
Abstract
Spike proteins protrude from the SARS-CoV-2 viral envelope and are responsible for initiating fusion into human epithelial cells by binding Angiotensin-Converting Enzyme 2 receptors on the host cell surface. Due to their exposed location on the outside of the virion and their key role in infection, SARS-CoV-2 spike proteins are an important target for vaccine development and drug design. Over the last two years, many spike protein structures have been experimentally determined, providing essential details into the complex structural rearrangements that occur after receptor binding and during fusion of the virion with the host cell, as well as into the interactions of spike protein molecules with antibodies. SARS-CoV-2 variants, particularly those associated with reduced vaccine efficacy, are strongly associated with mutations in two domains of the SARS-CoV-2 spike protein, namely the receptor binding domain and the N-terminal domain, which have both been structurally characterized. This review provides a comprehensive overview of the structural knowledge acquired over the past four years on the SARS-CoV-2 spike protein and its critical role in viral infection.
1. Introduction
The infection caused by the novel severe acute respiratory syndrome coronavirus SARS-CoV-2 was declared a pandemic by the WHO in March 2020 [Citation1], and continues to be a serious health concern worldwide to this day. Coronaviruses are named for the crown-like appearance of glycosylated fusion proteins (called ‘spike’ proteins, S proteins or surface glycoproteins) protruding from the surface of each virion [Citation2,Citation3]. In this review, the word ‘spike’ will always refer to the entire complex from SARS-CoV-2 including its glycosylation, while the protein itself is called ‘spike protein’ and one chain of spike protein in the context of the spike will be identified at a ‘spike protomer’. Spikes are trimeric and extend approximately 20 nm from the viral surface membrane [Citation4–6]. Observed virions contain small numbers of spikes [Citation7,Citation8] (one study reports 26 ± 15 [Citation9]), and far fewer than previously observed for SARS-CoV-1 [Citation9]. Spikes initiate entry into human host cells by interacting with the cell surface receptor Angiotensin-Converting Enzyme 2 (ACE2) [Citation10–13]. ACE2 is mainly expressed in lung and intestinal epithelial cells, but is also present on kidney, cardiovascular and gastrointestinal cells [Citation14]. After engaging ACE2, the spike is likely processed by proteases and parts of the complex dissociate before the remaining parts undergo major structural rearrangements necessary for membrane fusion [Citation2,Citation15–19]. As part of the refolding mechanism, the spike undergoes an irreversible transformation into a trimeric hairpin structure [Citation17,Citation18,Citation20–22]. This structural change brings the viral and cellular cell membranes into close proximity, ultimately initiating membrane fusion [Citation2,Citation17,Citation18,Citation20–22]. Important to note is that the precise mechanism by which a SARS-CoV-2 virion fuses with the host cell is not yet fully understood and our understanding relies mostly on research of other fusion proteins [Citation2,Citation20,Citation23–26].
Once the membranes are fused, the viral RNA is released into the cytosol of the host cell and its first open reading frame is translated by the host ribosome machinery to create the two variants of the viral polyprotein [Citation27]. Thus begins the cascade of events that results in the production of new viral particles. The spike is thereby key to invading the host cell and therefore crucial to the viral infection cycle [Citation2,Citation11,Citation28,Citation29]. Due to their exposed position on the viral surface membrane, spikes are also a major target for the host’s immune system and an important target for drug and vaccine development [Citation30–34]. However, as a way of reducing recognition by antibodies of the immune system, the surfaces of spikes are densely covered with complex carbohydrates, primarily N-glycans [Citation35–40]. The glycan shield, which masks between 60% to 90% of each spike's surface from antibodies [Citation41,Citation42], plays a crucial role in immune system evasion for exposed viral proteins [Citation37,Citation41,Citation43]. The attached glycans also aid the stability and maintain optimal activity of spikes [Citation41,Citation44].
2. Structural overview
The spike protein is one of four structural proteins encoded on the SARS-CoV-2 genome, with the others being a membrane glycoprotein (M), a nucleocapsid protein (N) and the small envelope protein (E) [Citation45–47], all four of which are present in the virion. The spike protein is encoded in the gene region between 21,563 and 25,384 bp, and is the largest of the four structural proteins [Citation48] ranging from 1250 to 1273 amino acids in length (Figure a), with the exact sequence and length depending on the virus strain [Citation49]. As a class I fusion protein, spike proteins form homotrimers, with each protomer comprising a large extracellular domain (EC) at the N-terminal end, a single α-helical transmembrane anchor (TM), and a short intracellular cytoplasmic tail (CT) at the C-terminus [Citation2,Citation9,Citation50]. The extracellular part of the spike protein can be divided into two subunits, S1 and S2 (Table ): Subunit S1 (also called the ‘head’) is responsible for receptor binding while S2 (the ‘stalk’) mediates membrane fusion [Citation13,Citation51,Citation52]. S1, the first 685 amino acids, is further subdivided into five segments: a short signal peptide (SS), the N-terminal domain (NTD), the receptor-binding domain (RBD), and two subdomains (SD1 and SD2). The S2 subunit, encompassing 588 amino acid residues, is subdivided into an N-terminal hydrophobic fusion peptide (FP), two heptad repeats (HR1 and HR2), the transmembrane domain and cytoplasmic tail (Figure a) [Citation53].
Figure 1. Overview of the SARS-CoV-2 spike protein. (a) SARS-CoV-2 genome with a sequence overview of domains and cleavage sites (S1/S2; S2´) of the spike protein. Abbreviations (as listed in Table ): ORF: open reading frame, N: nucleocapsid, M: M-protein, E: E-protein, S1 and S2: Subunit S1 and S2, NTD: N-terminal domain, RBD: receptor binding domain, SD1: sub-domain 1, SD2: sub-domain 2; FP1: fusion peptide region 1, FP2: fusion peptide region 2, HR1: heptad repeat 1, CH: central helix, CD: connector domain, HR2: heptad repeat 2., TM: transmembrane domain, CT: cytoplasmic tail. (b) Cryo-EM structure of a spike in its prefusion state (PDB 6XR8; coloured by monomer), with individual domains of subunit S1 and S2 labelled for one of the monomers (coloured by domain). (c) Trimeric spike after shedding of the S1 subunit and rearrangement into the post-fusion state (PDB 6XRA; coloured by monomer) with individual domains shown for one monomer (coloured by domain). The cytosolic and transmembrane domains are missing in both structures, and the FP is not resolved in the post-fusion state. This figure and the subsequent ones were created with Protein Imager [Citation54], Blender [Citation55], and Inkscape [Citation56]
![Figure 1. Overview of the SARS-CoV-2 spike protein. (a) SARS-CoV-2 genome with a sequence overview of domains and cleavage sites (S1/S2; S2´) of the spike protein. Abbreviations (as listed in Table 1): ORF: open reading frame, N: nucleocapsid, M: M-protein, E: E-protein, S1 and S2: Subunit S1 and S2, NTD: N-terminal domain, RBD: receptor binding domain, SD1: sub-domain 1, SD2: sub-domain 2; FP1: fusion peptide region 1, FP2: fusion peptide region 2, HR1: heptad repeat 1, CH: central helix, CD: connector domain, HR2: heptad repeat 2., TM: transmembrane domain, CT: cytoplasmic tail. (b) Cryo-EM structure of a spike in its prefusion state (PDB 6XR8; coloured by monomer), with individual domains of subunit S1 and S2 labelled for one of the monomers (coloured by domain). (c) Trimeric spike after shedding of the S1 subunit and rearrangement into the post-fusion state (PDB 6XRA; coloured by monomer) with individual domains shown for one monomer (coloured by domain). The cytosolic and transmembrane domains are missing in both structures, and the FP is not resolved in the post-fusion state. This figure and the subsequent ones were created with Protein Imager [Citation54], Blender [Citation55], and Inkscape [Citation56]](/cms/asset/c39eaf61-e9bf-4918-be1a-4dfb92c22a81/gcry_a_2363756_f0001_oc.jpg)
Table 1. Overview of subunits and domain structure of the spike protein. Major domains and subdomains are listed.
2.1. Subunit S1
The S1 subunit, which binds Angiotensin-Converting Enzyme 2, comprises a signal peptide (SP) and four domains, listed in Table . These four domains (of each S1 monomer) wrap around and cap the top of the helical S2 subunit (Figure b) [Citation64].
The spike protein is synthesized on the rough endoplasmic reticulum before being transported to the Golgi complex [Citation53,Citation65,Citation66]; the targeting to the rough endoplasmic reticulum is done by the N-terminal signal peptide (SP), which consists of thirteen amino acids with helix-forming, highly hydrophobic residues [Citation67–70]. Co-translational and post-translational processing result in the removal of this signal peptide in the ER lumen, trimerization of the spike protein, extensive glycosylation, and subunit cleavage by furin or furin-like proteases [Citation53,Citation71–75].
The N-terminal domain is similar to MERS- and bovine-CoV functionalized with eight N-linked glycan molecules [Citation76–78]. It shares only 52% sequence identity with SARS-CoV-1, and is highly similar to the equivalent domain in bat coronavirus RatG13 (>98%) [Citation79]. The N-terminal domain can be divided into three topological regions: top, core and bottom [Citation62,Citation80]. The exact role of the N-terminal domain is still not fully understood, but has been suggested that the sequence 72-GTNGTKR-78 (located in a loop connecting two core-region β-sheets), carrying a highly conserved N-glycosylation site at N74, can bind host-cell saccharides and/or additional receptors aside from ACE2 [Citation77,Citation79,Citation81–84]. Receptor binding through the N-terminal domain (NTD) has been previously observed in spike proteins of the mouse hepatitis virus, a member of the Betacoronaviridae family [Citation81,Citation83]. Additionally, the spike protein’s N-terminal domain may also be involved in interaction with the host cells via recognition of specific sugar molecules [Citation12,Citation84–86], as occurs in the transmissible gastroenteritis coronavirus and bovine coronavirus [Citation84,Citation86].
Structures of the prefusion spikes are often characterised by the conformation of their receptor binding domain (RBDs). In the trimeric spike, each RBD is located next to the N-terminal domain of the adjacent protomer and can adopt an ‘up’ and a ‘down’ conformation. It is in the ‘up’ state that the receptor binding domain interacts with host cell surface ACE2 receptors [Citation11,Citation87–89]. The RBD consists of a five-stranded antiparallel β-sheet core with short connecting α-helices, β-sheets and loops [Citation89]. The receptor-binding motif (RBM) is located as an extended insertion between two β-strands and contains most of the residues which mediate ACE2 binding [Citation89]. The RBD is stabilized by four disulphide bonds, three of which are located in the core (Cys336–Cys361, Cys379–Cys432, Cys391–Cys525), with the last stabilizing loops at the distal end of the RBM (Cys480-Cys488)[Citation89].
Both subdomains SD1 and SD2 (sometimes called C-terminal domain 1 and 2 or CTD1 and CTD2) are β-structure rich segments located after the RBD. The N-terminal end of S2 is adjacent to a cleavage site targeted by furin (or another protease) during pre-processing of the spike protein[Citation62].
2.2. Subunit S2
The central ‘stalk’ of the spike protein, responsible for viral fusion and entry, is formed of seven domains, listed in Table . The helical core of the S2 subunit is formed by the three-stranded coiled coil central helix domain, with most of the remaining polypeptide chain, including heptad repeat 1, the connector domain and the fusion peptide, wrapping around it (Figure b) [Citation18,Citation58,Citation62,Citation63]. HR1 and HR2 are characterized by seven-residue repeats of the sequence HPPHCPC in tandem (where H stands for a bulky hydrophobic residue, P is a polar or hydrophilic residue and C is any charged residue) [Citation57,Citation90,Citation91], which are important during later pre-fusion rearrangements (described later).
The exact sequence of the fusion peptide regions (FP1 and 2), which are inserted into the host cell membrane during the fusion process, have not yet been definitively determined [Citation67,Citation92–96]. The proposed fusion peptide regions encompassing FP1 and FP2 contain approximately 15–20 predominantly hydrophobic amino acids [Citation57], and are located downstream to the S2’ cleavage site at R815-S816 [Citation67,Citation92–96]. FP1 and FP2 were determined based on their relatively well-conserved sequences. FP1 comprises a conserved hydrophobic LLF motif (Leu821-Leu822-Phe823) while FP2 features a pair of highly conserved cysteine residues which both seem to play an important role during membrane fusion [Citation61,Citation93,Citation94,Citation96–98].
The transmembrane domain (TM) is highly conserved in all coronaviruses[Citation67,Citation99,Citation100]. It contains three regions: an aromatic region, a hydrophobic central region, and a cysteine-rich region [Citation67,Citation99,Citation100]. The central hydrophobic part of the transmembrane domain forms an α-helix that arranges as a trimer with its counterparts. Together with the spike protein’s cytoplasmic tail (CT), the transmembrane domain helps to stabilize the trimeric structure that is crucial for membrane fusion [Citation67,Citation100–102]. A study of the related coronavirus SARS-CoV-1 revealed that the rate of infection can be decreased by replacing hydrophobic residues in the central hydrophobic transmembrane section by hydrophilic ones such as lysine [Citation67]. Replacing cysteine residues in the TM domain proximal to the membrane, next to the central hydrophobic part, inhibits membrane fusion completely whereas mutation of other cysteines in the cysteine-rich region does not have an impact on fusion [Citation99,Citation103,Citation104]. In all currently available experimentally determined prefusion structures, the heptad repeat 2 domain, transmembrane domain and the cytoplasmic tail are not resolved.
The cytoplasmic tail (CT) of coronavirus spike proteins is known to contain localization signals for the endoplasmic reticulum-Golgi intermediate compartment [Citation105–111]. At the farthest C-terminal end, a novel dibasic motif within the sequence KLHYT (KXHXX) has been identified as a functional ER retrieval signal, where X denotes any amino acid residue [Citation105–107,Citation112]. This motif may also contribute to interactions of the spike protein with copies of the membrane (M) protein located in the virion envelope [Citation106] In both SARS-CoV-1 and SARS-CoV-2, the folded cytoplasmic tail domain may interact with the M protein to facilitate incorporation of a complete spike into the new virus particle [Citation110,Citation113].
2.3. Availability and consistency of structures of the spike
Previous outbreaks of related viruses, namely of SARS-CoV-1 in China and Canada in February 2003, and of the Middle East respiratory syndrome coronavirus (MERS-CoV) since April 2012, lead to depositions of 43 SARS-CoV–1 and 29 MERS-CoV structures of the spike in the Protein Data Bank (PDB) [Citation114]. Their availability accelerated scientific progress at the beginning of the COVID-19 pandemic in 2020, as the spikes of SARS-CoV-2 are highly similar to those from SARS-CoV-1 and MERS-CoV [Citation115,Citation116]; the spike protein of SARS-CoV-2 has up to 76% sequence identity and 86% sequence similarity with that of SARS-CoV-1, depending on the viral strain. However it has an even closer relationship with the spike protein of RaTG13, a SARS-like beta coronavirus found in bats, with up to 97.4% sequence identity and 98.4% sequence similarity [Citation117], indicating a close phylogenetic relationship between the two viruses. RaTG13 is less studied, with only three structures of its spike present in the PDB [Citation118–120].
Comparison of viral spike structures from SARS-CoV-2 (PDB ID: 6VSB [Citation64]) and SARS-CoV-1 (PDB ID: 6CRZ [Citation121]) in a recent publication [Citation64], gives a global Cα r.m.s.d. of 3.8 Å, indicating that both structures are broadly similar with their receptor binding domains in the ‘down’ conformation. One of the more significant differences is that in SARS-CoV-1, the receptor binding domain (RBD) packs more tightly against the N-terminal domain of the neighbouring protomer, as opposed to SARS-CoV-2 where it is located closer to the trimer’s central cavity [Citation64]. Comparing individual domains and subunits shows a higher degree of structural homology with Cα r.m.s.d. values of 2.6 Å (NTD), 3.0 Å (RBD), 2.7 Å (SD1 and SD2) and 2.0 Å (S2 subunit) [Citation64]. Another analysis compared only the receptor binding domains of both viruses (SARS-CoV-2: PDB ID 6M0J [Citation89]; SARS-CoV-1: PDB ID 2AJF [Citation28]), finding a lower Cα r.m.s.d. of 1.2 Å. The receptor binding motifs show a higher sequence variation but still high structural conservation (1.3 Å Cα r.m.s.d.), indicating that the overall binding mode between SARS-CoV-1 and SARS-CoV-2 is nearly identical [Citation89]. There are a number of other available services and studies of coronavirus structures [Citation122], including specific analysis of the spike protein variability [Citation123], and resources for accumulating data beyond experimental structures (https://covid.molssi.org/).
