183 – 190 .
- Beyer , T , Day , GM and Price , SL . 2001 . The Prediction, Morphology and Mechanical Properties of the Polymorphs of Paracetamol . J. Am. Chem. Soc. , 123 : 5086 – 5094 .
- Almenningen , A , Bastiansen , O , Fernholt , L , Cyvin , BN , Cyvin , SJ and Samdal , S . 1985 . Structure and Barrier of Internal Rotation of Biphenyl Derivatives in the Gaseous State: Part 1. The Molecular Structure and Normal Coordinate Analysis of Normal Biphenyl and Perdeuterated Biphenyl . J. Mol. Struct. , 128 : 59 – 76 .
- Baudour , JL and Sanquer , M . 1983 . Structural Phase Transition in Polyphenyls. VIII. The Modulated Structure of Phase III of Biphenyl (T ≈ 20 K) from Neutron Diffraction Data . Acta Crystallogr., Sect. B , B39 : 75 – 84 .
- Casalone , G , Mariani , C , Mugnoli , A and Simonetta , M . 1968 . The Molecular Structure of Biphenyl in the Gas and Solid Phases . Mol. Phys. , 15 : 339 – 348 .
- Busing , WR . 1983 . Modeling the Phase Change in Crystalline Biphenyl by using a Temperature-dependent Potential . Acta Crystallogr., Sect. A , A39 : 340 – 347 .
- Dzyabchenko , A and Scheraga , HA . 2004 . Model for the Crystal Packing and Conformational Changes of Biphenyl in Incommensurate Phase Transitions . Acta Crystallogr., Sect. B , B60 : 228 – 237 .
- Dzyabchenko , AV . 1984 . Theoretical Structures of Crystalline Benzene: The Search for a Global Minimum of the Lattice Energy in Four Space Groups . J. Struct. Chem. , 25 : 416 – 420 .
- Gavezzotti , A . 1991 . Generation of Possible Crystal Structures from Molecular Structure for Low-polarity, Organic Compounds . J. Am. Chem. Soc. , 113 : 4622 – 4629 .
- Gdanitz , RJ . 1992 . Prediction of Molecular Crystal Structures by Monte Carlo Simulated Annealing without Reference to Diffraction Data . Chem. Phys. Lett. , 190 : 391 – 396 .
- Holden , JR , Du , ZY and Ammon , HL . 1993 . Prediction of Possible Crystal Structures for C–, H–, N–, O–, and F-containing Organic Compounds . J. Comput. Chem. , 14 : 422 – 437 .
- Dzyabchenko , AV , Agafonov , V and Davydov , VA . 1999 . A Theoretical Study of the Pressure-induced Dimerization of C60 Fullerene . J. Phys. Chem. A , 103 : 2812 – 2820 .
- Brock , CP and Dunitz , JD . 1994 . Towards a Grammar of Crystal Packing . Chem. Mater. , 6 : 1118 – 1127 .
- Steiner , T . 2000 . Frequency of Z′ Values in Organic and Organometallic Crystal Structures . Acta Crystallogr., Sect. B , B56 : 673 – 676 .
- Spek , AL . 1980–2010 . PLATON , The Netherlands : Utrecht University . http://www.cryst.chem.uu.nl/platon (accessed 29 June 2010)
- Accelrys Inc., Cerius2, San Diego, USA, 2001
- Schmidt , MU and Englert , U . 1996 . Prediction of Crystal Structures . J. Chem. Soc., Dalton Trans. , : 2077 – 2082 .
- Schmidt, M.U.; Kalkhof, H. CRYSCA, Program for Crystal Structure Calculations of Flexible Molecules, Clariant GmbH, Frankfurt am Main, Germany, 1997
- Williams , DE . 1996 . Ab Initio Molecular Packing Analysis . Acta Crystallogr., Sect. A , A52 : 326 – 328 .
- Dzyabchenko , AV . 2004 . PMC , Moscow, , Russia : Karpov Institute of Physical Chemistry .
- van Eijck , BP and Kroon , J . 1999 . UPACK Program Package for Crystal Structure Prediction: Force Fields and Crystal Structure Generation for Small Carbohydrate Molecules . J. Comput. Chem. , 20 : 799 – 812 .
- Della Valle , RG , Venuti , E , Brillante , A and Girlando , A . 2003 . Inherent Structures of Crystalline Pentacene . J. Chem. Phys. , 118 : 807 – 815 .
