Abstract
Background: We have examined the relationship between the presence and numbers of methanogenic archaea in the faeces of humans and levels of the short chain fatty acids (SCFAs), especially butyrate, to gain insight into factors that may influence bowel health. In doing so, we have carried out the first cultivation-independent, molecular analysis of methanogen diversity and abundance in the human gastrointestinal tract. Materials and methods: The faeces of eight healthy individuals on their normal diets were collected weekly over a 12 week period. DNA was extracted from the faeces and PCR-based assays, using methanogen-specific 16S rRNA gene primer sets, were used in conjunction with denaturing gradient gel electrophoresis (DGGE) to identify and enumerate methanogens present. Results: Methanogens were detected in all faecal samples from five of the eight individuals tested. Three distinct methanogen phylotypes were found, with two or three phylotypes present in some individuals. DNA sequencing of DGGE bands indicated that all phylotypes were closely related to Methanobrevibacter smithii (99–100%). Real-time PCR analyses revealed that faecal methanogen numbers varied significantly between individuals and over time by up to two orders of magnitude. Concentrations of acetate, butyrate and propionate in faeces also varied significantly between individuals and with time. There was a negative correlation between mean faecal butyrate concentration and methanogen abundance (R = − 0.729, p<0.05, n=8), but no significant relationship existed for acetate, propionate or total SCFAs, and no relationship was found between total bacterial abundance and pH, SCFA concentration or methanogen abundance. Conclusion: As butyrate appears to be an important mediator of colonic health, the inverse relationship with methanogens uncovered here suggests that methanogens may be useful biomarkers of bowel health.
Introduction
Diets rich in complex carbohydrates have been shown to be associated with protection from diseases of the large bowel, including colorectal cancer, at least in part by increasing fermentation and subsequent production of the short chain fatty acids (SCFAs), especially butyrate Citation[1]. Butyrate is a primary energy source for cells lining the colon and appears to protect against activities associated with carcinogenesis by enhancing cell cycle arrest and apoptosis Citation[2], Citation[3].
A number of butyrate-producing bacteria have been identified in human faeces Citation[4]. A study of butyrate formation by Faecalibacterium prausnitzii and Roseburia intestinalis, two of the most predominant butyrate-producers in humans Citation[5], demonstrated that 85–90% of butyrate carbon was derived from external acetate Citation[6]. Also, a study by Barcenilla et al. Citation[7] demonstrated that 95% of isolated bacterial strains utilizing acetate were butyrate producers. This suggests that butyrate production by bacteria is heavily dependent on the availability of acetate, an SCFA that is also generated during colonic fermentation. Hence the availability of acetate and the activities of bacteria that influence that availability could also be expected to play a significant role in colonic health as rate-limiting steps in butyrate formation.
Methanogens are strict anaerobes that are physiologically and phylogenetically distinct members of the Euryarchaeta. In humans they are associated with the gastrointestinal tract, vagina and dental plaque Citation[8]. Cultivation-based studies of methanogen diversity in humans have indicated that the dominant methanogen is Methanobrevibacter smithii, with lower numbers of Methanosphaera stadtmanae Citation[9]. Members of the genus Methanobrevibacter including M. smithii, the primary methanogen associated with the human colon, require acetate as a major source of cell carbon. Given the apparent importance of acetate for butyrate production and large bowel health, it is plausible that the presence and activity of methanogens will have a significant influence on bowel health through their influence on acetate concentration. A number of publications have speculated about the role of methanogens in colonic pathology, including colorectal cancer, but no clear links have been established Citation[8], Citation[10–15].
Our understanding of the role of methanogens in the human colon is likely to have been limited by the methods used to detect them. The techniques used to detect methanogens in humans have so far relied on cultivation or measurement of methane emission by gas chromatography. The use of breath methane measurements for the detection of methanogens requires cell numbers in the order of 108 per gram of dry weight to be successful Citation[9], and is therefore unsuitable for analysis of potentially important changes in numbers below that level. In recent years advances in molecular biology techniques have been applied to microbiology and enabled the detection of bacteria, archaea and other organisms that may otherwise be difficult to culture or differentiate by conventional means.