Due to its key role in host-cell invasion, the spike is one of the most researched complexes of SARS-CoV-2 and one of the most common structures in the PDB: in total, more than 800 structures have been deposited. Structure-solution by single-particle cryo-electron microscopy and cryo-electron tomography has provided structures of both unbound trimers (e.g. PDB 6XR8 [Citation18], 6X6P [Citation124]) and trimers in complex with receptor domains and antibodies (e.g. PDB 7EH5 [Citation125,p.7], 7A97 [Citation10], 7L2F [Citation126]). Currently, there are over 600 cryo-EM structures available in the PDB ranging in resolution from 2.2 to 13.5 Å [Citation127–129]. The remaining structures have mostly been solved by X-ray crystallography, and are primarily of isolated receptor binding domains binding to Angiotensin-Converting Enzyme 2 receptor domains (e.g. PDB 6LZG [Citation130]), antibodies or small molecules, and range from 1.24 to 4.35 Å resolution [Citation89,Citation131–133]. NMR has been used to determine the structure of the transmembrane domain, the membrane-bound Heptad Repeat 1 domain as well as an fusion peptide domain inserted into a bilayered micelle [Citation134–136]. A summary of the most important experimentally determined structures available in the PDB is given in Table .
Table 2. A selection of high quality spike structures according to our evaluation[Citation137] available from the PDB, which may be useful for users to use in e.g. structure-based drug design. Please note that these are not the first or seminal structures of each type in the PDB, but rather a curated set used by the authors.
We highlight one story which demonstrates the incompleteness of structural models, and how users of the PDB should be careful and aware of what is not represented/resolved in the structure: The first model of a complete SARS-CoV-2 virion, published by the Centers for Disease Control and Prevention in April 2020, was created using a PDB structure that was believed to show the complete ectodomain of the spike. The selected PDB structure only contained the head and the top part of the stalk of the spike molecule instead of a full-length ectodomain [Citation147], and consequently, the first 3D model of a SARS-CoV-2 viral particle erroneously shows truncated spikes without full stalks; this error has been subsequently corrected [Citation147] but images of the incorrect model are still commonly published.
To date, all experimentally determined structures of the spike show the ectodomain with only the top of stalk resolved. Some components of the stalk, including the Heptad Repeat 2 domain, the transmembrane domain and the cytosolic tail have never been resolved in a prefusion structure, while conversely parts of the fusion peptide have only ever been resolved in prefusion state models. Other available structures refer only to single domains or the fusion core (e.g. PDB 6M1 V [Citation139] and 7L4Z [Citation148]). As of writing this review, there is only one experimental structure in the PDB that shows nearly the full-length post-fusion spike (Figure c), which was obtained via cryo–EM (PDB: 6XRA [Citation18]).
3. Structural changes in the spike protein during maturation and viral entry to the cell
SARS-CoV-2 cell entry begins with the binding of spikes to angiotensin converting enzyme 2 (ACE2) receptors [Citation11,Citation16,Citation89,Citation149]. It is suggested that upon engaging the ACE2 receptor, the fusion protein undergoes proteolytic cleavage at the ‘S2’ site’ (Figure b) [Citation15–17], which may facilitate the release of the fusion peptides [Citation93,Citation150–152]. The first subunit is shed, while the second subunit undergoes significant structural rearrangements to orient the fusion peptide towards and embed it into the host cell membrane. Further rearrangements of the S2 subunit result in the formation of a hairpin structure that brings the viral and host cell membranes into close apposition, ultimately leading to membrane fusion [Citation18,Citation95,Citation153–155].
Figure 2. Conformations of the receptor binding domains of the spike and binding to the ACE2 receptor. Abbreviations (as listed in Table ): RBD: receptor binding domain, SD2: sub-domain 2, FP2: fusion peptide region 2, TMPRSS2: type II transmembrane serin protease. (a) Cryo-EM structures of spikes with RBDs in down conformation (‘closed'; PDB 7N1U) and with one RBD in the up conformation (‘open'; PDB 7N1X). The three RBDs are coloured in light green, light blue and dark green. (b) Cryo-EM structure of spike (PDB 7N1X) with the location of the cleavage sites S1/S2 (furin) and S2´ (TMPRSS2) highlighted. (c) Complex structure (PDB 7DF4) of a spike bound to one ACE2 receptor N-terminal peptidase domain (ACE2 in purple, RBM highlighted in mint green). Inset images: Interactions in the interface between the RBM and ACE2 (PDB 6LZG).
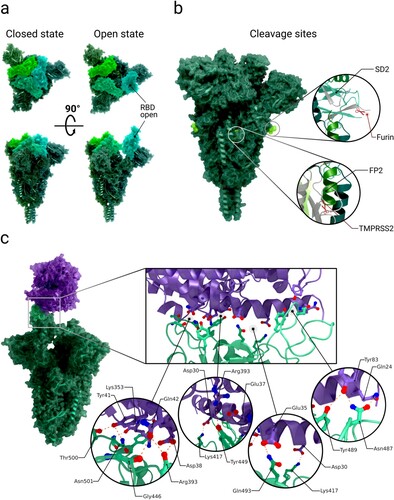
It is important to note that the precise mechanism of viral entry is not yet fully understood, and many steps described herein are inferred to occur based on analogy with other, well studied, viral fusion proteins such as spikes from HIV, influenza or MERS-CoV [Citation20,Citation23–26].
3.1. Spike maturation
For effective infection, the spike has been suggested to mature with a proteolytic cleavage event at the S1/S2 interface during synthesis of the virus (Table and Figure b) [Citation15–17]. In contrast to the coronavirus SARS-CoV-1, the interface of the S1/S2 subunit in SARS-CoV-2 contains a multibasic cleavage site (Table )[Citation72], the existence of which is a central virulence factor for many viruses [Citation72,Citation156]. The multibasic site exhibits a RRAR amino acid motif, which resembles the canonical recognition sequence RX(R/K)R for furin, a calcium-dependent protease (X can be any amino acid residue) [Citation138,Citation157–159]. Cleavage in the S1/S2 site by either furin or other proteases likely happens in the Golgi apparatus of the host cell during spike synthesis [Citation160,Citation161]. After the cleavage at S1/S2, every spike protomer consists of two subunits that remain connected solely by non-covalent interactions [Citation2,Citation138,Citation162]. This event is suggested to convert the spike into its fusion-competent form [Citation163] as it could potentially promote the mobility of S1 receptor-binding domains (RBDs) and N-terminal domains (NTDs), resulting in decreased thermal stability, thereby facilitating the adaptation of the open conformation necessary for receptor binding [Citation72,Citation119,Citation164,Citation165]. Full cleavage at the S1/S2 interface is also assumed to be necessary for the first subunit to dissociate before refolding into the post-fusion state[Citation166].
Table 3. Known cleavage sites on the spike protein and associated proteases.
3.2. Receptor recognition and binding
The receptor binding domains of the spike, which recognize and bind the ACE2 receptor, are located at the top of each spike in the S1 subunit [Citation28,Citation29,Citation89,Citation142]. Each RBD (one per protomer) can adopt two conformations, such that it is oriented in the ‘up’ or ‘down’ position (Figure a) [Citation18,Citation64,Citation167,Citation168]. When the domain adopts the ‘up’ conformation, the receptor binding motif (RBM) is exposed and is available to interact with the surface cell receptor ACE2 or antibodies [Citation167,Citation169]. In its ‘down’ conformation, the RBD interacts with neither ACE2 nor antibodies. The dynamics of this domain are thus relevant for immune evasion [Citation53,Citation64,Citation169,Citation170]. An analysis of atomic B-factors supports flexibility or instability of the ‘down’ state of the RBD through increased disorder of the RBD compared to surrounding domains [Citation171].
Structures of the receptor binding domain in complex with the human angiotensin converting enzyme 2 receptor reveal the interactions between the spike protein’s receptor binding motif and one of two binding-site lobes of the ACE2 N-terminal peptidase domain [Citation89] (c.f. PDB 7DQA [Citation130], 6LZG [Citation89], 6VW1 [Citation131]). The contact between the receptor binding domain (with an interaction surface area of 864 Å2) and ACE2 (with an interaction surface area of 823 Å2) is established at the bottom side of the ACE2 small lobe, with a concave outer surface in the receptor binding motif accommodating the N-terminal helix of the ACE2, generating a total buried surface area of 1687 Å2 in total [Citation89]. Buried surface areas above approximately 700 Å2 indicate a rather tight binding which is stable in solution [Citation172]. The RBM/RBD-ACE2 interface contains 13 hydrogen bonds and 2 salt bridges, with one salt-bridge outside of the RBM in the RBD between RBD Lys417 and ACE2 Asp30 [Citation89,Citation173] (see Figure c).
Even though the spike proteins of SARS-CoV-2 and SARS-CoV-1 share up to 76% sequence identity, their receptor binding motifs share only 47.8% sequence identity [Citation173]. Mutation studies performed to evaluate how the sequence differences impact binding affinity to human ACE2 revealed that the substitution of amino acid Pro499, Gln493, Phe486, Ala475 and Leu455 in SARS-CoV-2 enhances ACE2 binding compared to SARS-CoV-1 [Citation173,Citation174]. Substitution of Gln493 to glutamate leads to substantial structural changes, removing an internal salt bridge between ACE2 residues Lys31 and Glu35 that is buried in a hydrophobic environment and replacing it by two hydrogen bonds between each of the residues to Glu493. Lys31 of ACE2 forms another hydrogen bond with the non-polar Leu455 [Citation89,Citation173,Citation174].
After binding the host cell surface receptor ACE2, domains of subunit S1 undergo several rearrangements [Citation18]. One domain that is hypothesised to play a significant role within these rearrangements the fusion peptide proximal region (FPPR) (residues 828-853) which is situated next to SD1 [Citation18,Citation175–177]. The structure PDB 6XR8 [Citation18] with its RBD in ‘down’ conformation shows the FPPR in an ordered state [Citation18], packed around an internal disulphide bond between cysteines C840 and C851. This packing is reinforced by an extensive hydrogen network and an additional salt bridge between Lys835 and Asp848 [Citation18]. Superimposing the S2 domain of 6XR8 [Citation18], which contains the ordered FPPR, with a structure where the receptor binding domain is in the ‘up’ conformation (PDB 6VSB [Citation64]) results in clashes between the FPPR and an outwards-rotated sub-domain 1 (SD1; Figure a and 3b) [Citation18]. Thus, it is speculated that in the ‘up’ conformation, the FPPR is delocalized whereas in the closed state it helps to clamp down the receptor binding domain and stabilizes the ‘down’ conformation[Citation18,Citation59,Citation175–178]. SD1 is supposed to act as a relay between both domains (Figure b) [Citation18,Citation59].
Figure 3. Domain shifts between the up and down conformations of the spike protein receptor binding domain. Abbreviations (as listed in Table ): NTD: N-terminal domain, RBD: receptor binding domain, SD1: sub-domain 1, SD2: sub-domain 2; FPPR: fusion peptide proximal region. (a) Superimposed spike protein structures with receptor binding domains in up (PDB 6VSB; coloured by domain) and down (PDB 6VXX; coloured grey) conformations. The fusion peptide proximal region is not resolved in either structure. With the movement of the receptor binding domain into the ‘up' conformation, the SD1 domain of translates by approximately 1.9 Å. (b) Superimposed spike structures with receptor binding domains in up (PDB 6VSB; colours as in a) and down (PDB 6XR8; green) conformations; for 6XR8 only the fusion peptide proximal region is shown. In contrast to (a), the fusion peptide proximal region is resolved in 6XR8. When the receptor binding domain switches into ‘up' conformation, the SD1 domain rotates outwards and would then clash with an ordered fusion peptide proximal region (as shown), so the FPPR must delocalise in the up conformation.
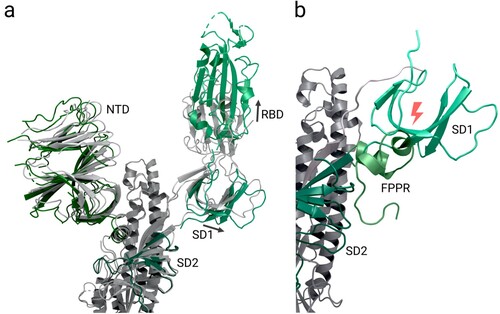
A proposed mechanism suggests that the stochastic thermal departure of FPPR from its stabilizing position enables the SD1 to rotate and the receptor binding domain to change to the up conformation to engage ACE2. The ‘up’ conformation is then fixed when the receptor binding motif binds the ACE2 receptor of the host cell, thereby leading to exposure of the S2’ cleavage site upstream of the fusion peptide [Citation18,Citation59,Citation178]. It's important to note that this proposed mechanism is based on theoretical considerations and requires further investigation for validation.
By comparing the native spike with one receptor binding domain in the ‘up’ position while either unbound or bound to ACE2, the binding to ACE2 appears to shift the position of the extended RBD, amounting to a rigid-body rotation that moves the RBD’s centre of mass approximately 5.5 Å away from the trimer axis [Citation10]. Additionally, the subdomains associated with the RBD (SD1) as well as the N-terminal domain (SD2) of the same monomer translate ∼1.9 Å and ∼2.3 Å, respectively. Within this translation, all three N-terminal domains move by roughly 1.5-3.0 Å [Citation10].
The flexibility of spikes has been proposed to promote receptor recognition [Citation179]. Cryo-electron tomography shows that spikes are highly variable with a standard deviation of 20° about the average deviation of 40° away from a perpendicular orientation to the viral membrane [Citation179,Citation180]. The sparse distribution of spikes on the viral envelope of SARS-CoV-2 particles may be crucial for allowing this flexibility, a characteristic that is markedly distinct to other class I fusion proteins [Citation179,Citation181]. Several Y-shaped spike assemblies were also observed on the viral surface [Citation179,Citation181]: due to the high degree of rotational freedom, adjacent spikes had intertwined their stems [Citation179,Citation181]. This feature may allow the spikes to better engage ACE2 receptors but a sparsely occupied surface may also make these molecules more vulnerable to antibody recognition [Citation179,Citation181].
3.3. Post-binding spike protein cleavage and activation
For the cleavage event that follows binding to the receptor, two different proteases are currently under consideration, depending on the viral cell entry route. Viral cell entry was observed to occur either through the plasma membrane pathway via the cell surface [Citation16,Citation157,Citation182–184] or the endosomal pathway after virus endocytosis [Citation184–188]. In the plasma membrane pathway, type II transmembrane serine protease (TMPRSS2) is thought to have a significant role by cleaving a monobasic site at location S2’ (Table ) [Citation15–17], possibly being necessary for releasing the fusion peptides [Citation93,Citation150–152]. This cell surface protein is widely expressed in epithelial cells of the respiratory and gastrointestinal tract [Citation14]. In studies where target cells lacked cell surface proteases like TMPRSS2, SARS-CoV-2 was internalized via endocytosis, with spike protein cleavage likely to be executed by cathepsin B or L [Citation157,Citation184,Citation186,Citation189]. The exact cleavage site/sites remain in discussion. Proposed locations are either directly at S2’ or upstream of this site [Citation190–192]. What cause (e.g. spike protein cleavage, dissociation of S1 or others) triggers the rearrangements from the prefusion to the post-fusion state is not fully clear and remains to be investigated.
The exact mechanism of the pre- to post-fusion transition of SARS-CoV-2 as well as membrane fusion itself is not yet fully understood. However, based on research of different fusion proteins including SARS-CoV-2 and 1, mechanisms for the rearrangements in the spike protein’s subunit S2 and the membrane fusion are proposed in the following three sections.
Hypothetical rearrangement in S2 to form the fusion peptide ‘complex’
Formation of the fusion peptide ‘complex’ after shedding of S1 entails the conformational rearrangements of the heptad repeat 1 (HR1) and heptad repeat 2 (HR2) domains, exposing the proposed fusion peptide regions (FP1 and FP2), which subsequently insert into the host cell membrane [Citation20,Citation155,Citation193–196]. As a first step in the formation of the complex, heptad repeat 1 rearranges to form a 180 Å-long three-stranded coiled-coil together with the central helix (CH) domain [Citation18,Citation62,Citation95,Citation155]. Simultaneously a section of heptad repeat 2 (I1169 to Q1180) adopts an extended structure with the remaining part of heptad repeat 2 forming a canonical α-helical structure [Citation139]. It is suggested that, similar to proposed mechanisms of other coronaviruses, the extension of the long central helix by inclusion of the N-terminal section of heptad repeat 1 (amino acids 990–1035) projects the fusion peptide in the direction of the host cell membrane [Citation10,Citation62,Citation95,Citation155].