- Karamertzanis , PG and Pantelides , CC . 2005 . Ab Initio Crystal Structure Prediction – I. Rigid Molecules . J. Comput. Chem. , 26 : 304 – 324 .
- Sobol’ , IM . 1967 . On the Distribution of Points in a Cube and the Approximate Evaluation of Integrals . Comput. Math. Math. Phys. , 7 : 86 – 112 .
- Accelrys Inc., Materials Studio, San Diego, USA
- Karfunkel , HR and Gdanitz , RJ . 1992 . Ab Initio Prediction of Possible Crystal Structures for General Organic Molecules . J. Comput. Chem. , 13 : 1171 – 1183 .
- Karfunkel , HR , Leusen , FJJ and Gdanitz , RJ . 1993 . The Ab Initio Prediction of yet Unknown Molecular Crystal Structures by Solving the Crystal Packing Problem . J. Comput. Aided Mater. Des. , 1 : 177 – 185 .
- Metropolis , N , Rosenbluth , AW , Rosenbluth , MN , Teller , AH and Teller , E . 1953 . Equation of State Calculation by Fast Computing Machines . J. Chem. Phys. , 21 : 1087 – 1092 .
- Oswald , IDH , Allan , DR , Day , GM , Motherwell , WDS and Parsons , S . 2004 . Realising Predicted Crystal Structures at Extreme Conditions: The Low Temperature and High-pressure Crystal Structures of 2-Chlorophenol and 4-Fluorophenol . Cryst. Growth Des. , 5 : 1055 – 1071 .
- Pillardy , J , Czaplewski , C , Wedemeyer , WJ and Scheraga , HA . 2000 . Conformation-Family Monte Carlo (CFMC): An Efficient Computational Method for Identifying the Low-energy States of a Macromolecule . Helv. Chim. Acta , 83 : 2214 – 2230 .
- Pillardy , J , Arnautova , YA , Czaplewski , C , Gibson , KD and Scheraga , HA . 2001 . Conformation-Family Monte Carlo: A New Method for Crystal Structure Prediction . Proc. Natl Acad. Sci. USA , 98 : 12351 – 12356 .
- Wawak , RJ , Pillardy , J , Liwo , A , Gibson , KD and Scheraga , HA . 1998 . Diffusion Equation and Distance Scaling Methods of Global Optimization; Applications to Crystal Structure Prediction . J. Phys. Chem. A , 102 : 2904 – 2918 .
- Bazterra , VE , Ferraro , MB and Facelli , JC . 2002 . Modified Genetic Algorithm to Model Crystal Structures: I. Benzene, Naphthalene and Anthracene . J. Chem. Phys. , 116 : 5984 – 5991 .
- Bazterra , VE , Ferraro , MB and Facelli , JC . 2002 . Modified Genetic Algorithm to Model Crystal Structures: II. Determination of a Polymorphic Structure of Benzene using Enthalpy Minimization . J. Chem. Phys. , 116 : 5992 – 5995 .
- Bazterra , VE , Ferraro , MB and Facelli , JC . 2004 . Modified Genetic Algorithm to Model Crystal Structures: III. Determination of Crystal Structures Allowing Simultaneous Molecular Geometry Relaxation . Int. J. Quantum Chem. , 96 : 312 – 320 .
- Bazterra , VE , Ona , O , Caputo , MC , Ferraro , MB , Fuentealba , P and Facelli , JC . 2004 . Modified Genetic Algorithms to Model Cluster Structures in Medium-sized Silicon Clusters . Phys. Rev. A , 69 : 053202
- Bazterra , VE , Thorley , M , Ferraro , MB and Facelli , JC . 2007 . A Distributed Computing Method for Crystal Structure Prediction of Flexible Molecules: An Application to N-(2-dimethyl-4,5-dinitrophenyl)acetamide . J. Chem. Theory Comput. , 3 : 201 – 209 .
- Motherwell , WDS . 2001 . Crystal Structure Prediction and the Cambridge Structural Database . Mol. Cryst. Liq. Cryst. , 356 : 559 – 567 .
- Martonák , R , Laio , A , Bernasconi , M , Ceriani , C , Raiteri , P , Zipoli , F and Parrinello , M . 2005 . Simulation of Structural Phase Transitions by Metadynamics . Z. Kristallogr. , 220 : 489 – 498 .