In the present study we set out to use molecular biology techniques to provide a clearer picture of the link between the presence and number of methanogenic archaea in the human colon and a range of bowel health-associated measures including SCFA concentration and faecal pH. To do this we collected the faeces of eight healthy individuals weekly over a 12 week period and developed PCR-based assays in conjunction with denaturing gradient gel electrophoresis (DGGE) to identify and enumerate any methanogens present. To our knowledge this represents the first cultivation-independent, molecular analysis of methanogen diversity and abundance in the human gastrointestinal tract.
Materials and methods
Sampling, DNA extraction and purification
Faecal samples were collected at weekly intervals, for 12 weeks, from each of eight individuals, five females and three males, with a median age of 45.5 years (±9.3), who ate their normal diet. In all, 84 samples were homogenized and duplicate 0.1 g samples were collected for DNA extraction in 2 ml microcentrifuge tubes (Eppendorf, USA). Samples were stored at −80°C until processing. Extraction of DNA from 0.1 g faecal samples was performed using the QIAmp® DNA Stool Mini Kit (Qiagen), following the manufacturer's protocol. Extracts were visualized on 1% (w/v) agarose gels containing 10 µg ml−1 ethidium bromide using UV transillumination to ensure that the extractions gave high quality high molecular weight DNA for analysis.
Faecal SCFAs and pH analysis
Faecal SCFA concentrations were determined using the method of Patten et al. Citation[16]. Faecal pH was measured using a Cyberscan 500 pH meter (Eutech Instruments, Singapore) by homogenizing 0.5–2.0 g of faecal sample in 3×w/v sterile H2O (pH 7.0) in a 10 ml centrifuge tube.
DGGE-PCR and phylogenetic analysis
Methanogen-specific 16S rRNA gene primer sets were used to amplify regions of the 16S rRNA gene. Methanogen-specific primers (850f and 1250r, 1250rc) were designed by aligning all full-length methanogen 16S rDNA gene sequences available on the NCBI database (http://www.ncbi.nlm.nih.gov/) using the ARB software package and database Citation[17], and using the Primer Design tool in ARB. Primers were checked in silico for cross-reactivity using the basic local alignment search tool program (BLAST) Citation[18] available through NCBI. Primers were checked against DNA extractions from pure cultures of a number of methanogens including Methanobrevibacter smithii, Methanobrevibacter ruminatium, Methanobrevibacter woesei, Methanosarcina barkeri and Methanosphaera stadtmanae, as well as faecal-derived bacterial strains representing a number of divisions. Both combinations of primers (850f-1250r and 850f-1250rc) demonstrated a high level of specificity to methanogens, with no cross-reactivity with non-methanogens detected.
Two sets of primers were used for the amplification of methanogens including the forward primers 850f (5′-GAGCACCACAACGCGU-3′) (this study) and 448f (5′-GGTGCCAGCCGCCGC-3′) Citation[19] and GC-clamped reverse primers 1260rc (5′- CGCCCGCCGCGCCCCGCGCCCGGCCCGCCGCCCCCGCCCCCTACGCATTCCAGCTTC-3′) (this study) and 1027rc (5′-CGCCCGCCGCGCCCCGCGCCCGGCCCGCCGCCCCCGCCCCGGTCTCGCTCGTTGCC-3′) (adapted from Wright and Pimm Citation[19]). The PCR was performed using BIOTAQ polymerase (Bioline) and 10 mM deoxynucleotide triphosphates (dNTPs), 10 ng genomic DNA and 12.5 pmol of both primers, in reactions made up to a volume of 25 µl with sterile MilliQ water. PCR was performed using a touch down protocol on a Hybaid PCR Express thermal cycler (Hybaid, UK), with 29 cycles of 1 min denaturation at 95°C, 30 s annealing using 0.5° steps from 65°C to 55°C and 2 min extension steps at 72°C, with a final 4 min extension step at 72°C. DGGE analysis was performed using the Ingeny phorU-2 (Ingeny International BV). Then 15 µl of PCR product was loaded per well and electrophoresed on a 6% acrylamide gel containing 20–80% denaturing gradient for 16.5 h at 110 V and 60°C. Gels were stained for 30 min in 100 ml of 1×Sybr-gold (Molecular Probes) in 40 mM Tris-acetate, 1 mM disodium EDTA at pH 8.0, and destained for 5 min in 100 ml of MilliQ H2O. Images were digitally captured under UV transillumination using DigiDoc Software (Bio-Rad Laboratories, Hercules, CA, USA).