The dissociation of S1 and cleavage of the S2´ site at 815R|S816 by type II transmembrane serine protease (TMPRSS2) is suggested to release the fusion peptides that contain several conserved amino acids as the new N-terminus of S2 [Citation67,Citation139,Citation197]. Located on the first consecutive section (FP1), there is an additional conserved hydrophobic LLF motif (L821/L822/F823) [Citation61,Citation93,Citation97,Citation197] which is thought to penetrate the target cell membrane and was previously shown to be crucial for interaction of the SARS-CoV-1 fusion peptides with lipid membranes [Citation61,Citation92]. The released fusion peptides trimerize to form a so-called ‘spearhead’ [Citation61,Citation95,Citation198] that anchors the spike in the target cell membrane [Citation2,Citation25,Citation61] through its hydrophobic residues [Citation199]. FP1 and FP2 are suggested to form a bipartite membrane interaction platform where certain residues enhance binding of the host cell membrane by interaction with Ca2+ ions [Citation61,Citation196,Citation200]. At the same time, the cysteine rich CT that anchors the C-terminal part of the spike in the viral membrane gets reinforced by a membrane-actin linker named ezrin [Citation67]. After conformational rearrangements in S2, the membranes are in close apposition for fusion [Citation61,Citation67].
Rearrangements in S2 leading to fusion and formation of post-fusion hairpin state.
Based on studies of other Class I virus fusion proteins it has been proposed that after inserting the spearhead into the host cell membrane, the two membranes are pulled together to initiate membrane fusion. As a result of this movement, the spike refolds into a closing hairpin structure [Citation18,Citation20–22]. Here, heptad repeats 1 and 2 play a crucial part [Citation18]: The repeated hydrophobic amino acids from the HPPHCPC motifs form a hydrophobic interface and drive heptad-repeat hairpin trimerization and furthermore six-helix bundle formation for membrane fusion with the host cell [Citation2,Citation18,Citation21,Citation57]. The post-fusion state of fusion domains, in particular the six-helix bundle or post-fusion core, has also been relatively well characterized through research of various viruses [Citation201] such as HIV gp41 [Citation202–204], parainfluenza virus F [Citation205], murine hepatitis virus [Citation20], and SARS-CoV [Citation22,Citation155,Citation195]. In the first post-fusion bundle of six helices, the hydrophobic groove of HR1 is bound in an antiparallel manner [Citation2] by the N-terminal section of HR2, which adopts a helical conformation with one turn [Citation18]. Together with the C-terminal domain of HR2, this organizes into a longer helix; the remaining part of the coiled coil of HR1 arranges in another bundle of six helices [Citation18,Citation206,Citation207] (see Figure a and 4b). Structural analysis of interactions between both HR domains reveals that, as well as hydrophobic interactions, multiple hydrogen bonds between the two domains occur (Asn925, Gln935, Gln949, Asn953, Asn960 of HR1 and Ala1174, Val1177, Ile1179, Gln1180, Ala1190, Asn1194, Ile1198 of HR2) [Citation139]. These multiple reinforcements of the central coiled coils result in a stable post-fusion structure [Citation18].
Figure 4. The post-fusion state of the spike and the fusion process. Abbreviations: S1: subunit 1, S2: subunit 2, HR1: heptad Repeat 1, CH: central helix, CD: connector domain, HR2: heptad repeat 2, FP: fusion peptides, TM: transmembrane domain, 6HB: six-helix bundle. (a) Spike structure in post-fusion state (PDB 6XRA). During the fusion process the spike folds into a hairpin structure. During transition to the post-fusion state, two six-helix bundles are formed. In the first, the hydrophobic groove of HR1 is bound in an antiparallel manner to the N-terminal section of HR2. (b) A 90° rotated image of the first six-helix bundle in (a). The second six-helix bundle is formed of amino acids 737–769 together with parts of the central helix (CH) domain. (c) Schematic illustration of the cell entry via the plasma membrane pathway. The spike receptor binding domain switches from closed into open state and binds to ACE2. This interaction likely allows the type II transmembrane serine protease (TMPRSS2) to cleave the spike protomer to release the fusion peptides. Additionally, the subunit S1 dissociates. The chronological sequence of these two events (cleavage and S1 dissociation) still needs to be examined. Subsequently, the heptad repeat 1 (HR1) domain of S2 extends the central helix (CH) domain to project the ‘fusion peptide-spearhead' towards the host cell membrane. After insertion of the spearhead, the spike rearranges into the post-fusion hairpin state bringing both membranes into close apposition for fusion. This figure was created with Protein Imager [Citation54], Blender [Citation55], and Inkscape [Citation56]
![Figure 4. The post-fusion state of the spike and the fusion process. Abbreviations: S1: subunit 1, S2: subunit 2, HR1: heptad Repeat 1, CH: central helix, CD: connector domain, HR2: heptad repeat 2, FP: fusion peptides, TM: transmembrane domain, 6HB: six-helix bundle. (a) Spike structure in post-fusion state (PDB 6XRA). During the fusion process the spike folds into a hairpin structure. During transition to the post-fusion state, two six-helix bundles are formed. In the first, the hydrophobic groove of HR1 is bound in an antiparallel manner to the N-terminal section of HR2. (b) A 90° rotated image of the first six-helix bundle in (a). The second six-helix bundle is formed of amino acids 737–769 together with parts of the central helix (CH) domain. (c) Schematic illustration of the cell entry via the plasma membrane pathway. The spike receptor binding domain switches from closed into open state and binds to ACE2. This interaction likely allows the type II transmembrane serine protease (TMPRSS2) to cleave the spike protomer to release the fusion peptides. Additionally, the subunit S1 dissociates. The chronological sequence of these two events (cleavage and S1 dissociation) still needs to be examined. Subsequently, the heptad repeat 1 (HR1) domain of S2 extends the central helix (CH) domain to project the ‘fusion peptide-spearhead' towards the host cell membrane. After insertion of the spearhead, the spike rearranges into the post-fusion hairpin state bringing both membranes into close apposition for fusion. This figure was created with Protein Imager [Citation54], Blender [Citation55], and Inkscape [Citation56]](/cms/asset/ab119122-a3fd-4d49-8cd5-cf29d439cad6/gcry_a_2363756_f0004_oc.jpg)
In the prefusion state, the connector domain folds collectively with a section of the S1/S2´ fragment (amino acid 718-729) into a three stranded β-sheet [Citation18,Citation62]. During the transition to the post-fusion state, the β-sheet does not alter position but instead is expanded by another strand formed of residues 1127–1135 (between connector domain and heptad repeat 2), as the C-terminal HR2 domain is projected towards the viral membrane [Citation18,Citation62]. Amino acids 737–769 from each spike protomer form a helical region which packs against the grooves of the CH forming another six-helix-bundle structure locked in place through disulphide bonds [Citation18,Citation62]. Both of these fragments are an integral part of the post fusion structure, and do not dissociate even after the cleavage event at the S2´ site [Citation18,Citation62].
3.4. Membrane fusion mediated by the fusion peptide
After insertion of the fusion peptide (FP) into the host cell membrane, and the formation of the hairpin post-fusion structure, both membranes are brought into close proximity [Citation61,Citation67]. The FP then mediates viral and host cell membrane fusion by disrupting and connecting lipid bilayers [Citation61,Citation199,Citation208]. During the fusion process, a fusion pore is formed enabling the transfer of the viral genetic material into the host cell [Citation163,Citation209]. To date, there is no molecular-level data about the structure of this spearhead or its interaction with the cell membrane [Citation61] and the mechanism leading to formation of the fusion pore is only poorly understood [Citation209]. Other regions which might also contribute to the fusion process have also not yet been sufficiently investigated, such as a conserved hydrophobic region next to the transmembrane domain that is necessary for trimerization of the spike protein and the connector domain region [Citation18]. Hence, a series of questions regarding the fusion process remain unanswered[Citation18]. A schematic illustration of the fusion process is given in Figure c.
4. Glycosylation
Spikes of enveloped viruses are heavily glycosylated [Citation210]. Viral glycosylation plays a major role in many different intrinsic and extrinsic properties of the complex, being essential to folding, trafficking and regulating interactions with lectins while also modulating binding to other receptors and glycans that mediate host cell entry [Citation38]. The characteristic dense coat of complex carbohydrates, also known as a glycan shield, retains a high degree of flexibility, dynamics and microheterogeneity [Citation210]. In addition to these functions, the highly dense glycan shield is known to effectively mask the virus from the immune system, protecting viral protein from recognition by cells and antibodies [Citation38,Citation41,Citation42].
Each SARS-CoV-2 spike protomer contains 22 N-glycosylation sites (sequons) [Citation36,Citation211,Citation212]. Of these, 18 are consistently occupied [Citation211], with predominantly complex N-glycan structures, and likely shield more than 60% of the surface of a spike’s head (S1) from antibody-sized molecules (i.e. scFVs, mAbs, nanobodies, etc; Figure a) [Citation41,Citation42,Citation213]. For smaller molecules, the shielding is suggested to be less effective [Citation41]. 17 O-glycosylation sites [Citation214] have also been identified, with a much lower degree of occupancy, averaging less than 2% [Citation215]. Of these 17 glycosylation sites, 11 are located on subunit S1 and the remaining 6 on subunit S2 [Citation214]. An essential aspect of this molecular disguise is that the spike protein is glycosylated by the host cell machinery, mimicking the natural glycosylation of the host’s proteins [Citation106,Citation160]. The spike protein’s N-glycans are linked to the protein surface in the endoplasmic reticulum and further refined in the Golgi apparatus of the infected host cell [Citation106,Citation160]. While the viruses move through the Golgi lumina, N-glycans undergo extensive processing by glycosylation enzymes. O-glycosylation also occurs in the Golgi [Citation160]. An overview over the types of N-glycans and O-glycans functionalizing the spike is given in Figure b.
Figure 5. Glycosylation of the spike protein. (a) Structures of glycosylated spikes in pre- and post-fusion conformation. Glycans are highlighted in blue. The prefusion structure shows a fully glycosylated SARS-CoV-2 spike ectodomain of the omicron variant BA2. The structure of the post-fusion conformation (PDB 6XRA) shows in contrast only fragments of glycans that could be resolved by cryo-EM. (b) Different types of N-glycan and O-glycan structures that occur in the S glycoproteins, namely oligomannose-, hybride- and complex type N-glycans [Citation219] as well as Core-1 and Core-2 O-glycans (less than 2%) [Citation220,Citation221] could be observed.
![Figure 5. Glycosylation of the spike protein. (a) Structures of glycosylated spikes in pre- and post-fusion conformation. Glycans are highlighted in blue. The prefusion structure shows a fully glycosylated SARS-CoV-2 spike ectodomain of the omicron variant BA2. The structure of the post-fusion conformation (PDB 6XRA) shows in contrast only fragments of glycans that could be resolved by cryo-EM. (b) Different types of N-glycan and O-glycan structures that occur in the S glycoproteins, namely oligomannose-, hybride- and complex type N-glycans [Citation219] as well as Core-1 and Core-2 O-glycans (less than 2%) [Citation220,Citation221] could be observed.](/cms/asset/5914a464-3404-4a34-8150-c48429255a62/gcry_a_2363756_f0005_oc.jpg)
The host protease furin, or some similar enzyme, cleaves the spike protomer at the S1/S2 interface before the virus gets secreted via lysosomes into the extracellular surroundings [Citation160,Citation161], and O-glycosylation has been shown to have both positive and negative impacts on proteolytic cleavage events [Citation216]. Therefore, differences in O-glycan expression may potentially modulate this protease activity and thus viral infectivity [Citation216].
The heterogeneous nature of the glycan shield also precludes experimental structural characterization [Citation210]. Thus, most structural studies of the viral glycan shield rely on molecular dynamics simulations [Citation210]. All-atom molecular simulations are highly complementary to glycoproteomics studies that indicate the type of glycan structures and the relative occupancy of the sites of glycosylation [Citation217], and to cryo-EM data. One limitation is that with cryo-EM only the most rigid (and commonly shared) part of the glycan structure [Citation218] can be resolved as shown in the post-fusion state in Figure a.
As a unique feature of the SARS-CoV-2 spike, the glycan shield is also mechanistically functional. Three important N-glycan sites, Asn165, Asn343 and Asn234 mediate the RBD conformational dynamics with a focus on stabilizing the RBD ‘up’ [Citation41,Citation222] conformation by a ‘load and lock’ mechanism [Citation41,Citation44,Citation222]. The mutations N165A/N234A and N234A lead to a significantly reduced ACE2 binding and cause a shift in the RBD conformational ensemble towards the ‘down’ conformation [Citation41].
5. Therapeutic interest
5.1. Antigenic epitopes in natural immune responses and vaccine development
As the largest protein on the viral surface [Citation31], the spike protein is the main antigen targeted by the immune system, particularly the receptor binding domain [Citation57,Citation223–225]. The spike can provoke both a strong T-cell response and a neutralizing immune response, inducing protective immunity against COVID-19 [Citation57]. This makes the surface protein attractive both as a target for development of vaccines and for drugs against SARS-CoV-2 [Citation31,Citation226]. One study of infected patients showed that monoclonal antibodies raised against the spike distributed reasonably well across the different regions. Of 377 antibodies isolated from 77 recovered COVID-19 patients, 34% bind to S1 and 53% bind to S2 [Citation227,Citation228]. The remaining 13% interact with none of the subdomains [Citation227]. Of the antibodies that bind to S1, 21% bind to the RBD and 11% to the NTD [Citation227]. Almost all inherently neutralising antibodies bind to the RBD, along with a smaller set which bind to an ‘antigenic supersite’ on the N-terminal domain located at the most radially distant loops from the centre of the spike [Citation227,Citation229]. However, one class of antibodies, originally discovered through cross-reactivity from SARS-CoV-1 antibody CR3022 [Citation230] and then rediscovered in patient-derived antibodies as EY6A [Citation231], achieves neutralization by forcing RBDs to an ‘up and out’ conformation which shows destabilization of the spike protein in cryo-EM images.
The immunodominance of the receptor binding domain is accounted for by the extensive shielding of most spike exodomains by N- or O-linked glycans [Citation85]. Additionally, as it mediates specificity in cell tropism, it elicits highly functionally relevant antibodies which prevent RBD-ACE2 interaction while avoiding the risk of antibody-dependent enhancement that could be mediated by weak or non-neutralizing antibodies [Citation85]. Rather than contributing to the immunity, such non-neutralizing antibodies facilitate viral entry and subsequent infection of the host cell, leading to an increased virulence [Citation85]. One example of such an epitope in SARS-CoV-1 is found nearby to the RBD: antibodies targeting this region markedly enhance the effectiveness infection [Citation85]. The RBD also contains epitopes for T-cell response [Citation232,p.19]. Most of the currently developed vaccines, encompassing both vector-based as well as mRNA-based vaccines, include the RBD in their immunogens [Citation233].
Other epitopes on the spike targeted by neutralizing antibodies are found on the NTD of the S1 subunit [Citation82,Citation85,Citation234,Citation235]. Antibodies targeting these regions do not directly prevent receptor binding but rather interfere with it or restrain the rearrangements from the prefusion to the post-fusion conformation [Citation85]. These antibodies show weaker neutralizing potency than those targeting the RBD [Citation82,Citation85]. Structural analysis of antibodies in complex with the N-terminal domain (NTD) revealed that the interaction takes place primarily within two glycan-free surfaces named the NTD-1 and NTD-2 regions (Figure a) [Citation62,Citation236]. NTD-1, located in the NTD top-core region, is targeted by most antibodies and is designated as the NTD-1-antigenic supersite [Citation62]. This site comprises five loop structures named N1-5 as well as an additional β-hairpin structure near N3. The loop structures span the amino acids 14–26 (N1), 67–79 (N2), 141–156 (N3), 177–186 (N4) and 246–260 (N5). Different reconfigurations of the loops are observed for different antibodies binding to the highly conserved NTD-1 supersite (Figure b and 6c) [Citation62].