- Raiteri , P , Martonák , R and Parrinello , M . 2005 . Exploring Polymorphism: The Case of Benzene . Angew. Chem. Int. Ed. , 44 : 3769 – 3773 .
- van Eijck , BP , Spek , AL , Mooij , WTM and Kroon , J . 1998 . Hypothetical Crystal Structures of Benzene at 0 and 30 kbar . Acta Crystallogr., Sect. B , B54 : 291 – 299 .
- Karamertzanis , PG , Raiteri , P , Parrinello , M , Leslie , M and Price , SL . 2008 . The Thermal Stability of Lattice-energy Minima of 5-fluorouracil: Metadynamics as an Aid to Polymorph Prediction . J. Phys. Chem. B , 112 : 4298 – 4308 .
- Gavezzotti , A . 1999–2000 . Zip-Promet, A Program for the Generation of Crystal Structures from Molecular Structure , Italy : University of Milano .
- Hofmann , DWM and Lengauer , T . 1997 . A Discrete Algorithm for Crystal Structure Prediction of Organic Molecules . Acta Crystallogr., Sect. A , A53 : 225 – 234 .
- Hofmann , DWM and Lengauer , T . 1999 . Prediction of Crystal Structures of Organic Molecules . J. Mol. Struct. , 474 : 13 – 23 .
- Hofmann , DWM and Apostolakis , J . 2003 . Crystal Structure Prediction by Data Mining . J. Mol. Struct. (Theochem) , 647 : 17 – 39 .
- Dzyabchenko , AV . 1994 . Method of Crystal-structure Similarity Searching . Acta Crystallogr., Sect. B , B50 : 414 – 425 .
- van Eijck , BP and Kroon , J . 1997 . Fast Clustering of Equivalent Structures in Crystal Structure Prediction . J. Comput. Chem. , 18 : 1036 – 1042 .
- Chisholm , JA and Motherwell , S . 2005 . COMPACK: A Program for Identifying Crystal Structure Similarity using Distances . J. Appl. Crystallogr. , 38 : 228 – 231 .
- Price , SL . 2000 . “ Toward More Accurate Model Intermolecular Potentials for Organic Molecules ” . In Reviews in Computational Chemistry , Edited by: Lipkowitz , KB and Boyd , DB . Vol. 14 , 225 – 289 . New York : Wiley-VCH, John Wiley and Sons .
- Gavezzotti , A . 2002 . Structure and Intermolecular Potentials in Molecular Crystals . Modell. Simul. Mater. Sci. Eng. , 10 : R1 – R29 .
- Stone , AJ . 1996 . The Theory of Intermolecular Forces , Vol. 32 , Oxford, , UK : Oxford University Press .
- van Eijck , BP . 2002 . Crystal Structure Predictions for Disordered Halobenzenes . Phys. Chem. Chem. Phys. , 4 : 4789 – 4794 .
- Jagielska , A , Arnautova , YA and Scheraga , HA . 2004 . Derivation of a New Force Field for Crystal-structure Prediction using Global Optimization: Nonbonded Potential Parameters for Amines, Imidazoles, Amides, and Carboxylic Acids . J. Phys. Chem. B , 108 : 12181 – 12196 .
- Arnautova , YA , Pillardy , J , Czaplewski , C and Scheraga , HA . 2003 . Global Optimization-based Method for Deriving Intermolecular Potential Parameters for Crystals . J. Phys. Chem. B , 107 : 712 – 723 .
- Arnautova , YA , Jagielska , A , Pillardy , J and Scheraga , HA . 2003 . Derivation of a New Force Field for Crystal-structure Prediction using Global Optimization: Nonbonded Potential Parameters for Hydrocarbons and Alcohols . J. Phys. Chem. B , 107 : 7143 – 7154 .
- Wheatley , RJ and Price , SL . 1990 . A Systematic Intermolecular Potential Method Applied to Chlorine . Mol. Phys. , 71 : 1381 – 1404 .
- Nobeli , I , Price , SL and Wheatley , RJ . 1998 . Use of Molecular Overlap to Predict Intermolecular Repulsion in N···H–O Hydrogen Bonds . Mol. Phys. , 95 : 523 – 537 .
- Nobeli , I and Price , SL . 1999 . A Non-empirical Intermolecular Potential for Oxalic Acid Crystal Structures . J. Phys. Chem. A. , 103 : 6448 – 6457 .
