Abstract
For preparation of pandemic plans for the H5N1 bird flu virus, it has been common practice among health authorities in several countries to use crude death figures from the Spanish flu pandemic as a worst case scenario. This has been done without taking into consideration either what is known about the molecular biology of malignant influenza viruses or the detailed statistical information that is available about the Spanish flu pandemic, as regards the variation of death risk as a function of age and also as regards the different behavior of the virus during successive pandemic waves. There is no scientific basis for the assumption that a new pandemic influenza virus cannot be worse than the Spanish flu virus. However, if we hypothetically assume recurrence of a pandemic influenza virus with virulence and transmissibility properties identical to the Spanish flu virus, but belonging to a subtype other than those that have been circulating in human populations since 1918, it will be unrealistic to assume that the older part of the population will be immune against a type of virus that their immune system has never encountered before. Therefore we have attempted to estimate what might have happened by counterfactually assuming that individuals in 1918 had not been immunized earlier against H1 (or H1N1) influenza viruses that were circulating before 1890. An overview is given of age-related changes in immunological factors that may affect the age-specific mortality in different age groups, and also expected consequences for the mortality in different age groups of mitochondrial DNA aging per se. Extrapolations for age-specific death rate have been made for four different scenarios, giving lower and upper boundaries for what might have happened if nobody had been immune when the 1918–1920 pandemic started. For the first (lower boundary) scenario the age-specific death rate is increasing up to the age of 30 and is assumed (very unrealistically) to be constant thereafter. For the second scenario the age-specific death rate is monotonically increasing as a function of age according to an almost cubic functionality. For the third (upper boundary) scenario the age-specific death rate is increasing exponentially with age. For the third scenario the mortality of the world population becomes 0.3. Thus 30% of the world population could have been killed by the Spanish flu virus, had nobody been immune when the pandemic started. For the fourth scenario the mortality by age due to the virus mimics the traditional mortality by age in a population. The fifth scenario is identical with scenario 3, except that it is also counterfactually assumed that the wave 2 virus came first. For this scenario, it is found that as much as 80% of the total world population in 1918 might have been killed by the Spanish flu virus.
Introduction
After the resurgence of highly pathogenic avian influenza virus as a serious public health threat, starting with the outbreak in Hong Kong and the resurgence of human cases of lethal H5N1 influenza from 2003 onwards, health authorities in several countries have made national pandemic influenza preparedness plans, in case there should be a pandemic outbreak of H5N1 or some other highly virulent form of avian influenza. But these plans have rarely been based on what is known about the virulence properties of what is commonly considered the most serious threat today, i.e. highly pathogenic H5N1 avian influenza. Instead they have been based on crude death statistics from the Spanish flu pandemic, which on an arbitrary basis has been assumed to represent a worst case scenario. In an assessment from 2005 of national pandemic influenza preparedness plans from 21 countries in Europe Citation[1], it was found that every country studied had been planning for a repetition of what happened in 1918, and no country had made a preparedness plan based on what is known about the lethality and biological properties (as based on epidemiological observations, molecular biological studies and animal experiments) of highly pathogenic H5N1 viruses now circulating in South-east Asia.
One might have hoped, when the Spanish flu has been used as a hypothetical worst case scenario, that this would have been based on a good historical and epidemiological understanding of what actually happened during the Spanish flu pandemic. But this appears not to be the case (the arguments on which this conclusion is based will be found in the discussion below).
The 1918–1920 Spanish influenza pandemic was a pandemic caused by an unusually severe and deadly virus belonging to the subtype H1N1 of influenza type A. Some of those who were killed during the pandemic must have succumbed to secondary bacterial infections, since no antibiotics were available Citation[2]. However, a subset was killed rapidly after the onset of symptoms in less than 5 days, and it is possible that a majority of those who died in 1918–1920 were killed by the influenza virus itself Citation[3].
Death numbers are better known from historical sources than the number of infected persons, since registration must have been much better in case of death than for patients with mild to moderate influenza symptoms. The surplus mortality rate for Norway was 0.6%, but for India it was about 6% of the whole population Citation[4], Citation[5]. A recurrent feature of the work carried out by historians on the pandemic over the last couple of decades has been the consistent upward revision of mortality figures Citation[4]. The last available compilation of historical work from all over the world, made by Johnson and Mueller, revises the figure for total numbers of death upwards to about 50 million (2.8% of the world population), while recognizing that even this figure may be substantially lower than the real death toll, perhaps being understated by as much as 100% Citation[4].
The Spanish influenza pandemic appeared in four ‘waves’, each of which should be considered an independent global influenza epidemic (or pandemic). The first cases were reported in March–April 1918, with the virus spreading fast and infecting a relatively large fraction of the population during the northern summer months, but relatively fewer patients died than during the next wave Citation[6] (even though the mortality during the first wave of the Spanish flu pandemic was much higher than for seasonal influenzas now). The virus apparently mutated during the summer, and at the end of August a second and highly virulent bout of influenza started Citation[6]. Two-thirds of all the deaths from Spanish influenza occurred during the months from October through December 1918 Citation[6]. A third bout of influenza started around the Christmas and New Year celebrations in the year shift 1918–1919 and did not subside before March–April 1919 Citation[6]. The third wave did not spread as fast as the first wave had done during the summer months of 1918 and was not as lethal as the second wave, probably because a large proportion of the population had gained immunity and frail individuals had already died from influenza or bacterial pneumonia in the previous waves Citation[6]. Thus, there were three major outbreaks within the duration of 1 year Citation[2], Citation[6–8]. But in some places, Spanish flu seems to have returned as a fourth wave in 1920 Citation[4], Citation[8].
The Spanish flu virus was unusual in killing many young and healthy victims, unlike the annual ordinary influenza epidemics, which kill mostly small children and the elderly Citation[6]. While curves for the influenza death rates with respect to age in annual influenza epidemics are normally U-shaped, with the highest mortality for the very young and the most elderly, during the Spanish flu pandemic a more W-shaped curve was observed instead Citation[5], Citation[6], with a sharp peak in age-specific mortality occurring between the age of 25 and 30 years Citation[5].
The causes of the peculiar shape of the curve for death risk as a function of age during the Spanish flu pandemic have not been well understood. The steep rise in age-specific mortality from age group 10–14 to age group 25–29 years may plausibly be explained as a consequence of host factors such as age-related changes in the number of T cells leaving the thymus and in the levels of immunostimulatory hormones. However, it is not possible to explain the steep decline in age-specific mortality from 25–29 years to higher age groups by any such mechanism. There is no known endogenous factor that varies as a function of age so as to show a sharp minimum or maximum at an age near 30 years Citation[5], and affecting women and men in the same way Citation[5].
Instead, the peak may plausibly be explained as a consequence of immunity from earlier influenza epidemics. The last one of these immunizing epidemics must then be postulated to have occurred about 30 years before 1918, which could have been just before the start of the ‘Russian flu’ pandemic that appears to have started in 1889 Citation[9]. In order to explain the strength of protective immunity among higher age groups during the Spanish flu pandemic Citation[5] and the flat minimum on the curve for age-specific mortality somewhere between 50 and 60 years Citation[5], we need to postulate that H1 (most likely H1N1) virus strains must have been circulating in human populations during the 1880s and earlier, at least back to the 1860s. To explain the lack of protective immunity among people younger than about 28–30 years, we need also to postulate that these influenza virus strains were wiped out following the Russian flu pandemic, similarly to the disappearance of H1N1 viruses in human populations after the Asian flu pandemic in 1957 and H2N2 viruses in human populations after the Hong Kong flu pandemic in 1968.
The World Health Organization (WHO) no longer considers death figures from the Spanish flu pandemic to represent a realistic worst case scenario. In a WHO report following the meeting of a working group on 21–22 September 2006, it is stated that should the virus improve its transmissibility in human populations by acquiring internal human genes through a reassortment event, then the lethality of the virus would most likely be reduced Citation[10]. However, should the virus improve its transmissibility through adaptation as a wholly avian virus (i.e. without reassortment), then the present high lethality could be maintained during a pandemic Citation[10]. Also, on its home page, WHO gives a statistical overview of all confirmed human H5N1 cases, by country and year, since the resurgence of H5N1 transmission to humans started in 2003. In 2006, average fatality among registered human H5N1 cases all over the world was 69%, but in Indonesia (the country that had the largest number of H5N1 cases in 2006) it was 82%. This is obviously much worse than the mortality observed in most countries during the Spanish flu pandemic.
The question still remains as to how much of this difference in mortality (if we compare sporadic H5N1 outbreaks with the Spanish flu pandemic) is a consequence of differences in the virulence properties of the virus itself, and to what extent it may be explained as a consequence of other circumstances at the time when the Spanish flu pandemic started. The best method to obtain a direct answer to this question is to compare the age-specific mortality among registered H5N1 cases since 2003 who are less than 28 years old and the registered surplus mortality for the same age groups through a time interval corresponding to the first three waves of the Spanish flu pandemic, assuming that 90% or more of the world population had been infected after the third pandemic wave. Another method for obtaining answers (albeit far less precise) to these questions is to carry out mathematical modeling experiments, making counterfactual historical assumptions. What could have happened if the same virus had started a pandemic in a world population where nobody enjoyed protective adaptive immunity before the pandemic started? And what would have happened if the mutated virus in wave 2 (which was more infective and more lethal than the wave 1 virus) had come first?
Changes in immunity as a function of age
Before turning to the mathematical modeling experiments, it may be useful to make a partial (but far from exhaustive!) survey of factors that change as a function of age in a way that may affect the chance that a person will die during an infectious disease episode, e.g. with some highly pathogenic form of influenza. Even if it is not possible to translate this biological information directly into mathematical curves, it should hopefully make it easier to evaluate the biological plausibility and relevance of the mathematical approach followed here. It is not our intention to present the correct curve (showing what actually would have happened, had historical circumstances been different). Instead we want to calculate an area of uncertainty, defined by curves representing upper and lower boundaries for what can be considered biologically plausible. We will first, however, attempt to give a simplified overview (but hopefully adequate for our mathematical purpose) of how the immune system changes as a function of age from cradle to grave, and next how aging itself may affect the risk of dying from an infectious disease episode.