The concentration of genomic DNA added to PCR reactions was varied to optimize the clarity of DGGE gels. Sequencing of DGGE bands was performed as described previously Citation[20], utilizing the primer sets listed above. DGGE band sequences were compared to sequences on the Genbank database using the BLAST tool. Sequences from this study were deposited in Genbank under accession numbers DQ304105–DQ304107.
Real-time PCR
Real-time PCR reactions were prepared in hard-shell thin-wall 96-well microplates and carried out in a Chromo4 thermocycler (MJ Research, USA). Results were analysed with the Opticon Monitor 3 software (V.3.1). Reactions (20 µl volumes) contained 1 µl of BIOTAQ (Bioline), 0.6 µl MgCl2 (50 mM), 0.4 µl 50 mM dUNTP mix (Bioline), 2 µl 10×NH4 reaction buffer, 0.2 µl of each of the primers 850f and 1260r (50 pmol/µl), 1 µl of 1× SYBR Green I nucleic acid stain (Molecular Probes) and 0.1–10 ng of template DNA and sterile MilliQ water. Primers used for amplification and detection of methanogen 16S rRNA genes included 850f and 1260r (5′-CTACGCATTCCAGCTTC-3′). Assays were performed using a thermocycling programme consisting of an initial 5 min step at 95°C followed by 35 cycles consisting of 95°C for 30 s, 56°C for 30 s and 72°C for 30 s with fluorescent acquisition, and a further fluorescent acquisition step at 80°C. A final melt curve analysis was performed after completion of all the amplification cycles with fluorescence acquired at 0.5°C intervals between 55°C and 100°C. Fluorescence analysis was performed at a temperature at which all primer dimers had melted, but the specific product had not. Amplified products from mixed template samples only contained a single peak, indicating that product length variability and G + C content did not have a significant effect on quantification. Positive control standards for the real-time PCR included a dilution series of DNA extracted from a known density of M. smithii culture.
The sensitivity and specificity of the assay were tested using a dilution series of methanogen-positive faecal DNA as well as DNA extracted from a culture of M. smithii of known density, both demonstrating a linear relationship between concentration and Ct value (R2>0.98). A series of 10-fold dilutions of the control, from 10 ng to 0.1 pg, were analysed in parallel with faecal DNA samples. Negative controls included samples lacking template DNA. All real-time PCR products were examined using agarose gel electrophoresis to ensure that products corresponded to the correct size and to ensure the absence of non-specific product. Values were corrected for their initial sample weight and averaged to calculate the total 16S rDNA amount per wet weight of faecal material. Samples that tested negative for methanogen DNA were assigned a methanogen DNA concentration of 0 ng/gww for the purpose of analysis. Analysis of bacterial abundance utilizing the primers 519f and 907r followed previous methods Citation[20].
Results
DGGE analysis
DGGE analysis of methanogen communities in 64 faecal samples, using two different 16S rDNA primer sets specific to members of the methanogenic archaea, demonstrated the presence of methanogen-associated phylotypes in all samples from five of the eight volunteers tested. The methanogens, detected in 55 of the 86 samples, had a dichotomous distribution, with variation of numbers in the methanogen-positive volunteers, but a universal absence of detection in the negative volunteers. All of the DNA extractions from the study were examined using bacterial-specific primers (907f and 1392r Citation[21], Citation[22], and PCR product confirmed the presence of PCR available high molecular weight bacterial DNA. Analysis of methanogen populations using DGGE demonstrated three distinct phylotypes, with some samples demonstrating two or three of these phylotypes. Subsequent band excision and sequencing revealed all to be closely related to M. smithii (99–100%). No other archaeal phylotypes were detected with the use of either methanogen-specific primer set or universal archaeal primers.