- Tsui , HHY and Price , SL . 1999 . A Non-empirical Method of Determining Atom-Atom Repulsion Parameters: Application to the Crystal Structure Prediction of an Oxyboryl Derivative . CrystEngComm , 1 : 24 – 32 .
- Mooij , WTM , van Duijneveldt , FB , van Duijneveldt-van de Rijdt , JGCM and van Eijck , BP . 1999 . Transferable Ab Initio Intermolecular Potentials: 1. Derivation from Methanol Dimer and Trimer Calculations . J. Phys. Chem. A , 103 : 9872 – 9882 .
- Mooij , WTM , van Eijck , BP and Kroon , J . 1999 . Transferable Ab Initio Intermolecular Potentials: 2. Validation and Application to Crystal Structure Prediction . J. Phys. Chem. A , 103 : 9883 – 9890 .
- Mitchell , JBO and Price , SL . 2000 . A Systematic Non-empirical Method of Deriving Model Intermolecular Potentials for Organic Molecules: Application to Amides . J. Phys. Chem. A , 104 : 10958 – 10971 .
- Day , GM and Price , SL . 2003 . A Nonempirical Anisotropic Atom-Atom Model Potential for Chlorobenzene Crystals . J. Am. Chem. Soc. , 125 : 16434 – 16443 .
- Misquitta , AJ , Welch , GWA , Stone , AJ and Price , SL . 2008 . A First Principles Prediction of the Crystal Structure of C6Br2ClFH2 . Chem. Phys. Lett. , 456 : 105 – 109 .
- Stone , AJ and Misquitta , AJ . 2007 . Atom-Atom Potentials from Ab Initio Calculations . Int. Rev. Phys. Chem. , 26 : 193 – 222 .
- van Eijck , BP , Mooij , WTM and Kroon , J . 2001 . Crystal Structure Prediction for Six Monosaccharides Revisited . J. Phys. Chem. B , 105 : 10573 – 10578 .
- van Eijck , BP , Mooij , WTM and Kroon , J . 2001 . Ab Initio Crystal Structure Predictions for Flexible Hydrogen-bonded Molecules. Part II. Accurate Energy Minimization . J. Comput. Chem. , 22 : 805 – 815 .
- Tremayne , M , Grice , L , Pyatt , JC , Seaton , CC , Kariuki , BM , Tsui , HHY , Price , SL and Cherryman , JC . 2004 . Characterization of Complicated New Polymorphs of Chlorothalonil by X-ray Diffraction and Computer Crystal Structure Prediction . J. Am. Chem. Soc. , 126 : 7071 – 7081 .
- Nyburg , SC and Faerman , CH . 1985 . A Revision of van der Waals Atomic Radii for Molecular Crystals: N, O, F, S, Cl, Se, Br and I Bonded to Carbon . Acta Crystallogr., Sect. B , B41 : 274 – 279 .
- Munowitz , MG , Wheeler , GL and Colson , SD . 1977 . A Critical Evaluation of Isotropic Potential Functions for Chlorine. Calculations on the Three Phases of p-Dichlorobenzene at 100 K . Mol. Phys. , 34 : 1727 – 1737 .
- Sigfridsson , E and Ryde , U . 1998 . Comparison of Methods for Deriving Atomic Charges from the Electrostatic Potential and Moments . J. Comput. Chem. , 19 : 377 – 395 .
- Williams , DE and Cox , SR . 1984 . Nonbonded Potentials for Azahydrocarbons: The Importance of the Coulombic Interaction . Acta Crystallogr., Sect. B , B40 : 404 – 417 .
- Williams , DE and Weller , RR . 1983 . Lone-pair Electronic Effects on the Calculated Ab Initio SCF-MO electric potential and the Crystal Structures of Azabenzenes . J. Am. Chem. Soc. , 105 : 4143 – 4148 .
- Karamertzanis , PG and Pantelides , CC . 2004 . Optimal Site Charge Models for Molecular Electrostatic Potentials . Mol. Simul. , 30 : 413 – 436 .
- Stone , AJ and Alderton , M . 1985 . Distributed Multipole Analysis: Methods and Applications . Mol. Phys. , 56 : 1047 – 1064 .