When a child is born, the adaptive part of its immune system may be considered a tabula rasa: the newborn child normally has no adaptive immunity of its own. However, the child is endowed with passive immune protection against a wide range of viruses and cellular pathogens because of passive transfer of immunoglobulins (IgGs) from its mother through the placenta Citation[11]. The IgGs have a fairly long half-life (about 3 weeks) compared with other types of immunoglobulins Citation[11], which may help to tide the child over the period it is presumably most vulnerable immediately after it has been born into a microbial world of which its own immune system has absolutely no experience Citation[11]. As soon as breastfeeding starts, the child will also receive immunoglobulins (with IgAs being especially important) through the mother's milk Citation[11–13]. Human milk contains several other factors that are also protective against a broad range of pathogens Citation[14], Citation[15], and it encourages growth of an intestinal flora with many bifidobacteria Citation[16], Citation[17] that are probably protective as well Citation[18], Citation[19].
These factors are not sufficient to confer total protection against either viral or bacterial pathogens, but they help to make the course of several of the infections concerned milder. In that way it is possible for the child's own immune system to develop resistance to several potentially dangerous new pathogens without fatal consequences. Immunological memory will be developed (that will make it possible in the future for the adaptive immune system to react very rapidly against the now new pathogen), but the child will not succumb to the infection.
When breastfeeding stops, it means that the child is deprived of the supply of immunoglobulins and other anti-infectious factors found in the human milk. Studies from hunter-gatherer societies indicate that it was normal for mothers to carry their youngest child on the back and breastfeed it very frequently until it was about 4 years old Citation[20]. The most important reason why this practice may have helped to enhance Darwinian fitness is probably that it helped to increase resistance against potentially dangerous infectious diseases, with malaria possibly being the most important one in much of Sub-Saharan Africa. The malaria mosquito is said to be attracted by the scent of faeces from breastfed infants Citation[21]. This might possibly represent an interesting example of co-evolution of parasite, parasite vector, and parasite host being mutually advantageous both to the parasite and to the human host – with the ‘intention’ (as seen from the host's perspective) being that the child will be infected as many times as possible at a time when the parasite is still non-lethal (because the child is protected against lethal infection by the combination of breastfeeding and an otherwise protective diet given when the child is sick). In that way, it may obtain immunological protection making the pathogen non-lethal also when it attacks during adult age.
The medical situation in Europe was not the same in 1918 as it is today. The mortality from several infectious diseases was much higher Citation[22], and this cannot be explained only as a consequence of the absence of effective antibacterial drugs. Poverty was more prevalent than now, which had important consequences for the nutritional status of part of the population, and the argument has been made that the rapid decrease of childhood infectious disease in Europe during the period 1860–1970 may in large measure be explained by the improvement in nutritional status for a large fraction of the population that took place during this period Citation[22].
If we compare the situation in developing countries, where poverty is far more prevalent even today, it is common that the mortality among infants and small children is much higher than for bigger children and young adults. One important reason for this must undoubtedly be the development of adaptive immunity. As a child grows older, it will encounter more and more new pathogens (including new strains of influenza virus), with each new encounter causing enlargement of the total number of pathogen species and strains that are remembered by the adaptive immune system. The chance of encountering some new pathogen that its adaptive immune system has not encountered before will therefore decrease as a function of age.
Once complementary feeding is introduced, infants and young children are more susceptible to malnutrition than older ones. This makes them more vulnerable if the diet is of poor quality, for example, regarding protein content, essential amino acids or important micronutrients such as zinc, iron or vitamin A. But the comparative lack of experience of the adaptive immune system in small children is also an important factor in making them more vulnerable to malnutrition, because it means that new infectious episodes will tend to occur more frequently in toddlers than in the older children – and each infectious episode will have an impact on the nutrition status of the host (because of enhanced degradation or excretion of several different nutrients when the child is sick) Citation[23], Citation[24].
This is usually not a serious problem in well-nourished populations – with a sufficient surplus in the ordinary diet of all the nutrients concerned to make it easy to compensate for the losses, e.g. within 1 or 2 weeks during the convalescence period. But it is completely different when the composition of the diet is marginal (e.g. regarding protein content, zinc, iron, folate, vitamin C or vitamin A) even for those who do not suffer from any infectious disease.
Finally, it should also be taken into consideration that several pathogens (e.g. rotavirus Citation[25] and malignant influenza virus Citation[26]) are much more lethal for malnourished hosts than for well-nourished ones. But this will be more important for infants and young children than for the older ones because they are more often malnourished.
The factors mentioned above do not completely explain the age distribution of infectious disease mortality among children in either the more highly developed or the underdeveloped world. But they are of great practical significance, since it should be a technologically, economically, and politically feasible task to overcome most of the poverty-related nutrition and health problems in the world if scientists, health personnel, and politicians alike share a scientifically correct understanding of their nature and often partly ecological and geochemical (and not purely social or economic) causation, being for instance related to factors such as deforestation, accelerated soil erosion, and too frequent anthropogenic fires.
The immune system is strongly influenced by hormones, with some hormones being mainly stimulatory and others (especially the glucocorticoids) being immunosuppressive. There are important changes in the secretion of several immunostimulatory hormones as a function of age. The thymus is important not only for production of T lymphocytes Citation[11], but also for the production of several peptide hormones with immunostimulatory effects Citation[27–29]; for example prothymosin alpha Citation[28] and the zinc-thymulin complex Citation[29]. The thymus is large in children, even if the density of T lymphocytes inside the thymus can be seen to start to decline when children are about 1 year old Citation[30]. At puberty, a more rapid process of thymus involution starts, where parenchymal tissue is lost and replaced with adipose tissue Citation[30]. The involution process is at first rapid and continues, albeit more slowly, among adults Citation[11], Citation[30]. The production of T cells in the thymus is much larger in children than in adults Citation[30], but the parenchymal tissue in the thymus is not completely lost even in old people Citation[30].
For other mammalian species, the concomitant decline in growth hormone and increase in sex steroid production with age has been thought to be responsible for the process of thymic involution that starts at puberty Citation[31], but experiments with gene-modified mice seem to indicate that there must also be other control mechanisms, since thymic involution progressed in a similar manner both in animals that had a genetic deficiency causing reduced production of growth hormone and in a hypogonadal strain that fails to produce thymocytotoxic sex steroids Citation[31]. The authors of this report concluded that their observations suggest that age-related changes in the endocrine system do not underlie thymic involution Citation[31]. However, it is difficult to explain the timing of the process in humans, especially when it starts, except by assuming that sex hormones must play an important role. And castration can slow down the rate of thymic involution Citation[30], Citation[32], even though in mice it does not influence the point in time where involution begins Citation[32]. It has, moreover, been reported that partial thymic regeneration could be induced in a 46-year-old human volunteer who was placed on a regimen of recombinant human growth hormone and pharmaceutical grade dehydroepiandrosterone for 1 month Citation[33].
The apparent conflict between different observations may possibly be resolved by an understanding of the more detailed mechanism of action of the thymocytotoxic sex steroids, and the very different rates of mitochondrial DNA aging, comparing humans and small rodents. Estrogen (at high levels) has been reported to induce thymic atrophy by eliminating early thymic progenitors and inhibiting proliferation of beta-selected thymocytes Citation[34]. In lizards it has also been reported that estradiol-17beta (E2) has an apoptotic effect involving caspase action on thymic cells Citation[34], but the apoptotic effect of E2 is effectively counteracted by progesterone Citation[35]. In rats it has been shown that E2-induced thymic atrophy increased the rates of apoptotic death, and enhanced the Fas/FasL mRNA levels in thymic cells Citation[36]. These findings suggest that Fas/FasL-mediated apoptosis is involved in the induction of thymic atrophy by E2 Citation[36].
But the expression of Fas ligand (FasL) is under positive control by the oxidatively regulated Citation[37] transcription factor NF-kappaB Citation[38], Citation[39]. FasL expression is also up-regulated by the transcription factor Sp-1 Citation[39]. Sp-1 (or Sp-1/AP-2 transcriptional factor complex) binding is enhanced by MAP kinases Citation[40–46] that can also be activated by oxidative stress Citation[45], Citation[47], Citation[48]. Sp-1-induced gene expression is therefore enhanced by oxidative stress Citation[45]. NF-kappaB activation has been shown to be enhanced as a function of age Citation[37], Citation[49], which may presumably be explained as being in large measure a consequence of enhanced production of reactive oxygen species (ROS) in the mitochondria because of mitochondrial DNA aging Citation[50]. Lifespan in mammals is strongly negatively correlated with the rate of ROS production in the mitochondria Citation[51], Citation[52], and also with the average number of double bonds per unsaturated fatty acid group in mitochondrial lipids Citation[52], Citation[53]. The rate of mitochondrial ROS production in short-lived species like mice and rats is therefore much larger than in long-lived humans. It might thus be hypothesized that enhancement of mitochondrial ROS production because of the aging process per se may be an important factor controlling the process of thymic involution (because it will increase the expression of FasL in thymic cells). But it may be possible that this factor is even more important in short-lived species such as mice and rats than in humans at the age of puberty – which could possibly mean that the balance between growth hormone and sex hormones may be the most important factor controlling the onset of thymic involution in humans at the age of puberty, while mitochondrial DNA aging could be the most important factor controlling the timing of thymic involution in rodents, even in young animals. However, it may be possible that it is a gradual enhancement of oxidative stress because of mitochondrial aging that is the most important factor in explaining why thymic involution continues to proceed in humans after the end of puberty.
There are also experimental observations suggesting that age-related changes in zinc metabolism, leading to diminished bioavailability of zinc in old mammals, may play an important role in the regulation of age-dependent thymic involution Citation[54]. We shall later return to the question of the possible mechanism(s) whereby aging may cause changes in zinc metabolism.