Figure 6. Structure of the N-terminal domain and interactions with antibodies. (a) Overview of the N-terminal domain of S1 (PDB 6XR8). Regions targeted by antibodies are highlighted in mint green (NTD-1) and pale green (NTD-2). b) Different reconfigurations within the NTD-1 antigenic supersite caused by interactions with the monoclonal antibody 4A8 are shown by superimposing PDB 7C2L with PDB 6XR8. PDB 7C2L refers to a NTD structure in complex with 4A8. The NTD-1 spans five loop structures labelled N1 to 5. (c) Structure of the NTD in complex with antibody 4A8 (PDB 7C2L). Important interactions take place between the heavy chain of 4A8 (yellow green) and residues within loop N3 and N5 (mint green)[Citation82].
![Figure 6. Structure of the N-terminal domain and interactions with antibodies. (a) Overview of the N-terminal domain of S1 (PDB 6XR8). Regions targeted by antibodies are highlighted in mint green (NTD-1) and pale green (NTD-2). b) Different reconfigurations within the NTD-1 antigenic supersite caused by interactions with the monoclonal antibody 4A8 are shown by superimposing PDB 7C2L with PDB 6XR8. PDB 7C2L refers to a NTD structure in complex with 4A8. The NTD-1 spans five loop structures labelled N1 to 5. (c) Structure of the NTD in complex with antibody 4A8 (PDB 7C2L). Important interactions take place between the heavy chain of 4A8 (yellow green) and residues within loop N3 and N5 (mint green)[Citation82].](/cms/asset/00e22dfa-d8d6-4130-9548-37e1147ba273/gcry_a_2363756_f0006_oc.jpg)
The part of subunit S2 proximal to the viral membrane also poses a potential target for neutralizing antibodies, even though it is more extensively shielded by glycans [Citation35,Citation85,Citation138], making it less immunogenic [Citation85]. It is possible that antibodies targeting S2 might interfere with the structural rearrangement of the spike protein and the insertion of the FP required for virus–host membrane fusion [Citation85]. Comparing neutralizing antibodies obtained from convalescent patients revealed that S2 targeting antibodies exhibit a lower neutralizing effect than those specific to the receptor binding domain in S1 [Citation85,Citation237].
5.2. Spike protein constructs in vaccine design
Current vaccines use different strategies to induce a robust immune response against the spike protein [Citation238]. The two most prevalent mRNA vaccines (‘BNT162b2’ by Pfizer–BioNTech and ‘mRNA-1273’ by Moderna) encode for a full-length spike protein stabilized in the prefusion state by two distinct mutations (K986P and V987P) [Citation238,Citation239], whilst another vaccine (‘CoronaVac’ by Sinovac) employs the strategy of a wild-type spike protein present on the viral surface of the inactivated virus vaccine [Citation238]. Novavax has recently completed a phase 3 trial showing strong efficacy for a vaccine (NVX-CoV2373) that only contains spikes as the antigen. The contained spikes were produced with the same two proline changes present in the mRNA sequences, but also contain three changes to the ‘furin’ cleavage site rendering it unrecognizable [Citation64].
Structures in the prefusion state with the RBD either in ‘up’ or ‘down’ conformation are a focus of vaccine development, due to immunogenicity of the RBD epitopes. Many of the constructs used for vaccine design contain two mutated amino acids at positions 986 and 987 [Citation18], which help to stabilize the spike in the prefusion state. However, these are now believed to alter the protein structure relative to the wild-type, as through the artificial mutation K986P a salt bridge between Lys986 and either Asp427 or Asp428 of an adjacent protomer is broken, leading to a relaxed apex [Citation18]. In a recent publication, the authors raised concerns that the altered structure could have an impact on vaccine efficiency, as antibodies induced by the spike with a relaxed apex may not recognize spikes presented on a viral surface [Citation18]. Hence, for vaccine design, structures of proteins in prefusion state without such kind of stabilizing mutations would presumably be preferred [Citation18]. Nevertheless, the currently used vaccines, Pfizer-BioNTech's ‘BNT162b2’ and Moderna's ‘mRNA-1273’, both using a stabilized prefusion spike, show a > 90% efficacy against wild-type SARS-CoV-2 after the second vaccination and six month follow up [Citation240–244]. Several studies highlight the contributions of structural biology in the ongoing efforts to combat COVID-19, addressing challenges related to vaccine design and emerging variants [Citation245–247].
5.3. Mutations and immune evasion
Since the first occurrence of SARS-CoV-2 in Wuhan in 2019, the virus genome has repeatedly changed due to genetic drift [Citation248–250]. A genomic analysis of 10,333 spike protein sequences taken from samples from across the globe revealed that there are 400 distinct mutation sites in the spike protein, and 44 in the receptor binding domain alone [Citation49]. Another site of frequent mutation is the region around the protease cleavage site (between residues 675-692). Structural data of the full-length prefusion ectodomain is also used to facilitate evaluation of mutations in different viral strains [Citation64] including notable variants of concern such as the Delta variant (B.1.1617.2) and the Omicron Variant (B.1.1.529). Depending on the location, each mutation impacts the spike conformational dynamics, receptor binding affinity or antigenicity, or combinations thereof [Citation251]. For Omicron and Delta, recent studies suggest that due to their mutations, both strains are less sensitive to neutralizing monoclonal antibodies from recovered patients and capable of evading immunity conferred through prior infections [Citation252,Citation253]. An overview of some of the latest variants of concern and interest and their mutations in the spike protein is given in Table . PDB codes related to latest virus variants are summarized in Table .
Table 4. Latest variants of concern and their mutations.
Table 5. PDB Codes related to named coronavirus variants.
The spike protein of the Delta variant contains eight mutations and two deletions and the variant is reported to be twice as infectious as the wild-type [Citation254,Citation255]. Mutation T19R and the deletion 156 and 157 are located in the N-termina domain ‘supersite’ which is the target of most anti-NTD antibodies [Citation256]. Mutation L452R and T478K, located in the receptor binding domain, have been identified as potentially significant for the infectivity and transmissibility of the virus [Citation252]. A simulation study noted a difference in the receptor-binding interface between the wild type and a L452R/T478K variant [Citation252]: A receptor-binding β-loop-β motif adopts an altered conformation, leading to shifts in epitope regions and a reduction in antibody recognition [Citation252].
In contrast to the Delta variant, the Omicron variant is much more heavily mutated and includes up to 30 mutations in the spike protein relative to the wild-type (Wuhan-Hu-1), depending on the precise sub-variant (BA.1 or BA.2) [Citation262]. Of those mutations, approximately half are located in the RBD and 11 are involved in receptor binding [Citation263]. These mutations substantially affect the binding mode to ACE2 and its interaction with antibodies and drugs developed against the wild-type spike protein [Citation251,Citation263,Citation264]. To date, it seems that Omicron is twice as infectious as Delta and more than ten times as infectious as the wild type [Citation264]. Key residues proposed to contribute in a critical way to the enhanced affinity of Omicron to ACE2 include mutations T478K and N501Y, which were also present in prior variants (T487K: Delta; N501Y: Alpha, Beta) and N440K [Citation264]. Mutations K417N, E484A and Y505H are involved in disrupting binding of antibodies to the spike, leading to an indicated 14-fold higher vaccine escape capability than Delta [Citation264]. If infection rates remain high, new variants of SARS-CoV-2 with further mutations, most likely in the RBD or NTD, will emerge quickly [Citation264,Citation265].
Antibodies which compete with RBD-ACE2 binding are under some of the strongest selection pressure from variants. Over the course of the pandemic, mutations have developed which favour either improved binding with the ACE2 receptor (often through electrostatic complementarity) [Citation266,Citation267], or escape from existing antibody responses [Citation268,Citation269] and many which do both (such as L452R, seen in the lineages B.1.427, B.1.429 (Epsilon), Delta and then again in the Omicron-variant (Table ) [Citation270]). Occasionally, mutations seem to balance improved infectivity with an increased susceptibility to the human immune system, such as K417N [Citation271]. Dramatically different sets of antibodies are raised against different variants. The original wild-type SARS-CoV-2 strain elicited a strong V3-53 (variable gene) sequence response [Citation227,Citation272] which was not seen against the Beta variant (Table ), which moderately favoured antibodies with V4-39 sequence [Citation273]. ACE2-competing antibodies raised against these variants, which only differed by K417N, E484K and N501Y in the RBD, were found to be completely distinguishable in cluster analysis using neutralization data. This demonstrates how sensitive the antibody response is to mutations in this region [Citation274].
A significant opportunity for spike binding inhibition is through the use of soluble human ACE2 derivates [Citation275–277], which bind to the viral RBDs and thereby prevent any interaction with target cell receptors (Figure b) [Citation277]. An advantage of this approach is its intrinsic resilience to viral mutation escape: though some viral mutations may result in a reduced antibody recognition, selection leading to a higher affinity towards ACE2 will increase the potency of these ‘ACE2 traps’ [Citation277]. As interaction with the spike mainly occurs at the N-terminal α-helices of ACE2, potential ACE2 derivates include those helices; such mimetics have already been shown to be successful in neutralizing SARS-CoV-1 [Citation277].
Figure 7. Alternative spike inhibition approaches. (a) Structure of the six-helix bundle formed by the spike’s HR1 domain (dark green) and inhibitor EK1 (blue) (PDB 7C53). Important interactions within the six-helix bundle are shown as sticks in inset images. (b) Structure of ACE2 bound to the receptor-binding domain (RBD) (PDB 6M0J). The ACE2-trap consists of the two N-terminal α-helices Helix-1 and 2 of ACE2 (highlighted in blue).
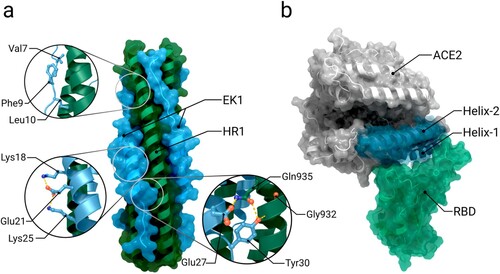
It has also been suggested that inclusion of the N-terminal domain in addition to the receptor binding domain as a target for neutralizing antibodies in vaccine development could broaden the number of neutralizing epitopes and reduce the potential of the virus to escape host immunity [Citation85]. Conversely, it seems that the NTD has been subjected to high evolutionary pressure as almost all variants of concern have extensive mutations around these sites [Citation278]. Analysis of sequence data from COG-UK (Covid-19 Genomics UK Consortium[Citation279,p.19]) shows that the NTD has an even higher mutation rate and that no clear consensus for specific mutations amongst notable variants has emerged, though there is a tendency towards reduction of the overall NTD mass. This feature would putatively make the NTD a poor target for escape-resistant therapeutics.
5.4. Molecular inhibitors
In addition to neutralizing antibodies, numerous molecular inhibitors are currently under development to block cell entry via the spike protein, encompassing fusion inhibitors and protease inhibitors. It's important to note that this review provides only a very limited selection of examples, while several publications offer a more comprehensive overview of different molecular inhibitors [Citation57,Citation280–283].
Given that the spike protein facilitates virus-cell fusion, fusion inhibitors present a promising approach for preventing SARS-CoV-2 cell entry [Citation58,Citation235,Citation284,Citation285]. To bring the viral and host cell membranes in close proximity for membrane fusion, spikes need to rearrange into a pre-hairpin intermediate [Citation53,Citation286]. During these structural rearrangements a six-helix bundle is formed between the trimeric coiled coil of HR1 and helical HR2 segments [Citation286]. One approach to inhibiting the fusion process is for peptides sharing sequence identity to the heptad repeat 1 and 2 domains to bind the pre-hairpin intermediate, thereby preventing formation of the six-helix bundle and subsequent membrane fusion (Figure a) [Citation235,Citation286].
Another major strategy for targeting the spike protein involves protease inhibitors [Citation16,Citation287–289]. The entry of SARS-CoV-2 into airway epithelial cells likely depends on proteolytic cleavage in the S1/S2 interface and within the S2′ site. Priming of the S2′ site is supposed to be performed by type II transmembrane serine protease (TMPRSS2) [Citation16,Citation290]. Different TMPRSS2 inhibitors, such as camostat mesylate, nafamostat mesylate, and peptidomimetics, are among those being investigated for the treatment of COVID-19.
Camostat mesylate and nafamostat mesylate are synthetic serine protease inhibitors targeting TMPRSS2 and related proteases. Camostat mesylate has been shown to significantly reduce but not completely abolish infection of Calu3 lung cells in vitro [Citation16,Citation138,Citation290]. The approval of camostat mesylate for human use in Japan highlights its potential as a promising drug [Citation290–292]. Important to note is that in cell culture, camostat mesylate is rapidly converted into GBPA (4-(4-guanidinobenzoyloxy) phenylacetic acid), which also has TMPRSS2 inhibitory activity, albeit to a lesser extent [Citation290,Citation293].
Potential and highly promising drug alternatives to camostat mesylate are peptidomimetics [Citation283,Citation294–296]. N-0385, a peptidomimetic with inhibitory activity against TMPRSS2 has shown to effectively block SARS-CoV-2 infection in Calu-3 cells [Citation294]. When administered to K18-hACE2 mice infected with SARS-CoV-2, N-0385 effectively prevented mortality caused by the virus and significantly mitigated pulmonary damage [Citation294]. In the same study, the authors state that N-0385 could be a promising drug for the early treatment of disease caused by the SARS-CoV-2 wild type and delta variant [Citation294]. The inhibitors mentioned here represent just a subset of the promising TMPRSS2 inhibitors available, with numerous others extensively documented in a comprehensive literature review by Wettenstein et al. [Citation296].
6. Summary
The SARS-CoV-2 spike plays a crucial role during pathogenesis of COVID-19, as it mediates entry of SARS-CoV-2 into host cells. It consists of two subunits, S1 and S2, each of which contains several domains needed for interaction with the host cell and subsequent membrane fusion. The hinge-like movement of the receptor binding domain from the ‘down’ to the ‘up’ conformation is crucial for receptor binding, since this movement exposes the interaction epitope. However, this epitope is also a main target for neutralizing antibodies. To evade the immune system and reduce interactions with antibodies, the RBD of the spike can switch into the down conformation. Cleavage with type II transmembrane serine protease (TMPRSS2) after receptor binding induces shedding of subunit S1 and triggers S2 to undergo major structural rearrangements that project the fusion peptide towards the host cell membrane, resulting in formation of a pre-hairpin intermediate that bridges viral and host cell membrane. The membranes are then brought into close proximity for fusion through formation of a six-helix bundle by the heptad repeat 1 and 2 domains. A striking feature of the spike is its glycan shield, which protects about 60% of its surface from being recognized by antibodies. Nevertheless, all currently used vaccines are based on the spike, which is the most attractive vaccine target, mainly due to its exposed location.
7. Discussion & outlook
Since 2019, over 800 protein structures related to the SARS-CoV-2 spike protein have been deposited in the PDB. This is emblematic of the amount of collective research that has been conducted worldwide on the coronavirus and its proteins during the pandemic. The contributions of structural biology and biophysics to the fight against COVID19 have been compiled and presented in a number of works [Citation123,Citation297–301]. The spike protein is one of the most studied proteins of SARS-CoV-2 and the collection of available structures provides a comprehensive overview of its function and antigenicity. They also serve as a fundamental basis for drug and vaccine development. Nevertheless, the spike is a large complex and many questions remain open regarding intra- and intermolecular interactions with different proteins, such as different proteases, cell membranes and the immune system. To date there is no full-length structure of the spike inserted into the membrane (with cytoplasmic tail and transmembrane domain) and every ectodomain structure contains unresolved sections. These presumably highly flexible/dynamic regions most commonly include the furin cleavage site, the heptad repeat 1 domain and especially the fusion peptide region. The absence of the latter limits the mechanistic understanding of intramolecular interactions as well as the understanding of the fusion process. In addition to coronaviridae, virus families that pose a danger to society such as orthomyxoviridae (influenza) and retroviridae (HIV) also utilise type-I-fusion proteins[Citation302]. Thus, the knowledge obtained through coronavirus research may be useful in gleaning further insights into the fusion mechanism of other viruses and thereby help to develop therapeutics and vaccines to prevent or limit future infections.
Since the first appearance of SARS-CoV-2, its genome has changed many times through mutations, resulting in multiple new variants of the virus. One of the proteins most affected by mutations is the spike protein, due to the combination of the high replication rate and high selection pressure from immunization by infection or vaccines. Under these circumstances, mutations emerge that balance affinity and binding of the human cell surface receptor ACE2 with evading the immune system. An example of this is the Omicron variant, whose heavily mutated spike protein results in the highest transmissibility and most effective immune evasion, where it is not uncommon for individuals to be reinfected [Citation303] within relatively short time periods. Given the importance of the up–down–up-down dynamics of the receptor binding domain for evading the immune system, there remains significant scope for characterising the structural effects of different mutations on these dynamics, which may reveal mechanisms for modifying them. What makes this difficult is that mutations may not ultimately have a significant impact on the average position of atoms within a model, but rather the relative stability of different (sub)states or interactions; identification of these features requires us to go beyond the analysis of average atomic positions.