- Stone , AJ . 1981 . Distributed Multipole Analysis, or How to Describe a Molecular Charge Distribution . Chem. Phys. Lett. , 83 : 233 – 239 .
- Mooij , WTM and Leusen , FJJ . 2001 . Multipoles Versus Charges in the (1999) Crystal Structure Prediction Test . Phys. Chem. Chem. Phys. , 3 : 5063 – 5066 .
- Brodersen , SW , Leusen , FJJ and Engel , G . 2003 . A Study of Different Approaches to the Electrostatic Interaction in Force Field Methods for Organic Crystals . Phys. Chem. Chem. Phys. , 5 : 4923 – 4931 .
- Buckingham , AD and Fowler , PW . 1985 . A Model for the Geometries of van der Waals Complexes . Can. J. Chem. , 63 : 2018 – 2025 .
- Buckingham , AD , Fowler , PW and Stone , AJ . 1986 . Electrostatic Predictions of Shapes and Properties of van der Waals Molecules . Int. Rev. Phys. Chem. , 5 : 107 – 114 .
- Day , GM , Price , SL and Leslie , ML . 2003 . Atomistic Calculations of Phonon Frequencies and Thermodynamical Quantities for Crystals of Rigid Organic Molecules . J. Phys. Chem. B , 107 : 10919 – 10933 .
- Day , GM , Price , SL and Leslie , M . 2000 . Elastic Constant Calculations for Molecular Organic Crystals . Cryst. Growth Des. , 1 : 13 – 27 .
- Willock , DJ , Price , SL , Leslie , M and Catlow , CRA . 1995 . The Relaxation of Molecular Crystal Structures using a Distributed Multipole Electrostatic Model . J. Comput. Chem. , 16 : 628 – 647 .
- Price , SL , Leslie , M , Welch , GWA , Habgood , M , Price , LS , Karamertzanis , PG and Day , GM . 2010 . Modelling Organic Crystal Structures using Distributed Multipole and Polarizability-based Model Intermolecular Potentials . Phys. Chem. Chem. Phys. , 12 : 8478 – 8490 .
- Karamertzanis , PG and Price , SL . 2006 . Energy Minimization of Crystal Structures Containing Flexible Molecules . J. Chem. Theory Comput. , 2 : 1184 – 1199 .
- Mooij , WTM , van Eijck , BP and Kroon , J . 2000 . Ab Initio Crystal Structure Predictions for Flexible Hydrogen-bonded Molecules . J. Am. Chem. Soc. , 122 : 3500 – 3505 .
- Welch , GWA , Karamertzanis , PG , Misquitta , AJ , Stone , AJ and Price , SL . 2008 . Is the Induction Energy Important for Modeling Organic Crystals? . J. Chem. Theory Comput. , 4 : 522 – 532 .
- Karamertzanis , PG , Day , GM , Welch , GWA , Kendrick , J , Leusen , FJJ , Neumann , MA and Price , SL . 2008 . Modelling the Interplay of Inter- and Intramolecular Hydrogen Bonding in Conformational Polymorphs . J. Chem. Phys. , 128 : 244708
- Vishweshwar , P , McMahon , JA , Oliveira , M , Peterson , ML and Zaworotko , MJ . 2005 . The Predictably Elusive Form II of Aspirin . J. Am. Chem. Soc. , 127 : 16802 – 16803 .
- Nowell , H and Price , SL . 2005 . Validation of a Search Technique for Crystal Structure Prediction of Flexible Molecules by Application to Piracetam . Acta Crystallogr., Sect. B , B61 : 558 – 568 .
- Day , GM and Cooper , TG . 2010 . Crystal Packing Predictions of the Alpha-Amino Acids: Methods Assessment and Structural Observations . CrystEngComm , 12 : 2443 – 2453 .
- Chisholm , JA , Motherwell , S , Tulip , PR , Parsons , S and Clark , SJ . 2005 . An Ab Initio Study of Observed and Hypothetical Polymorphs of Glycine . Cryst. Growth Des. , 5 : 1437 – 1442 .
- Rovira , C and Novoa , JJ . 2001 . A First-principles Computation of the Low-energy Polymorphic Forms of the Acetic Acid Crystal. A Test of the Atom-Atom Force Field Predictions . J. Phys. Chem. B , 105 : 1710 – 1719 .