The tight physiological regulation of the process of thymic involution shows that it is something that should be considered part of the normal physiological aging process and not a consequence of pathological aging (as in populations where it is common that low-density lipoprotein (LDL) cholesterol and blood pressure go up while insulin sensitivity goes down as a function of age – all of which should be considered as consequences mainly of pathological aging). At the same time, there can be little doubt that the rate of apoptosis associated with thymic involution must be influenced by dietary deficiencies and other factors (including infectious diseases and toxic substances with pro-oxidant effects) that can enhance the NF-kappaB- and Sp-1-mediated induction of Fas ligand. Theoretically it may thus be expected that factors such as selenium deficiency, low intake of sulfur amino acids, high burdens of toxic heavy metals, tobacco, and too much alcohol will increase the rate of thymic involution. In 1918, it may thus be possible that thymic involution may have been more pronounced in poor people, as a direct consequence of poor diet, than in the affluent ones. It may be possible that this could be important not only for the risk of dying from the Spanish flu, but also for dying from other dangerous infections such as tuberculosis. Today, there must be strong reason to suspect that regular consumption of estrogen-containing contraceptive pills could do the same (as a direct consequence of the anti-thymic effect of estrogens), and also anabolic steroids.
Not only the production of T cells, but also the production of thymic hormones is larger in children compared with young adults Citation[55]. In humans, the age-related decline in serum ‘facteur thymique serique’ (FTS) levels (another name for zinc-thymulin complex) begins after 20 years of age and FTS completely disappears from the blood between the fifth and sixth decade of life Citation[55]. In contrast, the serum levels of thymosin-alpha 1 and thymopoietin seem to decline earlier, starting as early as 10 years of age Citation[55]. The decline in thymic hormone production, comparing young adults with children, may presumably be explained not only as a consequence of thymic involution, but also of the decline of growth hormone secretion, since growth hormone has a stimulatory effect on the thymus Citation[56–60]. Daily growth hormone secretion is high in children, reaches maximal levels at puberty, and then decreases in an age-related manner in adulthood Citation[61].
In quasi-teleological language (instead of using the more correct, but also more cumbersome language of the evolutionary biologist), it might be speculated that the main ‘purpose’ of this arrangement might be to offer some kind of compensation for the lack of immunological experience of children, especially the youngest ones. The thymic hormones help to stimulate both innate Citation[28], Citation[62], Citation[63] and adaptive Citation[63], Citation[64] components of the immune system, thus making it easier for the innate immune system to make effective resistance against the enemy until the ‘rescue forces’ can arrive (i.e. when an effective adaptive immune response can be mounted), while the combination of larger thymic hormone production and much larger thymic production of T cells in children will make it possible for the ‘rescue forces’ to arrive earlier and be strong and numerous when they arrive.
When the production of sex hormones declines in women at the menopause, and also in old men Citation[65], it may be possible that this will have some protective effect on the thymus, which in turn could mean better protection against infectious diseases. One might speculate that it could be part of the wisdom of nature that old people will not die from infectious disease too early, because it may be important for their grandchildren to make good use of their accumulated knowledge, experience and wisdom – but they should not be able to reproduce (because the number of mutations increases in both the nuclear DNA and mitochondrial DNA of the germ cells as men and women grow older). A decline of sex hormone production in old age will help to accomplish both of these objectives simultaneously.
The age-specific mortality during the Spanish flu seems to have reached a minimum at an age just before the onset of puberty Citation[5]. It then rises steeply until a sharp maximum is reached at about the age of 30 (with a time resolution, however, not better than 5 year age intervals) Citation[5]. It was suggested earlier that this steep rise of mortality as a function of age from about the onset of puberty into young adult age (about 30 years) can be explained mainly as a consequence of the decline of thymic hormone secretion taking place within this age interval (from about 11–12 to about 30 years) Citation[26], Citation[66]. A closer consideration of what happens with the thymus during puberty and later and what is known about factors regulating thymic involution does not weaken the plausibility of this suggestion.
An important theoretical question as regards the effect of sex hormones, including testosterone Citation[55], on thymic involution is whether a constantly high level of sex hormones in young adults can directly explain why thymic involution continues to progress even after the end of puberty, or if the sex hormones only will down-regulate the mass of thymus parenchyma down to some plateau level, where their proapoptotic effect will be balanced by reduced production of another, even more important type of negative regulators, i.e. some form of thymus-specific chalone(s) Citation[67]. This question is of importance if we try to extrapolate the curve for age-specific mortality as a function of age beyond 30 years under the hypothetical (and counterfactual) assumption that nobody older than 30 years old was immune when the Spanish flu pandemic started. If the chalone effect is important enough, it may be possible that the real curve (which would be observed historically if nobody had been immune when the pandemic started) must be expected to have some form of knee, so that it will rise less steeply through parts of the interval between 30 and 50–55 years, whereupon it begins to rise more steeply again (and perhaps look more or less like an exponential curve for the higher age groups until it is cut off because almost everybody of higher age is dead). Without a pronounced chalone effect, it may be possible that there will not be any such knee on the curve, or it will at least not be equally pronounced.
The thymus is influenced not only by growth hormone and sex hormones, but also by several other extra-thymic hormones, including glucocorticoids Citation[55], dehydroepiandrosterone (DHEA) Citation[68] and its metabolites androstenedione Citation[69] and dehydroepiandrosterone sulfate (DHEA-S) Citation[70], thyroid hormones Citation[55], and melatonin Citation[71–76], which it can also produce itself Citation[77]. Glucocorticoids, like the sex steroids, contribute to thymic involution Citation[55], and DHEA has also been reported to have an apoptotic effect in thymic cells Citation[68], while its metabolites androstenedione and DHEA-S have been reported to have opposite effects, with DHEA-S being antiapoptotic Citation[70] and androstenedione stimulating thymocyte growth Citation[69]. The effect of thyroid hormone is biphasic with low doses being stimulatory Citation[55], while too high doses may cause thymic atrophy Citation[55]. Melatonin stimulates the secretion of thymic hormones Citation[71] and counteracts the apoptotic effects of glucocorticoids in thymic cells Citation[72–74], Citation[76].
The thymus is also stimulated by prolactin Citation[58], Citation[78], which means that the immune system of women of fertile age (notably when they are lactating) is probably better than that of men of the same age. From the point of evolutionary biology, it is not difficult to understand that this may make good sense. A small and vulnerable child is more strongly dependent on its mother than on its father for survival, which means it may be more important (in terms of Darwinian fitness for the whole population) to protect the mother against dying from some infectious disease than it is to protect the father. The mother might also need some form of compensation for the immunological handicap she might suffer as a consequence of nutrient losses associated with pregnancy and lactation.
It might be speculated that the involution of the thymus starting at puberty may be one of the main reasons why several infectious diseases appear to be less virulent and less lethal when affecting children than they do when they affect populations of adult people who have not been exposed to the same infectious agent before Citation[79]. This is something that has often been seen when populations that formerly had been completely isolated in relation to several bacterial and viral diseases common in the Old World have come into contact with Europeans and their diseases for the first time. The most dramatic example is the fate of the Amerindian populations in America following the discovery of America by Columbus Citation[79]. Recent estimates, based on sampling of tribute lists, missionary reports, and elaborate statistical arguments, have put the total Amerindian population on the eve of the conquest at about 100 million people, with 25 to 30 million of this total assignable to the Mexican civilizations and an approximately equal number to the Andean civilizations Citation[79]. Starting from such levels, population decay following the arrival of the Europeans was catastrophic Citation[79]. By 1568, the populations of central Mexico had shrunk to about three million, i.e. to about one-tenth of what had been there when Cortez landed Citation[79]. Decay continued, although at a reduced rate, for another 50 years, with the population reaching a low point of about 1.6 million by 1620 Citation[79]. Recovery remained very slow until the eighteenth century Citation[79]. The population decline was caused by a succession of epidemics with different infectious diseases, such as smallpox, measles, maybe typhus, and what was most likely a virulent form of influenza, perhaps worse than the Spanish flu pandemic, during the middle part of the sixteenth century Citation[79].
While some of the diseases concerned, such as measles, can be highly lethal for malnourished children, although not when affecting well-nourished children, there is little reason to believe that the high mortality among adult Amerindians can be explained mainly as a consequence of malnutrition. From what is known about the foods they were using before the arrival of the Europeans – including (for some of the American territories, but not everywhere) various protein-rich legume and pseudocereal crops with an essential amino acid composition complementary to maize, such as Amaranthus (various species that were used both in Central America and in the Andes region), quinoa (in the Andes region), Lupinus mutabilis (in the Andes region), various Phaseolus species (including semidesert-adapted ones in arid parts of North America), acorn flour treated with plant ashes (so as to neutralize the antinutritional effect of the tannin content of the acorn), plus (depending on the environment) protein from fish, game, and invertebrates – it is reasonable to speculate that the composition of their diets mostly must have been fairly good with regard to total protein, essential amino acids, and most of the micronutrients, with the probable exception of iodine in some areas and perhaps also selenium in parts of the territories concerned. Thymic involution may therefore offer a more plausible explanation for the high mortality in the adult Amerindian population when encountering several new infectious diseases in rapid succession, rather than malnutrition being a consequence of poverty and poor diets.
Melatonin is another important immunomodulating but mainly immunostimulating hormone. It acts at different levels in relation to the immune system with one important effect, as already mentioned, being its stimulatory and antiapoptotic effects in the thymus Citation[71–76]. It also counteracts apoptosis in bone marrow cells (most likely by an indirect mechanism of action) Citation[80], and stimulates their proliferation Citation[81], Citation[82]. Melatonin stimulates the production of progenitor cells for granulocytes-macrophages Citation[83]. It also stimulates the production of natural killer (NK) cells and CD4+ cells and inhibits CD8+ cells Citation[83]. The production and release of various cytokines from NK cells and T-helper lymphocytes are enhanced by melatonin Citation[82]. At the same time, melatonin also helps to improve the antioxidative defense system of the cells, not only because it functions as a good antioxidant itself Citation[84], Citation[85], but also – and possibly even more important – by enhancing the expression of several important antioxidative enzymes such as superoxide dismutase, glutathione peroxidase, glutathione reductase, and catalase Citation[86–88].