As the coronavirus SARS-CoV-2 will become endemic, mutations in the spike protein will likely have a long-term impact on societies as they continue to change SARS-CoV-2’s pathogenicity. Continuous study of the spike domains of new variants is therefore essential for pandemic preparedness, i.e. to adjust the composition of vaccines so that effective antibodies are created that efficiently interfere with membrane fusion of new SARS-CoV-2 variants and human airway epithelial cells. As long as there are reservoirs where SARS-CoV-2 can amplify, the spike protein will collect new mutations in antibody-binding sites, leading to changes in antibody recognition. Hence, it is the properties of the spike that may well determine the future course and the ultimate outcome of the pandemic. It is therefore vital that we keep up with structural changes in new variants as they emerge.
Subjects index
antibodies, 7, 10, 17, 19, 20, 21, 22, 25
cleavage site, 12, 13, 21, 25
drugs, 19, 22
furin, 5, 6, 11, 13, 17, 25
fusion, 3, 6, 10, 13, 14, 15, 16, 19, 23, 25
glycosylation, 5, 17, 18
hairpin, 9, 14, 15, 16, 19, 23
immune evasion, 10, 21, 25
mutations, 6, 21, 22, 25
six-helix bundle, 14, 16, 23, 24
TMPRSS2, 9, 11, 13, 14, 16, 23
vaccines, 19, 20, 21, 25
variants, 21, 22, 23, 25
Acknowledgements
The authors would also like to thank Rosemary Wilson for support and discussion. All figures are courtesy of the Coronavirus Structural Task Force (insidecorona.net), which retains copyright of text and figures. Authors with equal contributions have the right to put their name first/last in citations on their CVs. The prefusion structure of the SARS-CoV-2 omicron BA.2 spike ectodomain (Figure 5a) was provided by Akash Satheesan and Elisa Fadda.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
Notes on contributors

Sabrina Stäb
Sabrina Stäb is studying biochemistry (M.Sc.) and works as a research assistant for the Thorn Lab and the Coronavirus Structural Task Force. During her bachelor thesis on ‘Crystallization and Structure Solution of High-Quality Structures for MAD Experiments', she was able to gain a lot of experience in the field of crystallography and now brings this experience to the project.

Nicholas M. Pearce
Dr. Nicholas Pearce is an assistant professor and Data-Driven Life Sciences Fellow at Linköping University, Sweden, where he works on developing computational approaches to identify and model weak features and dynamics in crystallographic data. During his PhD he developed the PanDDA method, a statistical approach for identifying binding molecules in crystallographic fragment screening experiments, and modelling the resulting multi-state superpositions. Recently, he has worked on characterising the flexibility in protein structures to identify protein dynamics, including the main protease and spike proteins of the coronavirus.

Dale E. Tronrud
Dale Tronrud has both solved protein crystal structures and developed methods and software for the optimization of macromolecular models against X-ray data and known chemical structural information. He has had a long-standing interest in enzyme:inhibitor complexes and photosynthetic proteins, focusing on the Fenna-Matthews-Olson protein. In addition, he has also been involved in the validation and repair of many PDB models.

Helen Ginn
Dr Helen Ginn is a senior research scientist at Diamond Light Source in the UK and a computational methods developer in structural biology. She is currently working on Representation of Protein Entities (RoPE) for structural biologists to interpret subtle conformational changes in dynamic protein systems. She has developed Vagabond for torsion angle-driven model refinement and cluster4x for grouping datasets. Her research interests also include antibody and antigenic mapping for the spike protein of SARS-CoV-2.

Elisa Fadda
Elisa Fadda obtained a PhD in Chemistry at the Université de Montréal, Canada, in 2004, in Prof Dennis R. Salahub’s group on the development of approaches based on DFT for the prediction of NMR shieldings. Between 2004 and 2008 she worked as a Post-doctoral Researcher in Dr Régis Pomès’s group in Molecular Structure and Function at the Hospital for Sick Children in Toronto, Canada, where she studied biomolecular simulations of ions and protons translocation events through transmembrane channels and proton pumps. Between 2008 and 2013, Dr Elisa Fadda worked as a Research Fellow in Prof Robert J. Woods’s group at the National University of Ireland, Galway, Ireland, where she began working in computational glycoscience, studying the structure and function of glycoproteins and glycan-processing enzymes by high-performance computing molecular simulations. Since 2013 she is the principal investigator PI of the Computational Glycoscience research group in the Department of Chemistry and Hamilton Institute at Maynooth University, Ireland, where she is currently an Associate Professor (Senior Lecturer)

Gianluca Santoni
Gianluca Santoni’s work is focused on the development of methods for synchrotron serial crystallography, from the comparison of datasets for multi-crystal data collection methods to the implementation of new measurement techniques at synchrotron beamlines. He got his PhD in structural biology studying the structure of acetylcholinesterase in complex with organophosphate nerve agents, applying a mixture of crystallography and computational methods. More recently he has been involved in the data strategies implementation for open science, participating in activities concerning the storage of experimental metadata, the distribution of raw data along with publication according to FAIR principle, and the definition of the Gold standard format for protein diffraction data. His work with the coronavirus structural taskforce has been focused on the implementation of routines to evaluate the quality of both deposited datasets and models.

Andrea Thorn
Dr. Andrea Thorn is a specialist for structure solution by crystallography and Cryo-EM, having contributed to programs like SHELX, ANODE and PHASER in the past. Her group at the University of Hamburg develops AI-based methods in crystallography such as the diffraction diagnostics tool AUSPEX and a neural network for secondary structure annotation of Cryo-EM maps (HARUSPEX). Her methods enable other scientists to solve new structures and to answer challenging biological questions. Andrea is very passionate about structural biology and good at bringing people together. She started and leads the Coronavirus Structural Task Force.
References
- Coronavirus disease (COVID-19) pandemic [Internet]. [cited 2022 Jun 8]. Available from: https://www.who.int/europe/emergencies/situations/covid-19.
- Bosch BJ, van der Zee R, de Haan CAM, et al. The Coronavirus Spike Protein Is a Class I Virus Fusion Protein: Structural and Functional Characterization of the Fusion Core Complex. J Virol. 2003;77:8801–8811.
- Cavanagh D. The Coronavirus Surface Glycoprotein. In Siddell S G, editor. The Coronaviridae [Internet]. Boston, MA: Springer US; 1995 [cited 2023 Sep 2]. p. 73–113. Available from: http://link.springer.com/10.1007978-1-4899-1531-3_5.
- Liu C, Mendonça L, Yang Y, et al. The Architecture of Inactivated SARS-CoV-2 with Postfusion Spikes Revealed by Cryo-EM and Cryo-ET. Structure. 2020;28:1218–1224.
- Turoňová B, Sikora M, Schürmann C, et al. In situ structural analysis of SARS-CoV-2 spike reveals flexibility mediated by three hinges. Science. 2020;370:203–208.
- Davies HA, Macnaughton MR. Comparison of the morphology of three coronaviruses. Arch Virol. 1979;59:25–33.
- Laue M, Kauter A, Hoffmann T, et al. Morphometry of SARS-CoV and SARS-CoV-2 particles in ultrathin plastic sections of infected Vero cell cultures. Sci Rep. 2021;11:3515.
- Klein S, Winter CM, L S, et al. SARS-CoV-2 structure and replication characterized by in situ cryo-electron tomography. Nat Commun. 2020;11:5885.
- Beniac DR, Andonov A, Grudeski E, et al. Architecture of the SARS coronavirus prefusion spike. Nat Struct Mol Biol. 2006;13:751–752.
- Benton DJ, Wrobel AG, Xu P, et al. Receptor binding and priming of the spike protein of SARS-CoV-2 for membrane fusion. Nature. 2020;588:327–330.
- Li W, Moore MJ, Vasilieva N, et al. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature. 2003;426:450–454.
- Li F. Receptor Recognition Mechanisms of Coronaviruses: a Decade of Structural Studies. J Virol. 2015;89:1954–1964.
- Letko M, Marzi A, Munster V. Functional assessment of cell entry and receptor usage for SARS-CoV-2 and other lineage B betacoronaviruses. Nat Microbiol. 2020;5:562–569.
- Hamming I, Timens W, Bulthuis M, et al. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol. 2004;203:631–637.
- Belouzard S, Chu VC, Whittaker GR. Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites. Proc Natl Acad Sci USA. 2009;106:5871–5876.
- Hoffmann M, Kleine-Weber H, Schroeder S, et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell. 2020;181:271–280.
- Millet JK, Whittaker GR. Host cell proteases: Critical determinants of coronavirus tropism and pathogenesis. Virus Res. 2015;202:120–134.
- Cai Y, Zhang J, Xiao T, et al. Distinct conformational states of SARS-CoV-2 spike protein. Science. 2020;369:1586–1592.
- Dou D, Revol R, Östbye H, et al. Influenza A Virus Cell Entry, Replication, Virion Assembly and Movement. Front Immunol. 2018;9:1581.
- Xu Y, Liu Y, Lou Z, et al. Structural Basis for Coronavirus-mediated Membrane Fusion. J Biol Chem. 2004;279:30514–30522.
- Duquerroy S, Vigouroux A, Rottier PJM, et al. Central ions and lateral asparagine/glutamine zippers stabilize the post-fusion hairpin conformation of the SARS coronavirus spike glycoprotein. Virology. 2005;335:276–285.
- Supekar VM, Bruckmann C, Ingallinella P, et al. Structure of a proteolytically resistant core from the severe acute respiratory syndrome coronavirus S2 fusion protein. Proc Natl Acad Sci USA. 2004;101:17958–17963.
- Wu Zhang X, Leng Yap Y. Structural similarity between HIV-1 gp41 and SARS-CoV S2 proteins suggests an analogous membrane fusion mechanism. J Molecul Struc: THEOCHEM. 2004;677:73–76.
- Kliger Y, Levanon EY. Cloaked similarity between HIV-1 and SARS-CoV suggests an anti-SARS strategy. BMC Microbiol. 2003;3:20.
- Tang T, Bidon M, Jaimes JA, et al. Coronavirus membrane fusion mechanism offers a potential target for antiviral development. Antiviral Res. 2020;178:104792.
- White JM, Delos SE, Brecher M, et al. Structures and Mechanisms of Viral Membrane Fusion Proteins: Multiple Variations on a Common Theme. Crit Rev Biochem Mol Biol. 2008;43:189–219.
- Nakagawa K, Lokugamage KG, Makino S. Viral and Cellular mRNA Translation in Coronavirus-Infected Cells. Advances in Virus Research [Internet]. Elsevier; 2016 [cited 2021 Dec 17]. p. 165–192. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0065352716300409.
- Li F, Li W, Farzan M, et al. Structure of SARS Coronavirus Spike Receptor-Binding Domain Complexed with Receptor. Science. 2005;309:1864–1868.
- Li W, Zhang C, Sui J, et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005;24:1634–1643.
- Sikora M, von Bülow S, Blanc FEC, et al. Computational epitope map of SARS-CoV-2 spike protein. PLoS Comput Biol. 2021;17:e1008790.
- Papageorgiou AC, Mohsin I. The SARS-CoV-2 Spike Glycoprotein as a Drug and Vaccine Target: Structural Insights into Its Complexes with ACE2 and Antibodies. Cells. 2020;9:2343.
- Ahmed SF, Quadeer AA, McKay MR. Preliminary Identification of Potential Vaccine Targets for the COVID-19 Coronavirus (SARS-CoV-2) Based on SARS-CoV Immunological Studies. Viruses. 2020;12:254.
- Yang Y, Du L. SARS-CoV-2 spike protein: a key target for eliciting persistent neutralizing antibodies. Sig Transduct Target Ther. 2021;6:95.
- Buchholz UJ, Bukreyev A, Yang L, et al. Contributions of the structural proteins of severe acute respiratory syndrome coronavirus to protective immunity. Proc Natl Acad Sci USA. 2004;101:9804–9809.
- Watanabe Y, Allen JD, Wrapp D, et al. Site-specific glycan analysis of the SARS-CoV-2 spike. Science. 2020;369:330–333.
- Zhao X, Chen H, Wang H. Glycans of SARS-CoV-2 Spike Protein in Virus Infection and Antibody Production. Front Mol Biosci. 2021;8:629873.
- Doores KJ, Bonomelli C, Harvey DJ, et al. Envelope glycans of immunodeficiency virions are almost entirely oligomannose antigens. Proc Natl Acad Sci USA. 2010;107:13800–13805.
- Watanabe Y, Bowden TA, Wilson IA, et al. Exploitation of glycosylation in enveloped virus pathobiology. Biochimica et Biophysica Acta (BBA) - General Subjects. 2019;1863:1480–1497.
- Raman R, Tharakaraman K, Sasisekharan V, et al. Glycan–protein interactions in viral pathogenesis. Curr Opin Struct Biol. 2016;40:153–162.
- Stewart-Jones GBE, Soto C, Lemmin T, et al. Trimeric HIV-1-Env Structures Define Glycan Shields from Clades A, B, and G. Cell. 2016;165:813–826.
- Casalino L, Gaieb Z, Goldsmith JA, et al. Beyond Shielding: The Roles of Glycans in the SARS-CoV-2 Spike Protein. ACS Cent Sci. 2020;6:1722–1734.
- Grant OC, Montgomery D, Ito K, et al. Analysis of the SARS-CoV-2 spike protein glycan shield reveals implications for immune recognition. Sci Rep. 2020;10:14991.
- Bonomelli C, Doores KJ, Dunlop DC, et al. The Glycan Shield of HIV Is Predominantly Oligomannose Independently of Production System or Viral Clade. PLoS One. 2011;6:e23521.
- Harbison AM, Fogarty CA, Phung TK, et al. Fine-tuning the spike: role of the nature and topology of the glycan shield in the structure and dynamics of the SARS-CoV-2 S. Chem Sci. 2022;13:386–395.
- Naqvi AAT, Fatima K, Mohammad T, et al. Insights into SARS-CoV-2 genome, structure, evolution, pathogenesis and therapies: Structural genomics approach. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2020;1866:165878.
- Mortola E, Roy P. Efficient assembly and release of SARS coronavirus-like particles by a heterologous expression system. FEBS Lett. 2004;576:174–178.
- Qin L, Xiong B, Luo C, et al. Identification of probable genomic packaging signal sequence from SARS-CoV genome by bioinformatics analysis. Acta Pharmacol Sin. 2003;24:489–496.
- Rath SL, Kumar K. Investigation of the Effect of Temperature on the Structure of SARS-CoV-2 Spike Protein by Molecular Dynamics Simulations. Front Mol Biosci. 2020;7:583523.
- Guruprasad L. Human SARS CoV -2 spike protein mutations. Proteins. 2021;89:569–576.
- Gui M, Song W, Zhou H, et al. Cryo-electron microscopy structures of the SARS-CoV spike glycoprotein reveal a prerequisite conformational state for receptor binding. Cell Res. 2017;27:119–129.
- Wang Q, Qiu Y, Li J-Y, et al. A Unique Protease Cleavage Site Predicted in the Spike Protein of the Novel Pneumonia Coronavirus (2019-nCoV) Potentially Related to Viral Transmissibility. Virol Sin. 2020;35:337–339.
- Dong W, Bai B, Lin Y, et al. Detection of the mRNA expression of human angiotensin-converting enzyme 2 as a SARS coronavirus functional receptor in human femoral head. Nan Fang Yi Ke Da Xue Xue Bao. 2008;28:441–443.
- Duan L, Zheng Q, Zhang H, et al. The SARS-CoV-2 Spike Glycoprotein Biosynthesis, Structure, Function, and Antigenicity: Implications for the Design of Spike-Based Vaccine Immunogens. Front Immunol. 2020;11:576622.
- Tomasello G, Armenia I, Molla G. The Protein Imager: a full-featured online molecular viewer interface with server-side HQ-rendering capabilities. Bioinformatics. 2020;36:2909–2911.
- Community BO. Blender - a 3D modelling and rendering package [Internet]. Stichting Blender Foundation. Amsterdam: Blender Foundation; 2018. Available from: http://www.blender.org.
- Inkscape Project. Inkscape [Internet]. 2020. Available from: https://inkscape.org.