Real-time PCR analysis
Analysis of the sensitivity of the real-time PCR assay for members of the M. smithii group demonstrated a detection limit of 0.0025 ng DNA, and a corresponding concentration of 1.6×107 cells per gram of wet weight. Dilution series of sample and pure culture DNA extractions demonstrated no dilution effect on specificity of the assay with the standard curve of 1/10 dilutions above the detection limit demonstrating a strong linear relationship (R2>0.98) for both faecal sample and culture extractions. Real-time PCR analysis of methanogens in samples from five methanogen-positive individuals demonstrated a large range of methanogen DNA concentration per gram of wet weight faecal matter over time, with calculated methanogen numbers varying between one and two orders of magnitude within each individual (). Methanogen DNA concentrations in 55 methanogen-positive samples varied from 0.0143 mg/gww to 6.605 mg/gww.
Figure 1. Concentration of methanogen DNA (ng/gww) in human faecal samples over a 14 week period. •λ, volunteer 2; □, volunteer 6; ▴, volunteer 8; ▾, volunteer 5 and • volunteer 4.
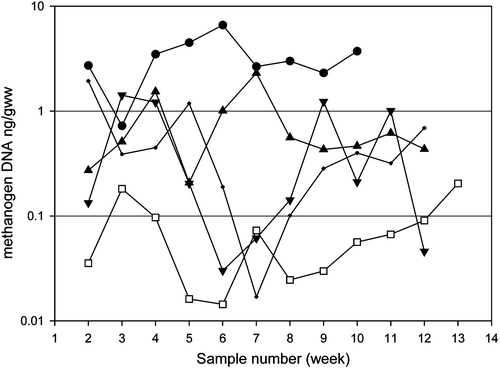
Analysis of the relative bacterial abundance using real-time PCR also demonstrated significant variation in the abundance of members of the domain bacteria in faecal samples (data not shown).
Faecal pH and SCFA concentrations
Analysis of SCFA concentrations in 86 individual samples demonstrated variations in total SCFAs of between 26.4 and 200.1 mmol/L, with butyrate (1.9–51.6 mmol/L), acetate (17.0–118.3 mmol/L) and propionate (3.5–51.6 mmol/L) varying to a similar degree ( and ). Faecal pH varied between 6.3 and 7.9 with mean faecal pH values for each volunteer varying between 6.68 and 7.62 ().
Figure 2. Relationship of methanogen 16S rDNA gene abundance and faecal butyrate concentration. Methanogen and butyrate concentration averaged over 12 weeks of the study. Error bars show standard error.
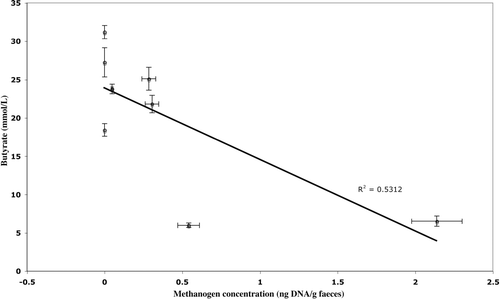
Figure 3. Relationship between faecal pH and faecal butyrate concentration. Error bars show standard error.
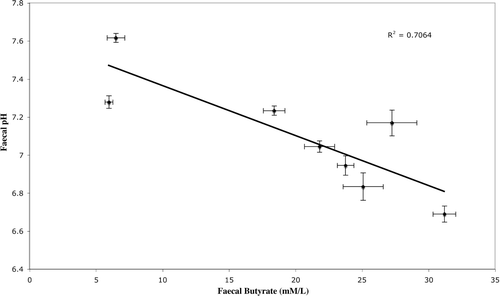
Figure 4. Relationship between methanogen 16S rRNA gene abundance and faecal pH for individuals in which methanogens were detected. Error bars show standard error.
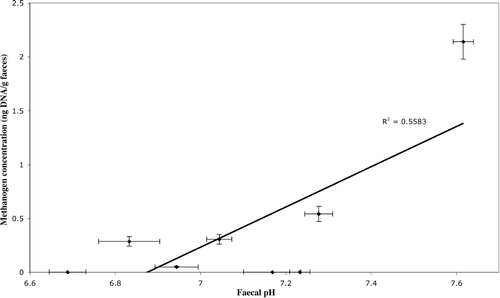
Analysis of faecal SCFA concentration and pH (mean values over 12 weeks) using the Pearson product-moment correlation demonstrated a positive relationship between each of the SCFAs measured (R > 0.86, p < 0.01) and a negative relationship between SCFAs and pH (R < –0.818, p < 0.01) (, Figure 3). A negative correlation was found between mean faecal butyrate concentration and methanogen abundance (R = − 0.729, p<0.05, n=8) (Figure 2), but no such relationship existed for acetate, propionate or total SCFAs (). A positive correlation between methanogen abundance and pH was also found (R = /0.747, p</0.05) (Table I, Figure 4). No significant relationship was found between total bacterial abundance and pH, SCFA concentration or methanogen abundance ().