The pineal gland (where much of the body's melatonin production takes place) does not undergo early atrophy as does the thymus. But melatonin is normally produced only when we sleep and it is dark. It is practically not produced when we are awake and flooded by light, natural or artificial. However, a baby normally sleeps much more than older children, and it is also normal that older children and adolescents sleep more than old people. It may thus be possible that the cumulative melatonin production per 24 hours will go down monotonically as a function of age, partly because the number of hours with sleep per night (or night plus day) tends to go down as a function of age and not only as a consequence of changes of the rate of melatonin production when we sleep. It is possible that this question has still not been studied in such detail as it probably deserves. How much melatonin is produced in babies when they are sleeping during the day-time when it is light? It has been observed, however, that melatonin production and circulating melatonin decrease with age Citation[83], Citation[89–91], which would appear to indicate that a decline in the rate of melatonin production when we sleep must also be an important factor. Melatonin production in the pineal gland declines progressively with age, such that in old animals and elderly humans the levels of melatonin available to the organism are only a fraction of that of young individuals Citation[90].
Melatonin synthesis proceeds via serotonin as an important intermediate Citation[61]. But the rate-controlling enzyme in the pathway of serotonin biosynthesis, i.e. tryptophan hydroxylase Citation[61], is inhibited by oxidative and nitrosative stress Citation[92–97]. It is therefore possible that melatonin production might go down with increasing age in part because enhanced mitochondrial ROS production in the pineal gland (as a consequence of mitochondrial DNA aging) could lead to progressive enhancement of oxidative inhibition of tryptophan hydroxylase in the pineal gland. Other mechanisms may, however, also play a role Citation[91]. It has been suggested that the age-related decline of pineal melatonin production is due to the degenerative changes of the neural structures (serotonergic and noradrenergic neuron systems) innervating the pineal gland and the suprachiasmatic nuclei rather than to the degeneration of the pineal tissue itself Citation[91]. Oxidative inhibition of tryptophan hydroxylase is not the same, however, as tissue degeneration.
DHEA and its active metabolites, such as DHEA-S, 5-androstenediol (AED), and androstenetriol (AET), comprise another group of hormones with important immunostimulatory and immunomodulatory (including antiglucocorticoid) effects Citation[26], Citation[98–106] that change with age. The pattern of secretion as a function of age is quite different for DHEA compared with the thymic hormones, with DHEA production normally reaching a maximum in the third decade of life (between 20 and 30 years) and then declining Citation[61]. Why the decline of DHEA production after an age of 30 years happens – with further decline as people are approaching old age Citation[107] – is not very easy to understand. As regards evolutionary explanations, it might be speculated that it happens not because it is advantageous as seen from the perspective of the individual, but because it might be advantageous both at a population level for the species concerned and at an ecosystem level – that it might be part of ecological regulatory mechanisms ensuring that we shall not survive too long (comparable, perhaps, to some extent to such forms of anti-virulence evolutionary adaptation that we can encounter among various pathogens including benign forms of influenza type A virus). It might be speculated that there may be a similar reason explaining why the thymus undergoes progressive involution from the onset of puberty until we reach old age: it is not in the long-term evolutionary interest of our species that we destroy our own habitat more than it is in the long-term evolutionary interest of a parasite that it causes the extinction of its own host.
The relevance of age-dependent changes in DHEA production as a possible contributory cause of age-dependent changes in the lethality of highly pathogenic influenza viruses (Spanish flu and H5N1) is illustrated by the results of animal experiments demonstrating the protective effect of DHEA and its metabolite AED against various highly lethal viruses, including influenza virus that was inoculated at a lethal dose Citation[108–115].
Consequences of mitochondrial DNA aging
Mitochondrial DNA aging should probably be regarded as the most universal and most fundamental of all aging mechanisms among multicellular animals. When mutations in mitochondrial DNA occur, this means that there will be a corresponding reduction in the number of ‘healthy’ mitochondrial DNA genes in relation to the total mass of mitochondrial DNA per cell. It may in principle be possible to compensate for this loss by enhancing the total number of mitochondrial chromosomes per cell (e.g. by making more mitochondria). But such compensation will not be possible indefinitely, as there is a limit to how many mitochondria and how much mitochondrial DNA there is room for per cell. Sooner or later, the number of mitochondrial DNA genes per cell will therefore start to go down, and this process will proceed monotonically (in the mathematical sense of this word) as a function of time until the individual dies.
Progressive reduction of the number of mitochondrial genes per cell means that, sooner or later, there will also be reduction of mitochondrial RNA production – which presumably will happen with mitochondrial ribosomal RNA (rRNA), mitochondrial tRNAs, and mitochondrial mRNAs. This reduction of mitochondrial RNA production means that there will also eventually be a reduction of the expression of mitochondrially encoded proteins. The curve for mitochondrially encoded protein production as a function of the average number of ‘healthy’ mitochondrial genes per cell will most likely not be a linear one, since the rate of protein synthesis inside the mitochondria will probably depend not only on the amount of mRNA as a rate-limiting factor, but also on the number of ribosomes per mitochondrion (which must depend on the supply of mitochondrially encoded ribosomal RNA) and the supply of aminoacyl-tRNA molecules (which must depend both on the rate of amino acid supply and on the rate of synthesis of mitochondrially encoded tRNA molecules). This means that aging per se and a low daily intake of protein must be expected to interact synergistically with each other as causes of reduced synthesis of mitochondrially encoded proteins.
Most of the mitochondrially encoded proteins function as necessary parts of protein complexes in the respiratory chain, with the greatest number (seven mitochondrially encoded subunits) found in complex I Citation[116]. Reduction of the rate of synthesis of these proteins means that there will be corresponding bottlenecks that impede the flow of electrons through the respiratory chain. This has two important consequences: it leads to reduction in the capacity for mitochondrial ATP production Citation[117], and it leads to enhancement of the rate of superoxide anion radical production by leakage of electrons from the respiratory chain before they have been consumed in the cytochrome c oxidase reaction for making water molecules plus ATP Citation[50], Citation[117].
However, enhancing the rate of superoxide anion radical production means that the rate of attack on mitochondrial DNA by ROS Citation[118–121] will be enhanced, which in turn will lead to more rapid accumulation of mutations in mitochondrial DNA. Mitochondrial DNA aging is therefore a self-accelerating process, and cannot be expected to proceed linearly as a function of the chronological age of an individual. It will proceed faster and faster when we are approaching the maximum normal lifespan of our species. Since the rate of mitochondrially encoded protein synthesis will also be non-linear when considered as a function of the number of ‘healthy’ mitochondrial genes, we must expect an even steeper decline of mitochondrial protein production as a function of chronological age when we become very old. This curve will be even more strongly non-linear than the curve for the number of ‘healthy’ mitochondrial genes per cell as a function of biological age.
There is more than one reason why mitochondrial DNA aging may have important consequences for an individual's chance of survival during an infectious episode, e.g. influenza. First it must be mentioned that leukocytes and their progenitor cells are subject to mitochondrial aging like all other nucleated cells in our body. The immune system will therefore ultimately become ‘worn out’ as a direct consequence of senescence. It may be possible that the mitochondrial DNA aging process will not always affect all types of leukocytes (or their progenitor cells) at the same rate. One might, for instance, speculate that CD4+ T cells under some circumstances might be more strongly affected (being more vulnerable to mitochondrial DNA damage) than most other types of leukocytes. It could also be speculated that this possibly might be the most fundamental reason for the long-term decline in the number of CD4+ cells that is observed in HIV patients Citation[122] – which, if correct, means that it should be possible to reduce the rate of progression of this disease when patients are in the asymptomatic stage by therapy specifically aimed at protecting the mitochondrial DNA of the CD4+ cells as well as possible. For instance, this might be attempted by ensuring a good supply of all nutrients that are important for scavenging superoxide anion radical and H2O2 in the mitochondria, by supplementation with carnitine to counteract acid sphingomyelinase activation in the CD4+ cells Citation[123], Citation[124], because sphingomyelinase activation will next lead to enhanced mitochondrial ROS production Citation[125], and also by ensuring adequate supply of all nutrients that are important for DNA repair (including, for instance zinc, sulfur amino acids, selenium, folate, vitamin B12, and niacin).
But mitochondrial DNA aging is also important because of the way certain cytokines including tumor necrosis factor (TNF)-alpha affect mitochondrial function. These cytokines can activate sphingomyelinases in their target cells Citation[125–129]. This will first lead to cleavage of sphingomyelin into ceramide and phosphocholine, which may be followed by the metabolism of ceramide to form other compounds (including sphingosine) that may function as intracellular messengers in their own right. Ceramide goes into mitochondria, where it causes partial block of the flow of electrons through the respiratory chain Citation[127], Citation[128], Citation[130]. The consequences will be the same as for mitochondrial DNA aging: reduction of the capacity for mitochondrial ATP production and enhancement of the rate of mitochondrial ROS production. It is more likely, however, that TNF-alpha and mitochondrial DNA aging will interact with each other synergistically (in a multiplicative fashion) rather than additively.
TNF-alpha, moreover, can cause a redistribution of ganglioside GD3 so that it goes from the plasma membrane into the mitochondria Citation[131]. This process is mimicked by acid sphingomyelinase and ionizing radiation, but not by neutral sphingomyelinase Citation[131]. Similarly to ceramide, this also causes enhancement of mitochondrial ROS generation Citation[132].
There is a limit as to how much mitochondrial ATP production can be depressed or mitochondrial ROS production can be enhanced in some organ vital for survival before a person will die. With a multiplicative interaction between impairment of mitochondrial protein synthesis happening as a consequence of mitochondrial DNA aging and impairment of electron flow through the respiratory chain as a consequence of cytokine action, it follows that a young person should be expected to tolerate much higher levels of cytokines such as TNF-alpha before death will occur as a direct consequence of mitochondrial dysfunction, compared with a very old person. A child will have much more mitochondrial ‘reserve capacity’ compared with a person who is 80 or 90 years old. However, even if this is fairly simple to understand in a more qualitative sense, we are not in the position that we have as good quantitative information about these processes as may be needed to make an accurate and realistic mathematical model describing how it happens.
Enhancement of the production of ROS in the mitochondria as a consequence of mitochondrial aging will also lead to more activation of various transcription factors (such as NF-kappaB) that affect the expression of a large number of genes and are themselves redox-regulated. This is the case with the genes for TNF-alpha Citation[133–136] and interleukin (IL)-6 Citation[137], Citation[138], both of which are major contributors to the so-called cytokine storm that appears to be very central in the pathogenesis of fatal Spanish flu and H5N1 influenza Citation[26].