- Byrd , EFC , Scuseria , GE and Chabalowski , CF . 2004 . An Ab Initio Study of Solid Nitromethane, HMX, RDX, and CL20: Successes and Failures of DFT . J. Phys. Chem. B , 108 : 13100 – 13106 .
- Neumann , MA and Perrin , M-A . 2005 . Energy Ranking of Molecular Crystals using Density Functional Theory Calculations and an Empirical van der Waals Correction . J. Phys. Chem. B , 109 : 15531 – 15541 .
- Civalleri , B , Zicovich-Wilson , CM , Valenzano , L and Ugliengo , P . 2008 . B3LYP Augmented with an Empirical Dispersion Term (B3LYP-D*) as Applied to Molecular Crystals . CrystEngComm , 10 : 405 – 410 .
- Grimme , S . 2004 . Accurate Description of van der Waals Complexes by Density Functional Theory Including Empirical Corrections . J. Comput. Chem. , 25 : 1463 – 1473 .
- Grimme , S . 2006 . Semiempirical GGA-type Density Functional Constructed with a Long-range Dispersion Correction . J. Comput. Chem. , 27 : 1787 – 1799 .
- Neumann , MA . 2008 . Tailor-made Force Fields for Crystal-structure Prediction . J. Phys. Chem. B , 112 : 9810 – 9829 .
- Gavezzotti , A . 2002 . Calculation of Intermolecular Interaction Energies by Direct Numerical Integration over Electron Densities: I. Electrostatic and Polarization Energies in Molecular Crystals . J. Phys. Chem. B , 106 : 4145 – 4154 .
- Gavezzotti , A . 2003 . Calculation of Intermolecular Interaction Energies by Direct Numerical Integration over Electron Densities: 2. An Improved Polarization Model and the Evaluation of Dispersion and Repulsion Energies . J. Phys. Chem. B , 107 : 2344 – 2353 .
- Gavezzotti , A . 2003 . Towards a Realistic Model for the Quantitative Evaluation of Intermolecular Potentials and for the Rationalization of Organic Crystal Structures: Part II. Crystal Energy Landscapes . CrystEngComm , 5 : 439 – 446 .
- Gavezzotti , A . 2003 . Towards a Realistic Model for the Quantitative Evaluation of Intermolecular Potentials and for the Rationalization of Organic Crystal Structures: Part I. Philosophy . CrystEngComm , 5 : 429 – 438 .
- Dzyabchenko , AV and Bailevskii , MV . 1985 . Theoretical Structure of Crystalline Benzene . J. Struct. Chem. , 26 : 553 – 558 .
- Shoda , T , Yamahara , K , Okazaki , K and Williamas , DE . 1994 . Molecular Packing Analysis: Prediction of Experimental Crystal Structures of Benzene Starting from Unreasonable Initial Structures . J. Mol. Struct. (Theochem) , 313 : 321 – 334 .
- Gavezzotti , A and Filippini , G . 1995 . Polymorphic Forms of Organic Crystals at Room Conditions: Thermodynamic and Structural Implications . J. Am. Chem. Soc. , 117 : 12299 – 12305 .
- Filippini , G and Gavezzotti , A . 1993 . Empirical Intermolecular Potentials for Organic-crystals: The ‘6-Exp’ Approximation Revisited . Acta Crystallogr., Sect. B , B49 : 868 – 880 .
- Dunitz , JD , Filippini , G and Gavezzotti , A . 2000 . Molecular Shape and Crystal Packing: A Study of C12H12 Isomers, Real and Imaginary . Helv. Chim. Acta , 83 : 2317 – 2335 .
- van Eijck , BP . 2001 . Ab Initio Crystal Structure Predictions for Flexible Hydrogen-bonded Molecules: Part III. Effect of Lattice Vibrations . J. Comput. Chem. , 22 : 816 – 826 .
- Anghel , AT , Day , GM and Price , SL . 2002 . A Study of the Known and Hypothetical Crystal Structures of Pyridine: Why are there Four Molecules in the Asymmetric Unit Cell? . CrystEngComm , 4 : 348 – 355 .
- Lewis , TC , Tocher , DA , Day , GM and Price , SL . 2003 . A Computational and Experimental Search for Polymorphs of Parabanic Acid: A Salutatory Tale Leading to the Crystal Structure of Oxo-Ureido-Acetic Acid Methyl Ester . CrystEngComm , 5 : 3 – 9 .