Table I. Pearson's moment correlation of faecal methanogen abundance, relative bacterial abundance and pH versus pH, bacterial abundance and SCFA concentrations.
Discussion
This study represents the first use of PCR-based methodology to directly examine methanogen diversity and population changes in human faecal samples. Using this methodology we have detected and quantified methanogens in faeces from five of eight individuals over a 12 week period and identified the methanogen phylotypes present. The values of faecal SCFA concentration from this study are comparable with previous studies Citation[1]. The study demonstrated that individuals with high mean faecal butyrate concentrations had correspondingly low or undetectable numbers of methanogens.
Additionally, we have used this methodology to demonstrate a negative association between methanogens and butyrate concentration in human faeces. Consequently, given the role that butyrate plays in maintaining colonic integrity, factors controlling the interplay between methanogenesis and butyrogenesis may have a strong bearing on colonic health.
Examination of 86 DNA extractions from faecal samples from 8 individuals, revealed detectable methanogen numbers in only 5 individuals (in 55 samples). The use of DGGE-PCR with two different primer sets targeted to members of the methanogenic archaea enabled us to demonstrate three separate phylotypes closely related to M. smithii, demonstrating > 99% similarity over the 420–565bp fragments sequenced. This low diversity of methanogens is consistent with previous studies Citation[9], Citation[23], which found the dominant phylotype to be M. smithii, with only one study reporting the presence of M. stadtmanae Citation[24]. Our study confirms M. smithii as being the predominant methanogen in the human colon.
Our study demonstrates that some individuals possess faecal methanogen numbers that are either lower than our ability to detect them or negligible. Previous studies of methanogen populations in humans have found cultivatable numbers of methanogens in faecal samples ranging from 108 to 1010 per gdw, whereas breath methane is thought to detect methanogen numbers above 108/gdw Citation[25]. We have demonstrated that our PCR method can detect methanogen numbers as low as 1.6×107 cells/gww. By using microscopy and cultivation methods, Miller and Wolin Citation[25] concluded that methanogens were present in all individuals on a western diet; however, the concentration varied over 10 logs between individuals. In light of this it is possible that the volunteers from this study that did not test positive for the presence of methanogens may still harbour methanogens at concentrations lower than 1.6×107 cells/gww. As individuals in this study were on their normal diets it is possible that differences in diet could account for differences in detectability of methanogens or that some individuals may simply have a low baseline level of methanogens. We found considerable differences in methanogen numbers from week to week within individuals, often with changes of an order of magnitude, and found corresponding variation in numbers between individuals.