Some of the oxidatively up-regulated genes concerned code for gene products that are themselves directly or indirectly immunosuppressive. This is the case with the COX-2 gene, which is positively regulated by the oxidatively activated transcription factor NF-kappaB Citation[139–142], and also the gene for TGF-beta, that is induced by the lipid peroxidation product 4-hydroxynonenal Citation[143–145]. TGF-beta has important immunosuppressive effects in its own right Citation[146–148], while enhanced expression of COX-2 will lead to enhanced production of prostaglandin E2, which is also mainly immunosuppressive Citation[149–151]. The cyclooxygenases (COX-1 and COX-2) must be oxidized for activation Citation[152–156], and they can use H2O2 Citation[157–160], organic hydroperoxides Citation[161–165], or peroxynitrite Citation[166–170] as activators. Age-related enhancement of mitochondrial ROS production must therefore be expected also to lead to more activation of the enzyme itself. When the cyclooxygenases are activated, a slower process of suicidal inactivation will also start Citation[161], Citation[171–174]; therefore on average each enzyme molecule can only make a limited number of prostaglandin molecules before the enzyme molecule itself has become irreversibly inactivated. But more rapid suicidal inactivation of COX-2 because of enhanced oxidative stress is compensated for by enhanced expression of the enzyme, also as a consequence of oxidative stress. Phospholipase A2, which liberates prostaglandin precursor fatty acids from membrane phospholipids, is also activated by oxidative stress Citation[175–180]. Thus there are a number of different mechanisms that all act in the same direction, so that enhanced oxidative stress (e.g. because of mitochondrial DNA aging) will lead to enhancement of the production of prostaglandins.
In connection with influenza, it may be important that NF-kappaB has been reported to be required for influenza virus replication Citation[181], Citation[182], in spite of influenza virus being an RNA virus, while NF-kappaB is a transcription factor normally acting on DNA. It is therefore reasonable to assume that enhancement of oxidative activation of NF-kappaB that occurs in aged individuals Citation[37], Citation[49] as a consequence of mitochondrial DNA aging could lead to enhancement of the rate of influenza virus replication, making the virus more dangerous for old people (even such influenza virus strains that are normally considered benign, when they infect young people).
Mitochondrial DNA aging will also affect nerve cells, including peripheral unmyelinated nerve fibers (C-fibers). One possible consequence could be an enhanced tendency for oxidative activation of protein kinase C isozymes alpha Citation[183], Citation[184], gamma Citation[185], Citation[186], and epsilon Citation[187] in the C-fibers, which will in turn lead to up-regulation of the sensibility of these nerves to stimulation via the vanilloid receptors and quite possibly also other forms of stimulation Citation[188–195]. This may in turn lead to enhancement of neurogenic inflammation (because of secretion of tachykinins and calcitonin gene-related peptide from the C-fibers), which may be an important part of the mechanisms leading to development of alveolar edema in experimental animals and human patients suffering from malignant forms of influenza Citation[26], Citation[196–213]. The C-fibers are also sensitized by prostaglandins and other eicosanoids (including PGE2, thromboxane A2, and leukotriene B4) Citation[214–224]. A tendency for enhanced COX-2 expression as a consequence of oxidative stress associated with mitochondrial aging may therefore have effects going in the same direction – with a tendency for strengthening of neurogenic inflammation as a function of age.
Mitochondrial DNA aging must be expected to lead to a general decline in the ATP production capacity of mitochondria, even though in some organs, such as skeletal muscle and the heart, it may be possible to compensate for this (at least when the process of mitochondrial DNA aging has not progressed too far) through physical exercise at some suitable level of intensity. When the ATP production capacity of the mitochondria goes down, this will commonly be compensated for by more glycolysis. It has been found that net acid-producing diets typical of many contemporary Western societies produce a low-grade systemic metabolic acidosis in otherwise healthy adult subjects, and that the degree of acidosis increases with age Citation[225]. It has also been suggested that the enhanced tendency for metabolic acidosis found among elderly people may be related to an age-related decline in renal functional capacity Citation[225]. However, it appears equally plausible to explain the tendency for metabolic acidosis associated with aging as a direct consequence of mitochondrial DNA aging, rather than as a consequence of age-related decline of renal function. It can be counteracted by increasing the intake of alkaline-ash foods such as potatoes, vegetables, fruits, and berries Citation[225], Citation[226].
It has been reported that low-grade metabolic acidosis (which may occur either as a consequence of an imbalanced diet or because of aging) may cause reduction of the secretion of growth hormone Citation[225] and enhanced secretion of glucocorticoids Citation[227]. Animal experiments have shown that the rate of ACTH secretion is in part controlled (via a reflex arc going through the central nervous system) by the level of C-fiber activity Citation[228–231]. It is therefore possible that acidosis may lead to enhanced secretion of glucocorticoids because it leads to enhanced stimulation of the C-fibers via vanilloid receptors Citation[232–236], which will in turn lead to enhanced secretion of ACTH. As a consequence of this mechanism, it must be expected that there will be a common tendency for the balance between glucocorticoid and antiglucocorticoid hormones to deteriorate with age, not only because DHEA production goes down after the third decade of life Citation[61], but also because there is a tendency for enhancement of glucocorticoid secretion. Painful conditions that are associated with C-fiber activation must be expected to lead to even more stimulation of ACTH secretion and therefore more immunosuppression, and painful diseases are also more common among old people than among young adults. Adipose tissue is an important source of blood lactic acid Citation[237–240]; it is therefore reasonable to believe that overweight may also be one of those factors that may contribute to immunosuppression because it may contribute to metabolic acidosis (when there is a surplus of acid-ash foods over alkaline-ash foods in the diet). This in turn would lead to enhanced vanilloid receptor activation in the C-fibers, followed by enhanced ACTH secretion in the hypophysis and enhanced glucocorticoid secretion in the adrenals.
However, patients suffering from hypervirulent influenza may also be affected by severe disturbance of pulmonary function Citation[10], Citation[26], Citation[241], especially when they develop alveolar edema Citation[26], Citation[241], which must be expected to lead to respiratory acidosis. In this case there will be a synergistic interaction between respiratory acidosis (reduction of blood plasma pH because the CO2 partial pressure in the blood is elevated Citation[226]) caused by severe influenza and metabolic acidosis (reduction of blood plasma pH because the HCO3– concentration in blood plasma is reduced Citation[226]) caused by mitochondrial DNA aging, diet, or overweight. It is a condition that can easily be treated in a modern hospital, and to some extent even in a poor village situation by recommending for the patients a diet rich in foods (if available in sufficient quantity) that are rich in alkaline-ash, but low in carbohydrates. But if left untreated, which must have been the case for practically all those patients who had a surplus of acid-ash foods in their diet during the Spanish flu pandemic, there can be little doubt that the synergistic interaction between influenza-induced respiratory acidosis and age plus diet-induced metabolic acidosis (which in combination must be expected to have led to strong enhancement of C-fiber activity not only in the lungs, but also in other organs such as skeletal muscle, which would in turn have led to strong reflex activation of the hypophysis and much ACTH secretion) must have been an important mechanism that would have led to enhanced mortality in elderly patients during the Spanish flu pandemic, had they not been so well protected by immunity from influenza epidemics before 1890.
This discussion is not meant to give an exhaustive overview of all known mechanisms whereby mitochondrial aging may affect the death risk associated with a certain type of infection. But one more possible mechanism deserves mention because it may help to explain one of those mechanisms by which immunostimulation from thymic hormones decreases in old people. One of these hormones, namely thymulin, is active only in the form of a zinc complex Citation[242–247] at the same time as its synthesis in the thymus also depends on zinc status Citation[248]. The activity of this hormone can be reduced because of dietary deficiency of zinc in experimental animals as well as humans Citation[249–253], but also because of diseases causing disturbance of zinc metabolism or disturbing thymus function and/or the activity of thymic hormones because of other mechanisms, such as cancer and leukemia Citation[244], Citation[254–257], cystic fibrosis Citation[258], Down's syndrome Citation[242], Citation[259–262], Crohn's disease Citation[263], Citation[264], HIV disease Citation[265], Citation[266], diabetes type 1 Citation[267], and chronic renal failure Citation[268].
But undersaturation of the same hormone with zinc is also commonly observed in aging experimental animals Citation[245], Citation[246], Citation[248], Citation[269], Citation[270] as well as in old people Citation[242], Citation[245], Citation[246], Citation[271], Citation[272], and it is reasonable to speculate that this does not happen only as a consequence of poor diet among some of the geriatric patients, but also as a consequence of the aging process itself leading to changes in the metabolism of zinc. Experimental evidence and observations in humans suggest that the disturbance of zinc metabolism observed in aging animals as well as in older humans may at least in part be explained as a consequence of enhanced zinc sequestration by metallothionein Citation[273–286]. The age-related disturbances in zinc metabolism and decline of the biologically active zinc-thymulin complex may at least partly be explained as a consequence of age-related changes in the levels of several hormones and at least one cytokine. There is evidence suggesting that age-related changes in the secretion of growth hormone Citation[243], Citation[247], Citation[287–289], prolactin Citation[58], Citation[78], Citation[289], somatomedin C Citation[270], melatonin Citation[247], Citation[257], Citation[290–294], glucocorticoids Citation[257], Citation[277–279] and IL-6 Citation[270], Citation[278–282], Citation[284] all may be of importance here, with growth hormone Citation[243], Citation[287], Citation[288], prolactin Citation[58], Citation[78], Citation[289], and melatonin Citation[247], Citation[257], Citation[290–294] being protective, while glucocorticoids Citation[257], Citation[277–279] and IL-6 Citation[270], Citation[278–282], Citation[284] function as negative regulators that may contribute to enhanced sequestration of zinc and impairment of thymic function in old experimental animals and humans.