- Huang Y, Yang C, Xu X, et al. Structural and functional properties of SARS-CoV-2 spike protein: potential antivirus drug development for COVID-19. Acta Pharmacol Sin. 2020;41:1141–1149.
- Xia S, Zhu Y, Liu M, et al. Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein. Cell Mol Immunol. 2020;17:765–767.
- Verkhivker GM. Molecular Simulations and Network Modeling Reveal an Allosteric Signaling in the SARS-CoV-2 Spike Proteins. J Proteome Res. 2020;19:4587–4608.
- Verkhivker GM, Agajanian S, Oztas DY, et al. Landscape-Based Mutational Sensitivity Cartography and Network Community Analysis of the SARS-CoV-2 Spike Protein Structures: Quantifying Functional Effects of the Circulating D614G Variant. ACS Omega. 2021;6:16216–16233.
- Khelashvili G, Plante A, Doktorova M, et al. Ca2+-dependent mechanism of membrane insertion and destabilization by the SARS-CoV-2 fusion peptide. Biophys J. 2021;120:1105–1119.
- Zhang J, Xiao T, Cai Y, et al. Structure of SARS-CoV-2 spike protein. Curr Opin Virol. 2021;50:173–182.
- Martí D, Torras J, Bertran O, et al. Temperature effect on the SARS-CoV-2: A molecular dynamics study of the spike homotrimeric glycoprotein. Comput Struct Biotechnol J. 2021;19:1848–1862.
- Wrapp D, Wang N, Corbett KS, et al. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science. 2020;367:1260–1263.
- Mulvey M, Brown DT. Assembly of the Sindbis Virus Spike Protein Complex. Virology. 1996;219:125–132.
- Green J, Griffiths G, Louvard D, et al. Passage of viral membrane proteins through the Golgi complex. J Mol Biol. 1981;152:663–698.
- Xia X. Domains and Functions of Spike Protein in SARS-Cov-2 in the Context of Vaccine Design. Viruses. 2021;13:109.
- Nothwehr SF, Gordon JI. Targeting of proteins into the eukaryotic secretory pathway: Signal peptide structure/function relationships. Bioessays. 1990;12:479–484.
- Zhang Z, Wan X, Li X, et al. Effects of a Shift of the Signal Peptide Cleavage Site in Signal Peptide Variant on the Synthesis and Secretion of SARS-CoV-2 Spike Protein. Molecules. 2022;27:6688.
- Hegde RS, Bernstein HD. The surprising complexity of signal sequences. Trends Biochem Sci. 2006;31:563–571.
- Braakman I, Hebert DN. Protein Folding in the Endoplasmic Reticulum. Cold Spring Harbor Perspect Biol. 2013;5:a013201–a013201.
- Hoffmann M, Kleine-Weber H, Pöhlmann S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol Cell. 2020;78:779–784.
- Fung TS, Liu DX. Post-translational modifications of coronavirus proteins: roles and function. Future Virol. 2018;13:405–430.
- Ritchie G, Harvey DJ, Feldmann F, et al. Identification of N-linked carbohydrates from severe acute respiratory syndrome (SARS) spike glycoprotein. Virology. 2010;399:257–269.
- Niemann H, Boschek B, Evans D, et al. Post-translational glycosylation of coronavirus glycoprotein E1: inhibition by monensin. EMBO J. 1982;1:1499–1504.
- Zhou H, Chen Y, Zhang S, et al. Structural definition of a neutralization epitope on the N-terminal domain of MERS-CoV spike glycoprotein. Nat Commun. 2019;10:3068.
- Peng G, Xu L, Lin Y-L, et al. Crystal Structure of Bovine Coronavirus Spike Protein Lectin Domain. J Biol Chem. 2012;287:41931–41938.
- Yang Q, Kelkar A, Sriram A, et al. Role for N -glycans and calnexin-calreticulin chaperones in SARS-CoV-2 Spike maturation and viral infectivity. Sci Adv. 2022;8:eabq8678.
- Behloul N, Baha S, Shi R, et al. Role of the GTNGTKR motif in the N-terminal receptor-binding domain of the SARS-CoV-2 spike protein. Virus Res. 2020;286:198058.
- Cheng Y, He B, Yang J, et al. Crystal structure of the S1 subunit N-terminal domain from DcCoV UAE-HKU23 spike protein. Virology. 2019;535:74–82.
- Peng G, Sun D, Rajashankar KR, et al. Crystal structure of mouse coronavirus receptor-binding domain complexed with its murine receptor. Proc Natl Acad Sci USA. 2011;108:10696–10701.
- Chi X, Yan R, Zhang J, et al. A neutralizing human antibody binds to the N-terminal domain of the Spike protein of SARS-CoV-2. Science. 2020;369:650–655.
- Kubo H, Yamada YK, Taguchi F. Localization of neutralizing epitopes and the receptor-binding site within the amino-terminal 330 amino acids of the murine coronavirus spike protein. J Virol. 1994;68:5403–5410.
- Krempl C, Schultze B, Laude H, et al. Point mutations in the S protein connect the sialic acid binding activity with the enteropathogenicity of transmissible gastroenteritis coronavirus. J Virol. 1997;71:3285–3287.
- Dai L, Gao GF. Viral targets for vaccines against COVID-19. Nat Rev Immunol. 2021;21:73–82.
- Lu G, Wang Q, Gao GF. Bat-to-human: spike features determining ‘host jump’ of coronaviruses SARS-CoV, MERS-CoV, and beyond. Trends Microbiol. 2015;23:468–478.
- Tipnis SR, Hooper NM, Hyde R, et al. A Human Homolog of Angiotensin-converting Enzyme. J Biol Chem. 2000;275:33238–33243.
- Donoghue M, Hsieh F, Baronas E, et al. A Novel Angiotensin-Converting Enzyme–Related Carboxypeptidase (ACE2) Converts Angiotensin I to Angiotensin 1-9. Circulation Research [Internet]. 2000 [cited 2023 Aug 10];87. Available from: https://www.ahajournals.org/doi/10.116101.RES.87.5.e1.
- Lan J, Ge J, Yu J, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature. 2020;581:215–220.
- Saponaro F, Rutigliano G, Sestito S, et al. ACE2 in the Era of SARS-CoV-2: Controversies and Novel Perspectives. Front Mol Biosci. 2020;7:588618.
- Chambers P, Pringle CR, Easton AJ. Heptad Repeat Sequences are Located Adjacent to Hydrophobic Regions in Several Types of Virus Fusion Glycoproteins. J Gen Virol. 1990;71:3075–3080.
- Schaefer SL, Jung H, Hummer G. Binding of SARS-CoV-2 Fusion Peptide to Host Endosome and Plasma Membrane. J Phys Chem B. 2021;125:7732–7741.
- Madu IG, Roth SL, Belouzard S, et al. Characterization of a Highly Conserved Domain within the Severe Acute Respiratory Syndrome Coronavirus Spike Protein S2 Domain with Characteristics of a Viral Fusion Peptide. J Virol. 2009;83:7411–7421.
- Guillén J, Kinnunen PKJ, Villalaín J. Membrane insertion of the three main membranotropic sequences from SARS-CoV S2 glycoprotein. Biochimica et Biophysica Acta (BBA) - Biomembranes. 2008;1778:2765–2774.
- Walls AC, Tortorici MA, Snijder J, et al. Tectonic conformational changes of a coronavirus spike glycoprotein promote membrane fusion. Proc Natl Acad Sci USA. 2017;114:11157–11162.
- Villalaín J. SARS-CoV-2 Protein S Fusion Peptide Is Capable of Wrapping Negatively-Charged Phospholipids. Membranes. 2023;13:344.
- Santamaria A, Batchu KC, Matsarskaia O, et al. Strikingly different roles of SARS-CoV-2 fusion peptides uncovered by neutron scattering [Internet]. Biophysics; 2021 [cited 2022 Jan 9]. Available from: http://biorxiv.org/lookup/doi/10.11012021.08.30.458099.
- Guillén J, Pérez-Berná AJ, Moreno MR, et al. Identification of the Membrane-Active Regions of the Severe Acute Respiratory Syndrome Coronavirus Spike Membrane Glycoprotein Using a 16/18-Mer Peptide Scan. Implications for the Viral Fusion Mechanism. J Virol. 2005;79:1743–1752.
- Corver J, Broer R, van Kasteren P, et al. Mutagenesis of the transmembrane domain of the SARS coronavirus spike glycoprotein: refinement of the requirements for SARS coronavirus cell entry. Virol J. 2009;6:230.
- Broer R, Boson B, Spaan W, et al. Important Role for the Transmembrane Domain of Severe Acute Respiratory Syndrome Coronavirus Spike Protein during Entry. J Virol. 2006;80:1302–1310.
- Xiao X, Feng Y, Chakraborti S, et al. Oligomerization of the SARS-CoV S glycoprotein: dimerization of the N-terminus and trimerization of the ectodomain. Biochem Biophys Res Commun. 2004;322:93–99.
- Song HC, Seo M-Y, Stadler K, et al. Synthesis and Characterization of a Native, Oligomeric Form of Recombinant Severe Acute Respiratory Syndrome Coronavirus Spike Glycoprotein. J Virol. 2004;78:10328–10335.
- Petit CM, Melancon JM, Chouljenko VN, et al. Genetic analysis of the SARS-coronavirus spike glycoprotein functional domains involved in cell-surface expression and cell-to-cell fusion. Virology. 2005;341:215–230.
- Petit CM, Chouljenko VN, Iyer A, et al. Palmitoylation of the cysteine-rich endodomain of the SARS–coronavirus spike glycoprotein is important for spike-mediated cell fusion. Virology. 2007;360:264–274.
- Lontok E, Corse E, Machamer CE. Intracellular Targeting Signals Contribute to Localization of Coronavirus Spike Proteins near the Virus Assembly Site. J Virol. 2004;78:5913–5922.
- McBride CE, Li J, Machamer CE. The Cytoplasmic Tail of the Severe Acute Respiratory Syndrome Coronavirus Spike Protein Contains a Novel Endoplasmic Reticulum Retrieval Signal That Binds COPI and Promotes Interaction with Membrane Protein. J Virol. 2007;81:2418–2428.
- Teasdale RD, Jackson MR. Signal-Mediated Sorting of Membrane Proteins Between the Endoplasmic Reticulum and the Golgi Apparatus. Annu Rev Cell Dev Biol. 1996;12:27–54.
- Bonifacino JS, Traub LM. Signals for Sorting of Transmembrane Proteins to Endosomes and Lysosomes. Annu Rev Biochem. 2003;72:395–447.
- Sadasivan J, Singh M, Sarma JD. Cytoplasmic tail of coronavirus spike protein has intracellular targeting signals. J Biosci. 2017;42:231–244.
- Kumar P, Bhardwaj T, Garg N, et al. Microsecond simulations and CD spectroscopy reveals the intrinsically disordered nature of SARS-CoV-2 spike-C-terminal cytoplasmic tail (residues 1242–1273) in isolation. Virology. 2022;566:42–55.
- Dey D, Singh S, Khan S, et al. An extended motif in the SARS-CoV-2 spike modulates binding and release of host coatomer in retrograde trafficking. Commun Biol. 2022;5:115.
- Cattin-Ortolá J, Welch LG, Maslen SL, et al. Sequences in the cytoplasmic tail of SARS-CoV-2 Spike facilitate expression at the cell surface and syncytia formation. Nat Commun. 2021;12:5333.
- Kumar P, Kumar A, Garg N, et al. An insight into SARS-CoV-2 membrane protein interaction with spike, envelope, and nucleocapsid proteins. J Biomol Struct Dyn. 2023;41:1062–1071.
- Berman HM, Battistuz T, Bhat TN, et al. The Protein Data Bank. Acta Crystallogr D Biol Crystallogr. 2002;58:899–907.
- Li Y-D, Chi W-Y, Su J-H, et al. Coronavirus vaccine development: from SARS and MERS to COVID-19. J Biomed Sci. 2020;27:104.
- Krammer F. SARS-CoV-2 vaccines in development. Nature. 2020;586:516–527.
- Mohammed MEA. The percentages of SARS-CoV-2 protein similarity and identity with SARS-CoV and BatCoV RaTG13 proteins can be used as indicators of virus origin. J Proteins Proteom. 2021;12:81–91.
- Zhang S, Qiao S, Yu J, et al. Bat and pangolin coronavirus spike glycoprotein structures provide insights into SARS-CoV-2 evolution. Nat Commun. 2021;12:1607.
- Wrobel AG, Benton DJ, Xu P, et al. SARS-CoV-2 and bat RaTG13 spike glycoprotein structures inform on virus evolution and furin-cleavage effects. Nat Struct Mol Biol. 2020;27:763–767.
- Liu K, Pan X, Li L, et al. Binding and molecular basis of the bat coronavirus RaTG13 virus to ACE2 in humans and other species. Cell. 2021;184:3438–3451.
- Kirchdoerfer RN, Wang N, Pallesen J, et al. Stabilized coronavirus spikes are resistant to conformational changes induced by receptor recognition or proteolysis. Sci Rep. 2018;8:15701.
- Gowthaman R, Guest JD, Yin R, et al. CoV3D: a database of high resolution coronavirus protein structures. Nucleic Acids Res. 2021;49:D282–D287.
- Stepanenko D, Budhan S, Simmerling C. SpikeScape: A Tool for Analyzing Structural Diversity in Experimental Structures of the SARS-CoV-2 Spike Glycoprotein. J Chem Inf Model. 2023;63:1087–1092.
- Herrera NG, Morano NC, Celikgil A, et al. Characterization of the SARS-CoV-2 S Protein: Biophysical, Biochemical, Structural, and Antigenic Analysis [Internet]. Biochemistry; 2020 [cited 2022 Mar 28]. Available from: http://biorxiv.org/lookup/doi/10.11012020.06.14.150607.
- Yang T-J, Yu P-Y, Chang Y-C, et al. Effect of SARS-CoV-2 B.1.1.7 mutations on spike protein structure and function. Nat Struct Mol Biol. 2021;28:731–739.
- Cerutti G, Guo Y, Zhou T, et al. Potent SARS-CoV-2 neutralizing antibodies directed against spike N-terminal domain target a single supersite. Cell Host Microbe. 2021;29:819–833.
- Zhou T, Teng I-T, Olia AS, et al. Structure-Based Design with Tag-Based Purification and In-Process Biotinylation Enable Streamlined Development of SARS-CoV-2 Spike Molecular Probes. SSRN Journal [Internet]. 2020 [cited 2022 Jan 3]; Available from: https://www.ssrn.com/abstract=3639618.
- Zhou T, Tsybovsky Y, Olia AS, et al. Cryo-EM Structures Delineate a pH-Dependent Switch that Mediates Endosomal Positioning of SARS-CoV-2 Spike Receptor-Binding Domains [Internet]. Immunology; 2020 [cited 2022 Jan 3]. Available from: http://biorxiv.org/lookup/doi/10.11012020.07.04.187989.
- Toelzer C, Gupta K, Yadav SKN, et al. Free fatty acid binding pocket in the locked structure of SARS-CoV-2 spike protein. Science. 2020;370:725–730.
- Wang Q, Zhang Y, Wu L, et al. Structural and Functional Basis of SARS-CoV-2 Entry by Using Human ACE2. Cell. 2020;181:894–904.
- Shang J, Ye G, Shi K, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–224.
- Huo J, Zhao Y, Ren J, et al. Neutralization of SARS-CoV-2 by Destruction of the Prefusion Spike. Cell Host Microbe. 2020;28:445–454.
- Han P, Su C, Zhang Y, et al. Molecular insights into receptor binding of recent emerging SARS-CoV-2 variants. Nat Commun. 2021;12:6103.
- Koppisetti RK, Fulcher YG, Van Doren SR. Fusion Peptide of SARS-CoV-2 Spike Rearranges into a Wedge Inserted in Bilayered Micelles. J Am Chem Soc. 2021;143:13205–13211.
- Chiliveri SC, Louis JM, Ghirlando R, et al. Transient lipid-bound states of spike protein heptad repeats provide insights into SARS-CoV-2 membrane fusion. Sci Adv. 2021;7:eabk2226.
- Fu Q, Chou JJ. A Trimeric Hydrophobic Zipper Mediates the Intramembrane Assembly of SARS-CoV-2 Spike. J Am Chem Soc. 2021;143:8543–8546.
- Croll TI, Diederichs K, Fischer F, et al. Making the invisible enemy visible. NSMB. 2021;28:404–408.
- Walls AC, Park Y-J, Tortorici MA, et al. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell. 2020;181:281–292.