The association of methanogens with decreased faecal butyrate concentration could occur via a number of mechanisms. Firstly, methanogens or syntrophically associated bacteria may represent net consumers of SCFAs in the gut. Previous studies Citation[26], Citation[27] investigating the syntrophic association of butyrate-degrading bacteria with methanogens have demonstrated that inhibitors of methanogens prevented butyrate degradation in thermophilic mixed cultures. Syntrophic strains convert butyrate into acetate and H2 under anaerobic conditions. It is possible that a similar syntrophic relationship between methanogens and butyrate-degrading bacteria is associated with SCFA degradation in the human gut. However, the lack of human faecal-derived sequences closely related to the syntrophomonodaceae on the GenBank database suggests this is unlikely. An alternative, and more likely, explanation for the observed relationship between butyrate and methanogens is competition for H2 between methanogens and hydrogenotrophic acetogens, both of which have been identified in the colon. Hydrogen assimilated by methanogens is utilized in the production of methane, which is not further utilized in the colon. Hydrogen assimilated by hydrogenotrophic acetogens is utilized in the reduction of CO2 to acetate via the acetyl-CoA pathway. It is likely that hydrogen assimilated in this manner contributes to faecal butyrate through the butyrate-producing bacteria. In vitro experiments involving the utilization of hydrogen by the methanogen M. smithii and the acetogen Ruminococcus hydrogenotrophicus demonstrated a synergistic increase in butyrate production by R. intestinalis when grown in co-culture with R. hydrogenotrophicus over co-culture with M. smithii Citation[28]. We hypothesize that competition for hydrogen between methanogens and acetogens results in reduced butyrate production in the presence of high numbers of methanogens, due to the lower H2 threshold of methanogens compared with acetogens when CO2 is the terminal electron acceptor Citation[29]. In addition to the competition for hydrogen with acetogens, methanogens use acetate as a major source of carbon, and as such would further reduce acetate available for butyrate production. The lack of a significant relationship between methanogens and acetate may partly be due to the effect of assimilation of acetate into butyrate, but also due to the effect of absorption of acetate on faecal acetate concentration. Vogt and Wolever Citation[30] found that unlike faecal butyrate and propionate, faecal acetate may reflect absorption rather than production in the colon. It is hypothesized that the inverse relationship observed here between faecal butyrate and methanogens will also occur between butyrate and the sulphate reducers, as methanogenesis and sulphidogenesis are primary users of hydrogen in the colon.
The negative relationship between pH and butyrate is consistent with the findings of Walker et al. Citation[31] who demonstrated an increase in butyrate production by human faecal microbial communities at lower pH. It has also been suggested that lower pH improves the competitiveness of acetogens for hydrogen Citation[29].
Data suggesting that archaea may be pathogenic Citation[8] could be explained by the relationships discussed here. That is, methanogens may not represent direct pathogens but rather may be associated with colorectal cancer indirectly by increasing in number when resistant starch fermentation and production of protective butyrate are low. Methanogens may therefore represent a potential biomarker for bowel health, particularly as their limited diversity makes them a good target for 16S rRNA gene-based techniques.
We thank Caroline Cooke and Kerry Nyland for assistance with sample processing, and Andre-Denis Wright (CSIRO Livestock Industries, WA) for strains and methanogen DNA, as well as assistance in proof-reading.
References
- Topping DL, Clifton PM. Short-chain fatty acids and human colonic function: roles of resistant starch and nonstarch polysaccharides. Physiol Rev 2001; 81: 1031–64
- Avivi-Green C, Polak-Charcon S, Madar Z, Schwartz B. Apoptosis cascade proteins are regulated in vivo by high intracolonic butyrate concentration: correlation with colon cancer inhibition. Oncol Res 2000; 12: 83–95
- Zgouras D, Wachtershauser A, Frings D, Stein J. Butyrate impairs intestinal tumor cell-induced angiogenesis by inhibiting HIF-1 alpha nuclear translocation. Biochem Biophys Res Commun 2003; 300: 832–8