A plausible hypothesis would be enhanced oxidative stress (because of mitochondrial DNA aging) leading by direct and indirect mechanisms to enhanced expression of zinc-sequestering proteins with antioxidant and sometimes also zinc storage functions, notably metallothionein and sensitive to apoptosis gene protein (SAG). It is also possible that alpha-2 macroglobulin may be of some importance for zinc sequestration in old age Citation[286]. Metallothionein can be induced by oxidative stress Citation[295–300], but also by IL-6 Citation[270], Citation[301–304]. The expression of IL-6 is enhanced, however, by the transcription factor NF-kappaB Citation[137], Citation[138], which is activated by oxidative stress Citation[37], Citation[305], Citation[306]. Metallothionein has important antioxidative effects Citation[300], Citation[307–310] in addition to being an important metal storage protein.
Sensitive to apoptosis gene protein (SAG) is a zinc-binding RING (really interesting new gene) finger protein Citation[311]. RING finger proteins containing a characteristic C3HC4 or C3H2C3 motif appear to act as E3 ubiquitin ligase and play important roles in many processes, including cell cycle progression, oncogenesis, signal transduction, and development Citation[311]. SAG and various related proteins are expressed in multiple mouse adult tissues, as well as early embryos Citation[311]. In humans, both SAG and the related protein ROC1 are ubiquitously expressed at a very high level in heart, skeletal muscle, and testis Citation[311]. Expression of both SAG and ROC1 is induced by mitogenic stimulation Citation[311]. SAG is also induced by a redox agent in cultured cells Citation[311], as well as in in vivo mouse brain upon ischemia/reperfusion Citation[311]. SAG binds to RNA, has metal ion-binding/free radical scavenging activity, and is redox-sensitive Citation[311]. In mammalian cells, SAG protects against apoptosis mainly through inhibition of cytochrome c release/caspase activation Citation[311].
SAG should probably not be regarded as a zinc storage protein. But it nevertheless binds zinc, at the same time as the expression of this antioxidant protein is enhanced by oxidative stress. It should also be noted that SAG is expressed at a high level in some organs, such as the heart, where metallothioneins are not very strongly expressed. Enhanced expression of these proteins, but especially metallothionein, must be expected to have adverse effects on the availability of zinc for incorporation into zinc-dependent enzymes and transcription factors. If metallothionein expression is enhanced in intestinal epithelial cells, it will lead to reduction of the net rate of zinc absorption in the intestine – because zinc that is bound to metallothionein in the enterocytes probably will tend to stay there until the cells are sloughed off and die. Zinc supplementation leads to up-regulation of the expression of metallothionein in the enterocytes at the same time as the expression of various other zinc-binding proteins (zinc transporter proteins) goes down Citation[312]. If metallothionein expression is up-regulated in the liver, it will make it more difficult for zinc to be mobilized from the liver for transport to other parts of the body, while up-regulation of the expression of zinc-binding proteins such as metallothionein or SAG in organs that receive zinc from the blood (such as the brain and the heart) will reduce the local availability of zinc that is imported to these organs (for incorporation into zinc-dependent enzymes and transcription factors in these organs).
It may thus be possible that a tendency for over-expression of metallothionein in the liver (and perhaps in the intestinal epithelium as well) as a direct consequence of the oxidative stress associated with old age might help to explain the tendency for undersaturation of thymulin with zinc that is often observed in older people. Again, it may be possible that we are dealing with some mechanism that might be considered harmful from the perspective of an individual approaching old age, but useful at a population level and also at the ecosystem level. It is not part of the economy of nature that we should become too old.
It is important to recognize that metallothioneins not only bind zinc, but also several other metals with a preference for coordination to sulfur atoms rather than oxygen – i.e. such metals that were classified as chalcophile (going into a copper-rich sulfide melt) by the Norwegian geochemist V.M. Goldschmidt Citation[313], Citation[314]. Among such metals that are strongly bound to metallothionein, we find not only toxic metals such as cadmium and mercury Citation[315], but also the essential trace element copper Citation[316]. CuS has a much lower solubility product than ZnS Citation[317], which tells us that cupric ions are bound to sulfur atoms (in the form of sulfide ions or thiol groups) even more strongly than zinc. Copper is therefore bound to metallothionein even more strongly than zinc.
It must be theoretically expected that an age-related enhancement of the expression of metallothioneins will not only affect zinc metabolism, but copper metabolism as well, and it may be possible that it will affect the availability of copper for incorporation into copper-dependent enzymes even more strongly than it affects the availability of zinc. It is also important to recognize that copper and zinc interact antagonistically with each other at the level of intestinal absorbtion Citation[318]; supplementing only one of these elements at a high dosage level may therefore lead to deterioration of deficiency of the other. Copper is important for immunological functions Citation[319], Citation[320]; it must therefore be expected that copper deficiency occurring as a direct consequence of the aging process (because of enhanced metallothionein expression) must lead to immunosuppression, similar to zinc deficiency. But copper deficiency has also been reported to have proinflammatory effects in relation to neutrophils and lung endothelial cells Citation[321], which must be expected to contribute to aggravation of all forms of influenza, but especially the hypervirulent ones such as Spanish flu and hypervirulent H5N1.
Copper deficiency must also be expected to lead to enhanced death risk from cardiovascular diseases. One reason for this is the function of copper as a necessary cofactor in the enzyme lysine oxidase, which is needed for normal cross-linking of elastin Citation[318], Citation[322], and copper deficiency has also been reported to lead to augmentation of metalloproteinase activity Citation[323]. Copper deficiency can therefore lead to deterioration of the tensile strength properties of blood vessel walls Citation[318], Citation[322], which in turn will lead to enhancement of the risk of death caused by fatal hemorrhage (e.g. from a ruptured aneurysm in the brain or in the aorta). Copper deficiency is, moreover, very dangerous for the heart, as it can lead to arrhythmias and most likely also myocardial infarction Citation[322]. It may thus be possible that aging-induced copper deficiency could be an important cause (or contributory cause) of malignant arrhythmia – which nevertheless should be considered one of the least painful and perhaps also one of the most natural ways to die when a person becomes very old without suffering from any form of pathological aging.
It should be emphasized that it must still be regarded more or less as an open question, how early mitochondrial DNA aging may start to affect lethality from infectious diseases. Is it something that might play a role as part of the explanation for the steep rise in death risk comparing 12-year-old and 28-year-old males during the Spanish flu pandemic? Or is this a factor coming into play only when people are more than 50 years old, or more than 60 years old? The best answer to these questions, for the time being, is probably that we still do not really know.
It may be concluded that there are several different mechanisms explaining why the elderly are more vulnerable to infectious diseases compared with young adults. We do not have enough quantitative knowledge about these processes to allow us to translate qualitative biological knowledge into mathematical curves. From what is known in a more qualitative sense, it must nevertheless be expected that many of these mechanisms will interact with each other in a synergistic fashion, the net effect of which will be something that might be expected to resemble an exponential curve for the lethality of several types of infection as a function of chronological age in the adult part of the population.
Another important conclusion that can be made is that immunosenescence appears to be a strongly physiologically regulated process. It is not only a question of age-related wear and tear affecting the immune system directly. But those changes that occur as a direct and inevitable consequence of aging per se appear to be strongly amplified by age-related changes in the secretion of various hormones with regulatory functions in relation to the immune system, by age-related changes in the production of prostaglandins and various cytokines, by age-related metabolic acidosis (if the diet does not contain an adequate surplus of alkaline-ash foods) and by age-related changes in the metabolism of zinc.
One gains the impression that we may be constructed – when living in the type of natural environment for which our ancestors had become adapted by evolutionary processes over a period of several million years before the agricultural revolution – to maintain a reasonably good functional overall capacity, physically and mentally, until we approach the limit of the natural lifespan of our species and some ‘point of no return’ beyond which degradation of immunological functions will take place at a very rapid and accelerating pace. It may also be concluded that dying from an infectious disease episode may be a very natural way of dying for our species. Dying from diseases related to pathological (or, by definition, unnatural) forms of aging is probably not.
It might be added that if societies and individuals could become much more clever than they are today in preventing all forms of pathological aging, this will still not be sufficient to eliminate natural aging and what appears to be one of the most important natural mechanisms of limiting the life expectancy of our species, i.e. the enhanced vulnerability to infectious disease when we become very old. It would not prevent us from dying when we become old enough. But it would help to save many of us from those debilitating diseases that should be considered consequences of pathological rather than of physiological aging – including diabetes type 2, osteoporosis, debilitating cerebrovascular disease, coronary heart disease, cancer, and Alzheimer's disease. For many of us as individuals, this would mean a much better quality of life when we get old. For society as a whole, it could mean enormous economic savings that could make it easier to solve some of those serious ecological challenges that are all too evident in the contemporary world.
The mathematical model
Let N(τ) be the number of individuals of age below or equal to τ. The number of individuals in a population can then be written as:Eq 1 where ρ is called the age density, i.e. ρCitation[50]Δτ, Δτ=1 becomes approximately equal to the number of individuals in the age of 50 years. We next define the following quantity
Eq 2 which we call the normalized age density. It follows directly that
. Thus
is the fraction of individuals in each age category. We will use data from the statistical central bureaus for this quantity in 1918.
Further define M(τ) as the total number of individuals that were killed due to the Spanish flu and had age below or equal to τ, to read:Eq 3 where m(τ) is called the mortality density, i.e. mCitation[50]Δτ, Δτ = 1 becomes approximately equal to the number of individuals of age 50 that were killed due to the virus. Thus
Eq 4 becomes the mortality of the population due to the virus. We further define
Eq 5 µ(τ) is simply the fraction of individuals that were killed in each age category, or the mortality by age. Thus if there were 1000 individuals of age 50 in the population, and 20 were killed due to the virus, µCitation[50]=20/1000 = 0.02. We will use data from the statistical central bureaus for this quantity in 1918.
Our logic then becomes:
(i) Achieve from the statistical data for 1918. (ii) Assume different relations for the mortality µ(τ) by assuming counterfactually history arguments. (iii) Calculate the total mortality by the formulae:
Eq 6
The mortality by age
The mortality by age µ(τ) is input to the calculations. Different scenarios will be considered.