- Born , M and Huang , K . 1954 . Dynamical Theory of Crystal Lattices , New York : Oxford University Press .
- Walmsley , SH . 1975 . “ Basic Theory of the Lattice Dynamics of Molecular Crystals ” . In Lattice Dynamics and Intermolecular Forces , Edited by: Corso , LV . 81 – 114 . New York : Academic Press .
- Walmsley , SH . 1968 . Lattice Vibrations and Elastic Constants of Molecular Crystals in the Pair Potential Approximation . J. Chem. Phys. , 48 : 1438 – 1444 .
- Califano , S , Schettino , V and Neto , N . 1981 . Lattice Dynamics of Molecular Crystals , Vol. 26 , Berlin : Springer-Verlag .
- Pawley , GS . 1972 . Analytic Formulation of Molecular Lattice Dynamics based on Pair Potential Functions . Phys. Status Solidi B. , 49 : 475 – 488 .
- Gramaccioli , CM , Simonetta , M and Suffritti , GB . 1973 . Lattice Dynamics in Crystals of ‘Rigid’ Hydrocarbons . Chem. Phys. Lett. , 20 : 23 – 28 .
- Bonadeo , H , D’Alessio , E , Halac , E and Burgos , E . 1978 . Lattice Dynamics, Thermodynamic Functions, and Phase Transitions of p-Dichloro- and 1,2,4,5-Tetrachlorobenzene . J. Chem. Phys. , 68 : 4714 – 4721 .
- Della Valle , RG and Brillante , A . 1994 . Lattice Dynamics of Halogenated Anthracene Derivatives under Pressure . J. Chem. Phys. , 100 : 7640 – 7647 .
- Brillante , A , Della Valle , RG , Farina , R and Venuti , E . 1995 . Pressure-induced Phase Transitions in 9,10-Anthracene Derivatives: Anthraquinone . Chem. Phys. , 191 : 177 – 184 .
- Della Valle , RG , Venuti , E and Brillante , A . 1995 . Pressure and Temperature Effects in Lattice Dynamics: The Case of Naphthalene . Chem. Phys. , 198 : 79 – 89 .
- Della Valle , RG , Venuti , E and Brillante , A . 1996 . Quasi Harmonic Lattice Dynamics: The Phase Diagram of Benzene . Chem. Phys. , 202 : 231 – 241 .
- Farina , L , Della Valle , RG and Brillante , A . 2000 . Pressure and Temperature Effects in Lattice Dynamics: 1,4-Dibromonaphthalene . Chem. Phys. , 262 : 437 – 444 .
- Luty , T and Pawley , GS . 1974 . On the Lattice Dynamics of Solid Nitrogen . Chem. Phys. Lett. , 28 : 593 – 596 .
- Reynolds , PA . 1973 . Lattice Dynamics of the Pyrazine Crystal Studied by Coherent Inelastic Neutron Scattering . J. Chem. Phys. , 59 : 2777 – 2786 .
- Gamba , Z and Bonadeo , H . 1981 . Lattice Dynamical Calculations on Azabenzene Crystals: The Distributed Dipole Model . J. Chem. Phys. , 75 : 5059 – 5066 .
- Richardson , MF , Tang , Q-C , Novotny-Bregger , E and Dunitz , JD . 1990 . Conformational Polymorphism of Dimethyl 3,6-dichloro-2,5-dihydroxyterephthalate: II. Structural, Thermodynamic, Kinetic and Mechanistic Aspects of Phase Transformations among the Three Crystal Forms . Acta Crystallogr., Sect. B , B46 : 653 – 660 .
- Ferretti , V , Gilli , P and Gavezzotti , A . 2002 . X-ray Diffraction and Molecular Simulation Study of the Crystalline and Liquid States of Succinic Anhydride . Chem. Eur. J. , 8 : 1710 – 1718 .
- Gray , AE , Day , GM , Leslie , M and Price , SL . 2004 . Dynamics in Crystals of Rigid Organic Molecules: Contrasting the Phonon Frequencies Calculated by Molecular Dynamics with Harmonic Lattice Dynamics for Imidazole and 5-Azauracil . Mol. Phys. , 102 : 1067 – 1083 .
- Mooij , WTM , van Eijck , BP , Price , SL , Verwer , P and Kroon , J . 1998 . Crystal Structure Predictions for Acetic Acid . J Comput. Chem. , 19 : 459 – 474 .