- Louis P, Duncan SH, McCrae SI, Millar J, Jackson MS, Flint HJ. Restricted distribution of the butyrate kinase pathway among butyrate-producing bacteria from the human colon. J Bacteriol 2004; 186: 2099–106
- Duncan SH, Barcenilla A, Stewart CS, Pryde SE, Flint HJ. Acetate utilization and butyryl coenzyme A (CoA): acetate-CoA transferase in butyrate-producing bacteria from the human large intestine. Appl Environ Microbiol 2002; 68: 5186–90
- Duncan SH, Holtrop G, Lobley GE, Calder AG, Stewart CS, Flint HJ. Contribution of acetate to butyrate formation by human faecal bacteria. Br J Nutr 2004; 91: 915–23
- Barcenilla A, Pryde SE, Martin JC, Duncan SH, Stewart CS, Henderson C, et al. Phylogenetic relationships of butyrate-producing bacteria from the human gut. Appl Environ Microbiol 2000; 66: 1654–61
- Cavicchioli R, Curmi PMG, Saunders N, Thomas T. Pathogenic archaea: do they exist?. Bioessays 2003; 25: 1119–28
- Weaver GA, Krause JA, Miller TL, Wolin MJ. Incidence of methanogenic bacteria in a sigmoidoscopy population: an association of methanogenic bacteria and diverticulosis. Gut 1986; 27: 698–704
- Lepp PW, Brinig MM, Ouverney CC, Palm K, Armitage GC, Relman DA. Methanogenic Archaea and human periodontal disease. Proc Natl Acad Sci U S A 2004; 101: 6176–81
- Jangid K, Rastogi G, Patole MS, Shouche YS. Methanobrevibacter: is it a potential pathogen?. Curr Sci 2004; 86: 1475–6
- Martin W. Pathogenic archaebacteria: do they not exist because archaebacteria use different vitamins?. Bioessays 2004; 26: 592–3
- Haines A, Metz G, Dilawari J, Blendis L, Wiggins H. Breath-methane in patients with cancer of the large bowel. Lancet 1977; 2: 481–3
- Pique JM, Pallares M, Cuso E, Vilar-Bonet J, Gassull MA. Methane production and colon cancer. Gastroenterology 1984; 87: 601–5
- Karlin DA, Jones RD, Stroehlein JR, Mastromarino AJ, Potter GD. Breath methane excretion in patients with unresected colorectal cancer. J Natl Cancer Inst 1982; 69: 573–6
- Patten GS, Bird AR, Topping DL, Abeywardena MY. Effects of convenience rice congee supplemented diets on guinea pig whole animal and gut growth, caecal digesta SCFA and in vitro ileal contractility. Asia Pac J Clin Nutr 2004; 13: 92–100
- Ludwig W, Strunk O, Westram R, Richter L, Meier H, Yadhukumar, et al. ARB: a software environment for sequence data. Nucleic Acids Res 2004; 32: 1363–71
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997; 25: 3389–402
- Wright ADG, Pimm C. Improved strategy for presumptive identification of methanogens using 16S riboprinting. J Microbiol Methods 2003; 55: 337–49
- Abell GCJ, Bowman JP. Ecological and biogeographic relationships of class Flavobacteria in the Southern Ocean. FEMS Microbiol Ecol 2005; 51: 265–77
- Santegoeds CM, Ferdelman TG, Muyzer G, de Beer D. Structural and functional dynamics of sulfate-reducing populations in bacterial biofilms. Appl Environ Microbiol 1998; 64: 3731–9
- Ferris MJ, Muyzer G, Ward DM. Denaturing gradient gel electrophoresis profiles of 16S rRNA-defined populations inhabiting a hot spring microbial mat community. Appl Environ Microbiol 1996; 62: 340–6
- Eckburg PB, Bik EM, Bernstein CN, Purdom E, Dethlefson L, Sargent M, et al. Diversity of the human intestinal microbial flora. Science 2005; 308: 1635–8
- Miller TL, Wolin MJ. Methanosphaera stadtmaniae gen. nov., sp. nov.: a species that forms methane by reducing methanol with hydrogen. Arch Microbiol 1985; 141: 116–22
- Miller TL, Wolin MJ. Methanogens in human and animal intestinal tracts. Syst Appl Microbiol 1986; 7: 223–9
- Ahring BK, Westermann P. Thermophilic anaerobic degredation of butyrate by a butyrate-utilizing bacterium in coculture and triculture with methanogenic bacteria. Appl Environ Microbiol 1987; 53: 429–33
- Zhang CY, Liu XL, Dong XX. Syntrophomonas erecta sp nov., a novel anaerobe that syntrophically degrades short-chain fatty acids. Int J Syst Evol Microbiol 2005; 55: 799–803
- Chassard C, Bernalier-Donadille A. H2 and acetate transfers during xylan fermentation between a butyrate-producing xylanolytic species and hydrogenotrophic microorganisms from the human gut. FEMS Microbiol Lett 2006; 254: 116–22
- Drake HL, Kusel K, Matthies C. Ecological consequences of the phylogenetic and physiological diversities of acetogens. Antonie Van Leeuwenhoek 2002; 81: 203–13
- Vogt JA, Wolever TM. Fecal acetate is inversely related to acetate absorption from the human rectum and distal colon. J Nutr 2003; 133: 3145–8
- Walker AW, Duncan SH, McWilliam Leitch EC, Child MW, Flint HJ. pH and peptide supply can radically alter bacterial populations and short-chain fatty acid ratios within microbial communities from the human colon. Appl Environ Microbiol 2005; 71: 3692–700