For the first scenario it is assumed that above the age of 27 all individuals have the same mortality by age and equal to the value for the age of 27. The rationale for this scenario is that no individuals were immunized due to earlier viruses in the population, and that the functionality of the immune systems of individuals of age 27 is the same as for age above 27.
The second scenario is to use a curve fit to the mortality by age between 10 and 20. The hypothesis is that the immune system of an individual is optimum at the age, say 10, and that it worsens thereafter. We hypothesize that newborn individuals must normally expect to meet most infections before the age of 10, and that the immune system is optimized for this by evolution. For higher ages the adaptive immune system comes into play and saves the older individuals. The common knowledge that newborn individuals (before age 1) have higher mortality by age we suggest is in part related to a ‘start problem’ for the immune system. We fit to a power function. The third scenario is also to use a curve fit to the mortality by age between 10 and 20. This time we use an exponential fit. For the fourth scenario the mortality by age mimics the common mortality by age in a population.
The results
shows the normalized age density as a function of age fitted to the statistical data material for Norway. The integral of the curve up to 100 years is 1.
Figure 1. The normalized age density as a function of the age τ in years.
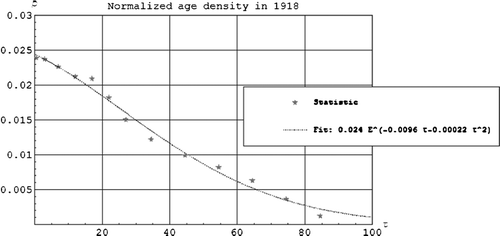
shows the mortality by age during the Spanish flu Citation[5]. The W is clearly seen. Between each data point we have used a quadratic polynomial which gives a good fit.
Figure 2. The mortality by age µ(τ) as a function of age τ in years.
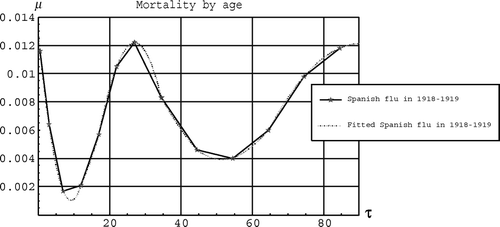
shows the mortality by age for the scenarios. Notice that both case 2 and case 3 are fitted to pass exactly thorough the two data points that we use. Observe in that the fitted curve for case 3 gives that at age 45 (or more) the mortality by age becomes 1.
Figure 3. The mortality by age µ(τ) as a function of age τ in years for the scenarios.
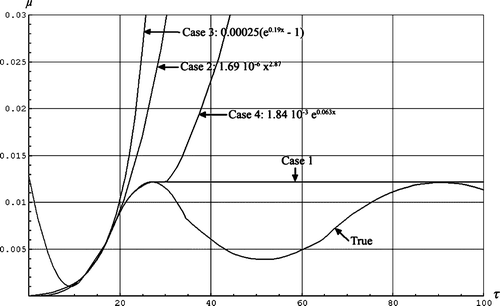
Figure 4. The mortality by age µ(τ) as a function of age τ in years for the scenarios.
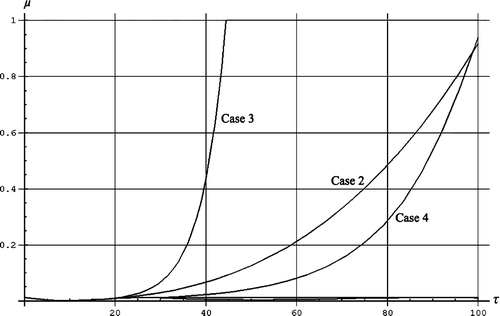
The benchmark test for the true curve gave a mortality of 0.65%. This is in good agreement with the experimental data of 0.63%. Case 1 gave a mortality of 0.9%. Case 2 gave a mortality of 7%, case 3 gave a mortality of 30% and case 4 gave a mortality of 3%. Thus for case 3, 30% of the world population would have been killed by the virus.
The Spanish flu came in three to four waves where the first wave may have immunized the population for the second wave Citation[26], Citation[66]. The death rate in the first wave was small. We have earlier found that the first wave may have affected 50% of the population and that the second wave affected 50% of the population that was not immunized, i.e. 25% of the population Citation[66]. If we assume that the second wave came first the situation will be worsening since the more deadly wave 2 virus will now be moving into a virgin population. shows the number of infected individuals for the second wave for two cases. The lower bulge to the right corresponds to what actually happened, while the larger bulge corresponds to what would have happened if the second wave came first. The total number of individuals that catch the flu is given in . We observe in that the number of affected individuals in the second wave may have increased roughly with a factor of 4 if the second wave came before the first wave Citation[26], which follows because the transmission rate is a factor of two higher during the second than during the first wave Citation[324], Citation[325] at the same time as the population density of susceptible individuals is a factor of two higher than when the first wave came first Citation[66]. To calculate the mortality we do not multiply with a factor of 4, but with a factor of 8/3, since the number of deaths for the Spanish flu also includes the deaths in the third wave that affected 1/8 of the population. Thus we achieve mortalities of 0.65%×8/3 = 1.7% (true), 0.9%×8/3 = 2.4% (case 1), 7%×8/3 = 19% (case 2), 30×8/3 = 80% (case 3) and 3×8/3 = 8% (case 4). Finally notice from and that if the second wave came before the first wave the time it takes before the pandemic is over decreases from around 125 days to 40 days.
Figure 5. The number of infected individuals in wave 2 relative to the population size as a function of time in days.
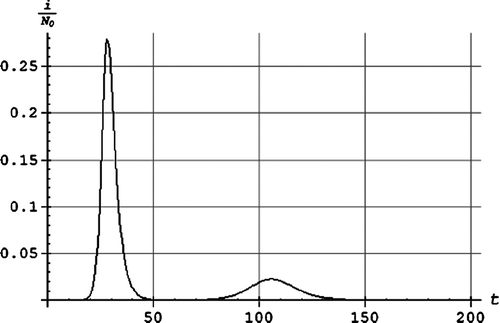
Figure 6. The number of affected individuals in wave 2 relative to the population size as a function of time in days. From above: (a) assuming the second wave of the Spanish flu came before the first wave, (b) the second wave of the Spanish flu.
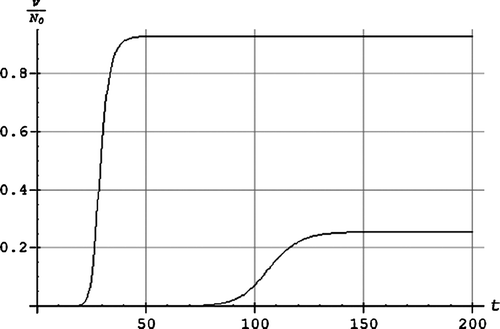
Conclusion
We have analyzed different worst case scenarios by assuming counterfactual history arguments.
For the worst case scenario we assume that no individuals had any adaptive immunity that was protective against the Spanish flu virus. We put forward a hypothesis for the worsening of the functionality of the immune system as a function of age. The mortality fraction then becomes 0.3. Thus 30% of the population could have been killed due to the Spanish virus during a first exposure. As an even worse worst case scenario we also assume, counterfactually, that the second wave of the Spanish flu came before the first wave. The mortality then becomes 80%. For the scenario where the mortality by age mimics the common mortality by age in a population the mortality becomes 8%.
We believe that our results are relevant inputs to making future plans and hopefully will lead to more realistic pandemic plans for the H5N1 bird virus.
Acknowledgements
We wish to thank Professor Nevin S. Scrimshaw for his generous assistance with a critical review of our article.
References
- Mounier-Jack S, Coker RJ. How prepared is Europe for pandemic influenza? Analysis of national plans. Lancet 2006; 367: 1405–11
- Taubenberger JK, Reid AH, Janczewski TA, Fanning TG. Integrating historical, clinical and molecular genetic data in order to explain the origin and virulence of the 1918 Spanish influenza virus. Philos Trans R Soc Lond B Biol Sci 2001; 356: 1829–39
- Osterholm MT. Preparing for the next pandemic. Foreign Affairs 2005; July/August:24–37.
- Johnson NP, Mueller J. Updating the accounts: global mortality of the 1918–1920 “Spanish” influenza pandemic. Bull Hist Med 2002; 76: 105–15
- Murray CJ, Lopez AD, Chin B, Feehan D, Hill KH. Estimation of potential global pandemic influenza mortality on the basis of vital registry data from the 1918–20 pandemic: a quantitative analysis. Lancet 2006; 368: 2211–18
- Mamelund S-E. Spanish Influenza and beyond: The case of Norway. Dr. polit. Thesis, Department of Economics, University of Oslo, September 2004.
- Oxford JS, Lambkin R, Sefton A, Daniels R, Elliot A, Brown R, et al. A hypothesis: the conjunction of soldiers, gas, pigs, ducks, geese and horses in Northern France during the Great War provided the conditions for the emergence of the “Spanish” influenza pandemic of 1918–1919. Vaccine 2005; 23: 940–5
- Mamelund S-E. Spanish influenza mortality of ethnic minorities in Norway 1918–1919. Eur J Population 2003; 19: 83–102
- Smith FB. The Russian influenza in the United Kingdom, 1889–1894. Soc Hist Med 1995; 8: 55–73
- World Health Organization. Influenza research at the human and animal interface. Report of a WHO working group. Geneva, Switzerland, 21–22 September 2006. WHO/EPR/GIP/2006.3. Available online on WHO home site.
- Sompayrac L. How the immune system works2nd edn. Blackwell Publishing, Oxford 2003
- Qureshi K, Molbak K, Sandström A, Kofoed PE, Rodrigues A, Dias F, et al. Breast milk reduces the risk of illness in children of mothers with cholera: observations from an epidemic of cholera in Guinea-Bissau. Pediatr Infect Dis J 2006; 25: 1163–6
- Ballabio C, Bertino E, Coscia A, Fabris C, Fuggetta D, Molfino S, et al. Immunoglobulin-A profile in breast milk from mothers delivering full term and preterm infants. Int J Immunopathol Pharmacol 2007; 20: 119–28
- Severin S, Wenshui X. Milk biologically active components as nutraceuticals: review. Crit Rev Food Sci Nutr 2005; 45: 645–56
- Morrow AL, Rangel JM. Breast milk reduces the risk of illness in children of mothers with cholera: observations from an epidemic of cholera in Guinea-Bissau. Pediatr Infect Dis J. 2006; 25: 1163–6
- Coppa GV, Bruni S, Morelli L, Soldi S, Gabrielli O. The first prebiotics in humans: human milk oligosaccharides. J Clin Gastroenterol 2004;38(6 Suppl):S80–S83.