- Sun H, Li Y, Liu P, et al. Structural basis of HCoV-19 fusion core and an effective inhibition peptide against virus entry. Emerg Microbes Infect. 2020;9:1238–1241.
- Xia S, Lan Q, Zhu Y, et al. Structural and functional basis for pan-CoV fusion inhibitors against SARS-CoV-2 and its variants with preclinical evaluation. Sig Transduct Target Ther. 2021;6:288.
- Xiao T, Lu J, Zhang J, et al. A trimeric human angiotensin-converting enzyme 2 as an anti-SARS-CoV-2 agent. Nat Struct Mol Biol. 2021;28:202–209.
- Yan R, Zhang Y, Li Y, et al. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science. 2020;367:1444–1448.
- Dejnirattisai W, Zhou D, Supasa P, et al. Antibody evasion by the P.1 strain of SARS-CoV-2. Cell. 2021;184:2939–2954.
- Yin W, Xu Y, Xu P, et al. Structures of the Omicron spike trimer with ACE2 and an anti-Omicron antibody. Science. 2022;375:1048–1053.
- Saville JW, Mannar D, Zhu X, et al. Structural and biochemical rationale for enhanced spike protein fitness in delta and kappa SARS-CoV-2 variants. Nat Commun. 2022;13:742.
- Han P, Li L, Liu S, et al. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell. 2022;185:630–640.
- Podbreagar N. Das Coronavirus zum Ausdrucken - Erstes biologisch korrektes Modell von SARS-CoV-2 für den 3D-Drucker - scinexx.de [Internet]. scinexx | Das Wissensmagazin. 2020 [cited 2022 Jan 13]. Available from: https://www.scinexx.de/news/biowissen/das-coronavirus-zum-ausdrucken/.
- Norman A, Franck C, Christie M, et al. Discovery of Cyclic Peptide Ligands to the SARS-CoV-2 Spike Protein Using mRNA Display. ACS Cent Sci. 2021;7:1001–1008.
- Shang J, Ye G, Shi K, et al. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;581:221–224.
- Yu J, Zhang Z-W, Yang H-Y, et al. Study of fusion peptide release for the spike protein of SARS-CoV-2. RSC Adv. 2023;13:16970–16983.
- Dodero-Rojas E, Onuchic JN, Whitford PC. Sterically confined rearrangements of SARS-CoV-2 Spike protein control cell invasion. eLife. 2021;10:e70362.
- Ou X, Zheng W, Shan Y, et al. Identification of the Fusion Peptide-Containing Region in Betacoronavirus Spike Glycoproteins. J Virol. 2016;90:5586–5600.
- Basso LGM, Vicente EF, Crusca E, et al. SARS-CoV fusion peptides induce membrane surface ordering and curvature. Sci Rep. 2016;6:37131.
- Liu S, Xiao G, Chen Y, et al. Interaction between heptad repeat 1 and 2 regions in spike protein of SARS-associated coronavirus: implications for virus fusogenic mechanism and identification of fusion inhibitors. Lancet. 2004;363:938–947.
- Fan X, Cao D, Kong L, et al. Cryo-EM analysis of the post-fusion structure of the SARS-CoV spike glycoprotein. Nat Commun. 2020;11:3618.
- Wu Y, Zhao S. Furin cleavage sites naturally occur in coronaviruses. Stem Cell Res. 2021;50:102115.
- Bestle D, Heindl MR, Limburg H, et al. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci Alliance. 2020;3:e202000786.
- Andersen KG, Rambaut A, Lipkin WI, et al. The proximal origin of SARS-CoV-2. Nat Med. 2020;26:450–452.
- Örd M, Faustova I, Loog M. The sequence at Spike S1/S2 site enables cleavage by furin and phospho-regulation in SARS-CoV2 but not in SARS-CoV1 or MERS-CoV. Sci Rep. 2020;10:16944.
- Brun J, Vasiljevic S, Gangadharan B, et al. Assessing Antigen Structural Integrity through Glycosylation Analysis of the SARS-CoV-2 Viral Spike. ACS Cent Sci. 2021;7:586–593.
- Follis KE, York J, Nunberg JH. Furin cleavage of the SARS coronavirus spike glycoprotein enhances cell–cell fusion but does not affect virion entry. Virology. 2006;350:358–369.
- De Haan CAM, Te Lintelo E, Li Z, et al. Cooperative Involvement of the S1 and S2 Subunits of the Murine Coronavirus Spike Protein in Receptor Binding and Extended Host Range. J Virol. 2006;80:10909–10918.
- White JM, Whittaker GR. Fusion of Enveloped Viruses in Endosomes. Traffic. 2016;17:593–614.
- Berger I, Schaffitzel C. The SARS-CoV-2 spike protein: balancing stability and infectivity. Cell Res. 2020;30:1059–1060.
- Zabiegala A, Kim Y, Chang K-O. Roles of host proteases in the entry of SARS-CoV-2. Animal Diseases. 2023;3:12.
- Yan R, Wang R, Ju B, et al. Structural basis for bivalent binding and inhibition of SARS-CoV-2 infection by human potent neutralizing antibodies. Cell Res. 2021;31:517–525.
- Yuan M, Liu H, Wu NC, et al. Recognition of the SARS-CoV-2 receptor binding domain by neutralizing antibodies. Biochem Biophys Res Commun. 2021;538:192–203.
- Yan R, Zhang Y, Li Y, et al. Structural basis for the different states of the spike protein of SARS-CoV-2 in complex with ACE2. Cell Res. 2021;31:717–719.
- Brotzakis ZF, Löhr T, Vendruscolo M. Determination of intermediate state structures in the opening pathway of SARS-CoV-2 spike using cryo-electron microscopy. Chem Sci. 2021;12:9168–9175.
- Barnes CO, Jette CA, Abernathy ME, et al. SARS-CoV-2 neutralizing antibody structures inform therapeutic strategies. Nature. 2020;588:682–687.
- Pearce NM, Gros P. A method for intuitively extracting macromolecular dynamics from structural disorder. Nat Commun. 2021;12:5493.
- Rupp B. Biomolecular crystallography: principles, practice, and application to structural biology. New York: Garland Science; 2010.
- Yi C, Sun X, Ye J, et al. Key residues of the receptor binding motif in the spike protein of SARS-CoV-2 that interact with ACE2 and neutralizing antibodies. Cell Mol Immunol. 2020;17:621–630.
- Wan Y, Shang J, Graham R, et al. Receptor Recognition by the Novel Coronavirus from Wuhan: an Analysis Based on Decade-Long Structural Studies of SARS Coronavirus. J Virol. 2020;94:e00127.
- Xiong X, Qu K, Ciazynska KA, et al. A thermostable, closed SARS-CoV-2 spike protein trimer. Nat Struct Mol Biol. 2020;27:934–941.
- Qu K, Chen Q, Ciazynska KA, et al. Engineered disulfide reveals structural dynamics of locked SARS-CoV-2 spike. PLoS Pathog. 2022;18:e1010583.
- Sorokina M, Belapure J, Tüting C, et al. An Electrostatically-steered Conformational Selection Mechanism Promotes SARS-CoV-2 Spike Protein Variation. J Mol Biol. 2022;434:167637.
- Zhang J, Cai Y, Xiao T, et al. Structural impact on SARS-CoV-2 spike protein by D614G substitution. Science. 2021;372:525–530.
- Yao H, Song Y, Chen Y, et al. Molecular Architecture of the SARS-CoV-2 Virus. Cell. 2020;183:730–773.
- Bar-On YM, Flamholz A, Phillips R, et al. SARS-CoV-2 (COVID-19) by the numbers. eLife. 2020;9:e57309.
- Wang B, Zhong C, Tieleman DP. Supramolecular Organization of SARS-CoV and SARS-CoV-2 Virions Revealed by Coarse-Grained Models of Intact Virus Envelopes. J Chem Inf Model. 2022;62:176–186.
- Glowacka I, Bertram S, Muller MA, et al. Evidence that TMPRSS2 Activates the Severe Acute Respiratory Syndrome Coronavirus Spike Protein for Membrane Fusion and Reduces Viral Control by the Humoral Immune Response. J Virol. 2011;85:4122–4134.
- Iwata-Yoshikawa N, Okamura T, Shimizu Y, et al. TMPRSS2 Contributes to Virus Spread and Immunopathology in the Airways of Murine Models after Coronavirus Infection. J Virol. 2019;93:e01815–e01818.
- Koch J, Uckeley ZM, Doldan P, et al. TMPRSS2 expression dictates the entry route used by SARS-CoV-2 to infect host cells. The EMBO Journal [Internet]. 2021 [cited 2022 Jan 5];40. Available from: https://onlinelibrary.wiley.com/doi/10.15252embj.2021107821.
- Liu T, Luo S, Libby P, et al. Cathepsin L-selective inhibitors: A potentially promising treatment for COVID-19 patients. Pharmacol Ther. 2020;213:107587.
- Simmons G, Gosalia DN, Rennekamp AJ, et al. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc Natl Acad Sci USA. 2005;102:11876–11881.
- Zhao M-M, Yang W-L, Yang F-Y, et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Sig Transduct Target Ther. 2021;6:134.
- Padmanabhan P, Desikan R, Dixit NM. Targeting TMPRSS2 and Cathepsin B/L together may be synergistic against SARS-CoV-2 infection. PLoS Comput Biol. 2020;16:e1008461.
- Huang I-C, Bosch BJ, Li F, et al. SARS Coronavirus, but Not Human Coronavirus NL63, Utilizes Cathepsin L to Infect ACE2-expressing Cells. J Biol Chem. 2006;281:3198–3203.
- Zhao M-M, Zhu Y, Zhang L, et al. Novel cleavage sites identified in SARS-CoV-2 spike protein reveal mechanism for cathepsin L-facilitated viral infection and treatment strategies. Cell Discov. 2022;8:53.
- Bosch BJ, Bartelink W, Rottier PJM, Cathepsin L. Functionally Cleaves the Severe Acute Respiratory Syndrome Coronavirus Class I Fusion Protein Upstream of Rather than Adjacent to the Fusion Peptide. J Virol. 2008;82:8887–8890.
- Bollavaram K, Leeman TH, Lee MW, et al. Multiple sites on SARS-CoV -2 spike protein are susceptible to proteolysis by cathepsins B, K, L, S, and V. Protein Sci. 2021;30:1131–1143.
- Harrison SC. Viral membrane fusion. Nat Struct Mol Biol. 2008;15:690–698.
- Furuta RA, Wild CT, Weng Y, et al. Capture of an early fusion-active conformation of HIV-1 gp41. Nat Struct Mol Biol. 1998;5:276–279.
- Xu Y, Lou Z, Liu Y, et al. Crystal Structure of Severe Acute Respiratory Syndrome Coronavirus Spike Protein Fusion Core. J Biol Chem. 2004;279:49414–49419.
- Lai AL, Millet JK, Daniel S, et al. The SARS-CoV Fusion Peptide Forms an Extended Bipartite Fusion Platform that Perturbs Membrane Order in a Calcium-Dependent Manner. J Mol Biol. 2017;429:3875–3892.
- Schaefer SL, Jung H, Hummer G. Binding of SARS-CoV-2 fusion peptide to host membranes [Internet]. Biophysics; 2021 [cited 2022 Jan 7]. Available from: http://biorxiv.org/lookup/doi/10.11012021.05.10.443474.
- Zhang Q, Xiang R, Huo S, et al. Molecular mechanism of interaction between SARS-CoV-2 and host cells and interventional therapy. Sig Transduct Target Ther. 2021;6:233.
- Millet JK, Whittaker GR. Physiological and molecular triggers for SARS-CoV membrane fusion and entry into host cells. Virology. 2018;517:3–8.
- Lai AL, Freed JH. SARS-CoV-2 Fusion Peptide has a Greater Membrane Perturbating Effect than SARS-CoV with Highly Specific Dependence on Ca2+ [Internet]. Molecular Biology; 2021 [cited 2023 Sep 8]. Available from: http://biorxiv.org/lookup/doi/10.11012021.01.04.425297.
- Hakansson-McReynolds S, Jiang S, Rong L, et al. Solution Structure of the Severe Acute Respiratory Syndrome-Coronavirus Heptad Repeat 2 Domain in the Prefusion State*. J Biol Chem. 2006;281:11965–11971.
- Chan DC, Fass D, Berger JM, et al. Core Structure of gp41 from the HIV Envelope Glycoprotein. Cell. 1997;89:263–273.
- Tan K, Liu J, Wang J, et al. Atomic structure of a thermostable subdomain of HIV-1 gp41. Proc Natl Acad Sci USA. 1997;94:12303–12308.
- Weissenhorn W, Dessen A, Harrison SC, et al. Atomic structure of the ectodomain from HIV-1 gp41. Nature. 1997;387:426–430.
- Yin H-S, Paterson RG, Wen X, et al. Structure of the uncleaved ectodomain of the paramyxovirus (hPIV3) fusion protein. Proc Natl Acad Sci USA. 2005;102:9288–9293.
- Melikyan GB, Markosyan RM, Hemmati H, et al. Evidence That the Transition of HIV-1 Gp41 into a Six-Helix Bundle, Not the Bundle Configuration, Induces Membrane Fusion. J Cell Biol. 2000;151:413–424.
- Russell CJ. Membrane fusion machines of paramyxoviruses: capture of intermediates of fusion. EMBO J. 2001;20:4024–4034.
- Tamm LK, Han X. Viral Fusion Peptides: A Tool Set to Disrupt and Connect Biological Membranes. Biosci Rep. 2000;20:501–518.
- Tai L, Zhu G, Yang M, et al. Nanometer-resolution in situ structure of the SARS-CoV-2 postfusion spike protein. Proc Natl Acad Sci USA. 2021;118:e2112703118.
- Fadda E. Understanding the Structure and Function of Viral Glycosylation by Molecular Simulations: State-of-the-Art and Recent Case Studies. Comprehensive Glycoscience [Internet]. Elsevier; 2021 [cited 2022 Jan 10]. p. 405–415. Available from: https://linkinghub.elsevier.com/retrieve/pii/B9780128194751000560.
- Watanabe Y, Berndsen ZT, Raghwani J, et al. Vulnerabilities in coronavirus glycan shields despite extensive glycosylation. Nat Commun. 2020;11:2688.
- Chawla H, Fadda E, Crispin M. Principles of SARS-CoV-2 Glycosylation. Curr Opin Struct Biol. 2022: 102402.
- Urbanowicz RA, Wang R, Schiel JE, et al. Antigenicity and Immunogenicity of Differentially Glycosylated Hepatitis C Virus E2 Envelope Proteins Expressed in Mammalian and Insect Cells. J Virol. 2019;93:e01403–e014018.
- Tian W, Li D, Zhang N, et al. O-glycosylation pattern of the SARS-CoV-2 spike protein reveals an “O-Follow-N” rule. Cell Res. 2021;31:1123–1125.
- Bagdonaite I, Thompson AJ, Wang X, et al. Site-Specific O-Glycosylation Analysis of SARS-CoV-2 Spike Protein Produced in Insect and Human Cells. Viruses. 2021;13:551.
- Zhang L, Mann M, Syed ZA, et al. Furin cleavage of the SARS-CoV-2 spike is modulated by O-glycosylation. Proc Natl Acad Sci USA. 2021;118:e2109905118.
- Bielik AM, Zaia J. Historical Overview of Glycoanalysis. In: Li J, editor. Functional Glycomics [Internet]. Totowa, NJ: Humana Press; 2010 [cited 2022 Jan 11]. p. 9–30. Available from: http://link.springer.com/10.1007978-1-60761-454-8_2.
- Atanasova M, Bagdonas H, Agirre J. Structural glycobiology in the age of electron cryo-microscopy. Curr Opin Struct Biol. 2020;62:70–78.
- Wu ZL, Ertelt JM. Fluorescent glycan fingerprinting of SARS2 spike proteins. Sci Rep. 2021;11:20428.
- Gong Y, Qin S, Dai L, et al. The glycosylation in SARS-CoV-2 and its receptor ACE2. Sig Transduct Target Ther. 2021;6:1–24.
- Roberts DS, Mann M, Li BH, et al. Distinct core glycan and O-glycoform utilization of SARS-CoV-2 Omicron variant Spike protein RBD revealed by top-down mass spectrometry. Chem Sci. 2022;13:10944–10949.
- Sztain T, Ahn S-H, Bogetti AT, et al. A glycan gate controls opening of the SARS-CoV-2 spike protein. Nat Chem. 2021;13:963–968.