- Gavezzotti , A . 2000 . A Molecular Dynamics Test of the Different Stability of Crystal Polymorphs under Thermal Strain . J. Am. Chem. Soc. , 122 : 10724 – 10725 .
- Bennema , P , Meekes , H , Boerrigter , SXM , Cuppen , HM , Deij , MA , van Eupen , J , Verwer , P and Vlieg , E . 2004 . Crystal Growth and Morphology: New Developments in an Integrated Hartman-Perdok-connected Net-roughening Transition Theory, Supported by Computer Simulations . Cryst. Growth Des. , 4 : 905 – 913 .
- Donnay , JDH and Harker , D . 1937 . A New Law of Crystal Morphology, Extending the Law of Bravais . Am. Mineral. , 22 : 446 – 467 .
- Hartman , P and Perdok , W . 1955 . On the Relations between Structure and Morphology of Crystals: I . Acta Crystallogr. , 8 : 49 – 52 .
- Hartman , P and Bennema , P . 1980 . The Attachment Energy as a Habit Controlling Factor I. Theoretical Considerations . J. Cryst. Growth , 49 : 145 – 156 .
- Coombes , DS , Catlow , CRA , Gale , JD , Rohl , AL and Price , SL . 2005 . Calculation of Attachment Energies and Relative Volume Growth Rates as an Aid to Polymorph Prediction . Cryst. Growth Des. , 5 : 879 – 885 .
- Boerrigter , SXM , Cuppen , HM , Ristic , RI , Sherwood , JN , Bennema , P and Meekes , H . 2002 . Explanation for the Supersaturation-dependent Morphology of Monoclinic Paracetamol . Cryst. Growth Des. , 2 : 357 – 361 .
- Cuppen , HM , Day , GM , Verwer , P and Meekes , H . 2004 . Sensitivity of Morphology Prediction to the Force Field: Paracetamol as an Example . Cryst. Growth Des. , 4 : 1341 – 1349 .
- Dunitz , JD and Scheraga , HA . 2004 . Exercises in Prognostication: Crystal Structures and Protein Folding . Proc. Natl Acad. Sci. , 101 : 14309 – 14311 .
- Day , GM and Motherwell , WDS . 2006 . An Experiment in Crystal Structure Prediction by Popular Vote . Cryst. Growth Des. , 6 : 1985 – 1990 .
- Sarma , JARP and Desiraju , GR . 2002 . The Supramolecular Synthon Approach to Crystal Structure Prediction . Cryst. Growth Des. , 2 : 93 – 100 .
- Dey , A , Kirchner , MT , Vangala , VR , Desiraju , GR , Mondal , R and Howard , JAK . 2005 . Crystal Structure Prediction of Aminols: Advantages of a Supramolecular Synthon Approach with Experimental Structures . J. Am. Chem. Soc. , 127 : 10545 – 10559 .
- Dey , A , Pati , NN and Desiraju , GR . 2006 . Crystal Structure Prediction with the Supramolecular Synthon Approach: Experimental Structures of 2-Amino-4-ethylphenol and 3-Amino-2-Naphthol and Comparison with Prediction . CrystEngComm , 8 : 751 – 755 .
- Neumann , MA , Leusen , FJJ and Kendrick , J . 2008 . A Major Advance in Crystal Structure Prediction . Angew. Chem. Int. Ed. , 47 : 2427 – 2430 .
- Gavezzotti , A . 2002 . Ten Years of Experience in Polymorph Prediction: What Next? . CrystEngComm , 4 : 343 – 347 .
Subject Index
Atom-atom method 26
Blind tests 5, 10, 15, 35–38, 40, 42
Cambridge Structural Database 8
Crystal growth 23, 27, 32, 33, 35, 40, 42
Density functional theory 7
Dynamics 5, 13, 14, 19, 27, 29, 31, 32, 42
Electrostatics 18–22, 25, 26, 29
Energy minimization 7, 8, 12, 13, 15, 16, 20–24, 26, 29, 34, 40
Hydrogen bonding 13, 22, 23, 25, 30
Lattice energy 6–8, 12–33, 40, 42
Molecular flexibility 8, 40, 41
Morphology 32, 33
PIXEL method (see semi-classical density sums method) 26
Polymorphism 4, 17, 26, 27, 32
Semi-classical density sums method 24, 26