- Deshpande A, Harris BD, Martinez-Sobrido L, et al. Epitope Classification and RBD Binding Properties of Neutralizing Antibodies Against SARS-CoV-2 Variants of Concern. Front Immunol. 2021;12:691715.
- Prajapat M, Sarma P, Shekhar N, et al. Drug for corona virus: A systematic review. Indian J Pharmacol. 2020;52:56.
- Forni G, Mantovani A. COVID-19 vaccines: where we stand and challenges ahead. Cell Death Differ. 2021;28:626–639.
- Hu X, Zhou Z, Li F, et al. The study of antiviral drugs targeting SARS-CoV-2 nucleocapsid and spike proteins through large-scale compound repurposing. Heliyon. 2021;7:e06387.
- Dejnirattisai W, Zhou D, Ginn HM, et al. The antigenic anatomy of SARS-CoV-2 receptor binding domain. Cell. 2021;184:2183–2200.
- Tang J, Lee Y, Ravichandran S, et al. Epitope diversity of SARS-CoV-2 hyperimmune intravenous human immunoglobulins and neutralization of variants of concern. iScience. 2021;24:103006.
- McCallum M, Marco AD, Lempp F, et al. N-terminal domain antigenic mapping reveals a site of vulnerability for SARS-CoV-2 [Internet]. Immunology; 2021 [cited 2022 Jan 28]. Available from: http://biorxiv.org/lookup/doi/10.11012021.01.14.426475.
- Meulen J, van den Brink EN, Poon LLM, et al. Human Monoclonal Antibody Combination against SARS Coronavirus: Synergy and Coverage of Escape Mutants. PLoS Med. 2006;3:e237.
- Zhou D, Duyvesteyn HME, Chen C-P, et al. Structural basis for the neutralization of SARS-CoV-2 by an antibody from a convalescent patient. Nat Struct Mol Biol. 2020;27:950–958.
- Kleanthous H, Silverman JM, Makar KW, et al. Scientific rationale for developing potent RBD-based vaccines targeting COVID-19. npj Vaccines. 2021;6:128.
- Kim Y-I, Kim D, Yu K-M, et al. Development of Spike Receptor-Binding Domain Nanoparticles as a Vaccine Candidate against SARS-CoV-2 Infection in Ferrets. mBio. 2021;12:e0023.
- Brouwer PJM, Caniels TG, van der Straten K, et al. Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability. Science. 2020;369:643–650.
- Jiang S, Zhang X, Du L. Therapeutic antibodies and fusion inhibitors targeting the spike protein of SARS-CoV-2. Expert Opin Ther Targets. 2021;25:415–421.
- Tong P, Gautam A, Windsor IW, et al. Memory B cell repertoire for recognition of evolving SARS-CoV-2 spike. Cell. 2021;184:4969–4980.
- Wec AZ, Wrapp D, Herbert AS, et al. Broad neutralization of SARS-related viruses by human monoclonal antibodies. Science. 2020;369:731–736.
- Watanabe Y, Mendonça L, Allen ER, et al. Native-like SARS-CoV-2 Spike Glycoprotein Expressed by ChAdOx1 nCoV-19/AZD1222 Vaccine. ACS Cent Sci. 2021;7:594–602.
- Martínez-Flores D, Zepeda-Cervantes J, Cruz-Reséndiz A, et al. SARS-CoV-2 Vaccines Based on the Spike Glycoprotein and Implications of New Viral Variants. Front Immunol. 2021;12:701501.
- Polack FP, Thomas SJ, Kitchin N, et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine. N Engl J Med. 2020;383:2603–2615.
- Thomas SJ, Moreira ED, Kitchin N, et al. Safety and Efficacy of the BNT162b2 mRNA Covid-19 Vaccine through 6 Months. N Engl J Med. 2021;385:1761–1773.
- El Sahly HM, Baden LR, Essink B, et al. Efficacy of the mRNA-1273 SARS-CoV-2 Vaccine at Completion of Blinded Phase. N Engl J Med. 2021;385:1774–1785.
- Baden LR, El Sahly HM, Essink B, et al. Efficacy and Safety of the mRNA-1273 SARS-CoV-2 Vaccine. N Engl J Med. 2021;384:403–416.
- Fiolet T, Kherabi Y, MacDonald C-J, et al. Comparing COVID-19 vaccines for their characteristics, efficacy and effectiveness against SARS-CoV-2 and variants of concern: a narrative review. Clin Microbiol Infect. 2022;28:202–221.
- Zhao F, Zai X, Zhang Z, et al. Challenges and developments in universal vaccine design against SARS-CoV-2 variants. npj Vaccines. 2022;7:167.
- Chavda VP, Apostolopoulos V. COVID-19 vaccine design and vaccination strategy for emerging variants. Expert Rev Vaccines. 2022;21:1359–1361.
- Zhang J, Xia Y, Liu X, et al. Advanced Vaccine Design Strategies against SARS-CoV-2 and Emerging Variants. Bioengineering. 2023;10:148.
- Rausch JW, Capoferri AA, Katusiime MG, et al. Low genetic diversity may be an Achilles heel of SARS-CoV-2. Proc Natl Acad Sci USA. 2020;117:24614–24616.
- Castonguay N, Zhang W, Langlois M-A. Meta-Analysis and Structural Dynamics of the Emergence of Genetic Variants of SARS-CoV-2. Front Microbiol. 2021;12:676314.
- Harvey WT, Carabelli AM, Jackson B, et al. SARS-CoV-2 variants, spike mutations and immune escape. Nat Rev Microbiol. 2021;19:409–424.
- Kumar S, Thambiraja TS, Karuppanan K, et al. Omicron and Delta variant of SARS-CoV-2: A comparative computational study of spike protein. J Med Virol. 2021: jmv.27526.
- Baral P, Bhattarai N, Hossen ML, et al. Mutation-induced changes in the receptor-binding interface of the SARS-CoV-2 Delta variant B.1.617.2 and implications for immune evasion. Biochem Biophys Res Commun. 2021;574:14–19.
- McCallum M, Czudnochowski N, Rosen LE, et al. Structural basis of SARS-CoV-2 Omicron immune evasion and receptor engagement. Science. 2022;375:864–868.
- Reardon S. How the Delta variant achieves its ultrafast spread. Nature. 2021: d41586-021–d4101986-w.
- Tzou PL, Tao K, Pond SLK, et al. Coronavirus Resistance Database (CoV-RDB): SARS-CoV-2 susceptibility to monoclonal antibodies, convalescent plasma, and plasma from vaccinated persons. PLoS One. 2022;17:e0261045.
- Planas D, Veyer D, Baidaliuk A, et al. Reduced sensitivity of SARS-CoV-2 variant Delta to antibody neutralization. Nature. 2021;596:276–280.
- Wrobel AG, Benton DJ, Roustan C, et al. Evolution of the SARS-CoV-2 spike protein in the human host. Nat Commun. 2022;13:1178.
- McCallum M, Bassi J, De Marco A, et al. SARS-CoV-2 immune evasion by the B.1.427/B.1.429 variant of concern. Science. 2021;373:648–654.
- Wang Y, Liu C, Zhang C, et al. Structural basis for SARS-CoV-2 Delta variant recognition of ACE2 receptor and broadly neutralizing antibodies. Nat Commun. 2022;13:871.
- Geng Q, Shi K, Ye G, et al. Structural Basis for Human Receptor Recognition by SARS-CoV-2 Omicron Variant BA.1. J Virol. 2022;96:e00249.
- Stalls V, Lindenberger J, Gobeil SM-C, et al. Cryo-EM structures of SARS-CoV-2 Omicron BA.2 spike [Internet]. bioRxiv; 2022 [cited 2022 Apr 27]. p. 2022.04.07.487528. Available from: https://www.biorxiv.org/content/10.11012022.04.07.487528v1.
- Lupala CS, Ye Y, Chen H, et al. Mutations on RBD of SARS-CoV-2 Omicron variant result in stronger binding to human ACE2 receptor. Biochem Biophys Res Commun. 2022;590:34–41.
- Shah M, Woo HG. Omicron: A Heavily Mutated SARS-CoV-2 Variant Exhibits Stronger Binding to ACE2 and Potently Escapes Approved COVID-19 Therapeutic Antibodies. Front Immunol. 2022;12:830527.
- Chen J, Wang R, Gilby NB, et al. Omicron Variant (B.1.1.529): Infectivity, Vaccine Breakthrough, and Antibody Resistance. J Chem Inf Model. 2022;62:412–422.
- Lauring AS, Hodcroft EB. Genetic Variants of SARS-CoV-2—What Do They Mean? JAMA. 2021;325:529.
- Zhou D, Dejnirattisai W, Supasa P, et al. Evidence of escape of SARS-CoV-2 variant B.1.351 from natural and vaccine-induced sera. Cell. 2021;184:2348–2361.
- Zahradník J, Marciano S, Shemesh M, et al. SARS-CoV-2 variant prediction and antiviral drug design are enabled by RBD in vitro evolution. Nat Microbiol. 2021;6:1188–1198.
- Dejnirattisai W, Huo J, Zhou D, et al. Omicron-B.1.1.529 leads to widespread escape from neutralizing antibody responses [Internet]. Microbiology; 2021 [cited 2022 Jan 28]. Available from: http://biorxiv.org/lookup/doi/10.11012021.12.03.471045.
- Motozono C, Toyoda M, Zahradnik J, et al. SARS-CoV-2 spike L452R variant evades cellular immunity and increases infectivity. Cell Host Microbe. 2021;29:1124–1136.
- The CITIID-NIHR BioResource COVID-19 Collaboration, The COVID-19 Genomics UK (COG-UK) Consortium, Collier DA, et al. Sensitivity of SARS-CoV-2 B.1.1.7 to mRNA vaccine-elicited antibodies. Nature. 2021;593:136–141.
- Chen RE, Zhang X, Case JB, et al. Resistance of SARS-CoV-2 variants to neutralization by monoclonal and serum-derived polyclonal antibodies. Nat Med. 2021;27:717–726.
- Yuan M, Liu H, Wu NC, et al. Structural basis of a shared antibody response to SARS-CoV-2. Science. 2020;369:1119–1123.
- Liu C, Zhou D, Nutalai R, et al. The antibody response to SARS-CoV-2 Beta underscores the antigenic distance to other variants. Cell Host Microbe. 2022;30:53–68.
- Yi C, Sun X, Lin Y, et al. Comprehensive mapping of binding hot spots of SARS-CoV-2 RBD-specific neutralizing antibodies for tracking immune escape variants. Genome Med. 2021;13:164.
- Feng F, Chen J, Zhao J, et al. Killing Two Birds with One Stone by Administration of Soluble ACE2: A Promising Strategy to Treat Both Cardiovascular Diseases and SARS-CoV-2 Infection. Viruses. 2021;13:2243.
- Jing W, Procko E. ACE2-based decoy receptors for SARS coronavirus 2. Proteins. 2021;89:1065–1078.
- Pomplun S. Targeting the SARS-CoV-2-spike protein: from antibodies to miniproteins and peptides. RSC Med Chem. 2021;12:197–202.
- Dubey A, Choudhary S, Kumar P, et al. Emerging SARS-CoV-2 Variants: Genetic Variability and Clinical Implications. Curr Microbiol. 2022;79:20.
- COVID-19 Genomics UK Consortium | UK-Wide Genomic Sequencing. [cited 2022 May 9]; 2021. Available from: https://www.cogconsortium.uk/.
- Alipoor R, Ranjbar R. Small-molecule metabolites in SARS-CoV-2 treatment: a comprehensive review. Biol Chem. 2023;404:569–584.
- Zhong L, Zhao Z, Peng X, et al. Recent advances in small-molecular therapeutics for COVID-19. Precision Clinical Medicine. 2022;5:pbac024.
- McKee DL, Sternberg A, Stange U, et al. Candidate drugs against SARS-CoV-2 and COVID-19. Pharmacol Res. 2020;157:104859.
- Wettstein L, Knaff PM, Kersten C, et al. Peptidomimetic inhibitors of TMPRSS2 block SARS-CoV-2 infection in cell culture. Commun Biol. 2022;5:1–12.
- Kandeel M, Yamamoto M, Tani H, et al. Discovery of New Fusion Inhibitor Peptides against SARS-CoV-2 by Targeting the Spike S2 Subunit. Biomol Ther (Seoul). 2021;29:282–289.
- Xia S, Liu M, Wang C, et al. Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion. Cell Res. 2020;30:343–355.
- Zhu Y, Yu D, Yan H, et al. Design of Potent Membrane Fusion Inhibitors against SARS-CoV-2, an Emerging Coronavirus with High Fusogenic Activity. Pfeiffer JK, editor. J Virol [Internet]. 2020 [cited 2022 Jan 12];94. Available from: https://journals.asm.org/doi/10.1128JVI.00635-20.
- Mahoney M, Damalanka VC, Tartell MA, et al. A novel class of TMPRSS2 inhibitors potently block SARS-CoV-2 and MERS-CoV viral entry and protect human epithelial lung cells. Proc Natl Acad Sci USA. 2021;118:e2108728118.
- Cheng Y-W, Chao T-L, Li C-L, et al. Furin Inhibitors Block SARS-CoV-2 Spike Protein Cleavage to Suppress Virus Production and Cytopathic Effects. Cell Rep. 2020;33:108254.
- Rahbar Saadat Y, Hosseiniyan Khatibi SM, Zununi Vahed S, et al. Host Serine Proteases: A Potential Targeted Therapy for COVID-19 and Influenza. Front Mol Biosci. 2021;8:725528.
- Hoffmann M, Hofmann-Winkler H, Smith JC, et al. Camostat mesylate inhibits SARS-CoV-2 activation by TMPRSS2-related proteases and its metabolite GBPA exerts antiviral activity. EBioMedicine. 2021;65:103255.
- Ohshio G, Saluja AK, Leli U, et al. Esterase inhibitors prevent lysosomal enzyme redistribution in two noninvasive models of experimental pancreatitis. Gastroenterology. 1989;96:853–859.
- Gibo J, Ito T, Kawabe K, et al. Camostat mesilate attenuates pancreatic fibrosis via inhibition of monocytes and pancreatic stellate cells activity. Lab Invest. 2005;85:75–89.
- Hempel T, Raich L, Olsson S, et al. Molecular mechanism of inhibiting the SARS-CoV-2 cell entry facilitator TMPRSS2 with camostat and nafamostat. Chem Sci. 2021;12:983–992.
- Shapira T, Monreal IA, Dion SP, et al. A TMPRSS2 inhibitor acts as a pan-SARS-CoV-2 prophylactic and therapeutic. Nature. 2022;605:340–348.
- Cao J-F, Yang X, Xiong L, et al. Mechanism of N-0385 blocking SARS-CoV-2 to treat COVID-19 based on molecular docking and molecular dynamics. Frontiers in Microbiology [Internet]. 2022 [cited 2023 Nov 2];13. Available from: https://www.frontiersin.org/articles/10.3389fmicb.2022.1013911.
- Wettstein L, Kirchhoff F, Münch J. The Transmembrane Protease TMPRSS2 as a Therapeutic Target for COVID-19 Treatment. IJMS. 2022;23:1351.
- Burley SK, Bhikadiya C, Bi C, et al. RCSB Protein Data Bank: powerful new tools for exploring 3D structures of biological macromolecules for basic and applied research and education in fundamental biology, biomedicine, biotechnology, bioengineering and energy sciences. Nucleic Acids Res. 2021;49:D437–D451.
- Bárcena M, Barnes CO, Beck M, et al. Structural biology in the fight against COVID-19. Nat Struct Mol Biol. 2021;28:2–7.
- Barrantes FJ. The Contribution of Biophysics and Structural Biology to Current Advances in COVID-19. Annu Rev Biophys. 2021;50:493–523.
- Zhang J, Chen B. Fighting SARS-CoV-2 with structural biology methods. Nat Methods. 2022;19:381–383.
- Lynch ML, Snell EH, Bowman SEJ. Structural biology in the time of COVID-19: perspectives on methods and milestones. IUCrJ. 2021;8:335–341.
- Kielian M, Rey FA. Virus membrane-fusion proteins: more than one way to make a hairpin. Nat Rev Microbiol. 2006;4:67–76.
- Bastard J, Taisne B, Figoni J, et al. Impact of the Omicron variant on SARS-CoV-2 reinfections in France, March 2021 to February 2022. Eurosurveillance [Internet]. 2022 [cited 2022 May 11];27. Available from: https://www.eurosurveillance.org/content/10.28071560-7917.ES.2022.27.13.2200247.