- Coppa GV, Zampini L, Galeazzi T, Gabrielli O. Prebiotics in human milk: a review. Dig Liver Dis 2006; 38(Suppl 2)S291–S294
- Picard C, Fioramonti J, Francois A, Robinson T, Neant F, Matuchansky C. Review article: bifidobacteria as probiotic agents – physiological effects and clinical benefits. Aliment Pharmacol Ther 2005; 22: 495–512
- Bruzzese E, Volpicelli M, Squaglia M, Tartaglione A, Guarino A. Impact of prebiotics on human health. Dig Liver Dis 2006; 38(Suppl 2)S283–S287
- Leakey R, Lewins R. People of the lake. Mankind & its beginnings. Dutton, New York 1977
- Woldemaria A, Norwegian University of Life Sciences (personal communication).
- McKeon T. The modern rise of population. Academic Press, New York 1976
- Scrimshaw NS, Taylor CE, Gordon JE. Interactions of nutrition and infection. WHO, Geneva 1968
- Scrimshaw NS, SanGiovanni JP. Synergism of nutrition, infection, and immunity: an overview. Am J Clin Nutr 1997; 66: 464S–477S
- Leung AK, Kellner JD, Davies HD. Rotavirus gastroenteritis. Adv Ther 2005; 22: 476–87
- Christophersen OA, Haug A. More about hypervirulent avian influenza: is the world now better prepared? Microb Ecol Health Dis 2007;19:78–121.
- Hannappel E, Huff T. The thymosins. Prothymosin alpha, parathymosin, and beta-thymosins: structure and function. Vitam Horm 2003; 66: 257–96
- Cordero OJ, Sarandeses CS, Lopez JL, Nogueria M. Prothymosin alpha enhances human natural killer cell cytotoxicity: role in mediating signals for NK activity. Lymphokine Cytokine Res 1992; 11: 277–85
- Saha AR, Hadden EM, Hadden JW. Zinc induces thymulin secretion from human thymic epithelial cells in vitro and augments splenocyte and thymocyte responses in vivo. Int J Immunopharmacol 1995; 17: 729–33
- Geneser F. Histologi – på molekylærbiologisk grundlag. [Histology – on basis of molecular biology.] 1st edn, 4th reprint. København: Munksgaard, 2004 (in Danish).
- Min H, Montecino-Rodriguez E, Dorshkind K. Reassessing the role of growth hormone and sex steroids in thymic involution. Clin Immunol 2006; 118: 117–23
- Bellamy D, Hinsull SM, Phillips JG. Factors controlling growth and age involution of the rat thymus. Age Ageing 1976; 5: 12–19
- Fahy GM. Apparent induction of partial thymic regeneration in a normal human subject: a case report. J Anti Aging Med 2003; 6: 219–27
- Zoller AL, Kersh GJ. Estrogen induces thymic atrophy by eliminating early thymic progenitors and inhibiting proliferation of beta-selected thymocytes. J Immunol 2006; 176: 7371–8
- Hareramadas B, Rai U. Cellular mechanism of estrogen-induced thymic involution in wall lizard: caspase-dependent action. J Exp Zool A Comp Exp Biol 2006; 305: 396–409
- Yao G, Hou Y. Thymic atrophy via estrogen-induced apoptosis is related to Fas/FasL pathway. Int Immunopharmacol 2004; 4: 213–21
- Kim DH, Kim CH, Kim MS, Kim JY, Jung KJ, Chung JH, et al. Suppression of age-related inflammatory NF-kappaB activation by cinnamaldehyde. Biogerontology 2007; 8: 545–54
- Peng Y, Gallagher SF, Haines K, Baksh K, Murr MM. Nuclear factor-kappaB mediates Kupffer cell apoptosis through transcriptional activation of Fas/FasL. J Surg Res 2006; 130: 58–65
- Yao PL, Lin YC, Sawhney P, Richburg JH. Transcriptional regulation of FasL expression and participation of sTNF-alpha in response to Sertoli cell injury. J Biol Chem 2007; 282: 5420–31
- Davis BH, Chen A, Beno DW. Raf and mitogen-activated protein kinase regulate stellate cell collagen gene expression. J Biol Chem 1996; 271: 11039–42
- Berra E, Pages G, Pouyssegur J. MAP kinases and hypoxia in the control of VEGF expression. Cancer Metastasis Rev 2000; 19: 139–45
- Pages G, Milanini J, Richard DE, Berra E, Gothie E, Vinals F, et al. Signaling angiogenesis via p42/p44 MAP kinase cascade. Ann N Y Acad Sci 2000; 902: 187–200
- Moon SK, Jung SY, Kim CH. Transcription factor Sp1 mediates p38MAPK-dependent activation of the p21WAF1 gene promoter in vascular smooth muscle cells by pyrrolidine dithiocarbamate. Biochem Biophys Res Commun 2004; 316: 605–11
- D'Addario M, Arora PD, McCulloch CA. Role of p38 in stress activation of Sp1. Gene 2006; 379: 51–61
- Dasari A, Bartholomew JN, Volonte D, Galbiati F. Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements. Cancer Res 2006; 66: 10805–14
- Gee K, Angel JB, Mishra S, Blahoianu MA, Kumar A. IL-10 regulation by HIV-Tat in primary human monocytic cells: involvement of calmodulin/calmodulin-dependent protein kinase-activated p38 MAPK and Sp-1 and CREB-1 transcription factors. J Immunol 2007; 178: 798–807
- Tu VC, Bahl JJ, Chen QM. Distinct roles of p42/p44(ERK) and p38 MAPK in oxidant-induced AP-1 activation and cardiomyocyte hypertrophy. Cardiovasc Toxicol 2003; 3: 119–33
- Zdanov S, Debacq-Chainiaux F, Remacle J, Toussaint O. Identification of p38MAPK-dependent genes with changed transcript abundance in H2O2-induced premature senescence of IMR-90 hTERT human fibroblasts. FEBS Lett 2006; 580: 6455–63
- Kim DH, Kim HK, Park S, Kim JY, Zou Y, Cho KH, et al. Short-term feeding of baicalin inhibits age-associated NF-kappaB activation. Mech Ageing Dev 2006; 127: 719–25
- Lee HC, Wei YH. Oxidative stress, mitochondrial DNA mutation, and apoptosis in aging. Exp Biol Med (Maywood) 2007; 232: 592–606
- Barja G. Mitochondrial free radical production and aging in mammals and birds. Ann N Y Acad Sci 1998; 854: 224–38
- Barja G. Rate of generation of oxidative stress-related damage and animal longevity. Free Radic Biol Med 2002; 33: 1167–72
- Pamplona R, Portero-Otin M, Riba D, Ledo F, Gredilla R, Herrero A, et al. Heart fatty acid unsaturation and lipid peroxidation, and aging rate, are lower in the canary and the parakeet than in the mouse. Aging (Milano) 1999; 11: 44–9
- Bodey B, Bodey B, Jr, Siegel SE, Kaiser HE. The role of zinc in pre- and postnatal mammalian thymic immunohistogenesis. In Vivo 1998; 12: 695–722
- Bodey B, Bodey B, Jr, Siegel SE, Kaiser HE. Involution of the mammalian thymus, one of the leading regulators of aging. In Vivo 1997; 11: 421–40
- Goya RG, Gagnerault MC, De Moraes MC, Savino W, Dardenne M. In vivo effects of growth hormone on thymus function in aging mice. Brain Behav Immun 1992; 6: 341–54
- Yamada M, Hato F, Kinoshita Y, Tominaga K, Tsuji Y. The indirect participation of growth hormone in the thymocyte proliferation system. Cell Mol Biol (Noisy-le-grand) 1994; 40: 111–21
- De Mello-Coelho V, Savino W, Postel-Vinay MC, Dardenne M. Role of prolactin and growth hormone on thymus physiology. Dev Immunol 1998; 6: 317–23
- Vigano A, Saresella M, Trabattoni D, Giacomet V, di Natale B, Merlo M, et al. Growth hormone in T-lymphocyte thymic and postthymic development: a study in HIV-infected children. J Pediatr 2004; 145: 542–8
- Polgreen L, Steiner M, Dietz CA, Manivel JC, Petryk A. Thymic hyperplasia in a child treated with growth hormone. Growth Horm IGF Res 2007; 17: 41–6
- Brunton LL, Lazo JS, Parker KL. Goodman & Gilman's The pharmacological basis of therapeutics, 11th edn. New York: McGraw-Hill, 2006.
- Huang Y, Chen Z, Zhou C, Yao H, Li M, Xu C. The modulation of thymosin alpha 1 in the maturation, differentiation and function of murine bone marrow-derived dendritic cells in the absence or presence of tumor necrosis factor-alpha. Int Immunopharmacol 2004; 4: 539–46
- Yang YM, Lu XY, Huang WD, Shen MY. [Effect of thymosin alpha 1 on cellular immune function in elderly patients with malignant tumor.]. Zhejiang Da Xue Xue Bao Yi Xue Ban 2003; 32: 339–41 (in Chinese)
- Lauria F, Raspadori D, Tura S. Effect of a thymic factor on T lymphocytes in B cell chronic lymphocytic leukemia: in vitro and in vivo studies. Blood 1984; 64: 667–71
- Wespes E, Schulman CC. Male andropause: myth, reality, and treatment. Int J Impot Res 2002; 14(Suppl 1)S93–S98
- Moxnes JE, Christophersen OA. Counter-attacking pandemic H5N1 bird influenza by counter-pandemic. Microb Ecol Health Dis 2006; 18: 4–25
- Ronichevskaya GM, Smirnov PN, Goncharova NB, Zlobina GA. [The biological activity of chalone-like proteoglycans isolated from the spleen and thymus at various periods of ontogeny.]. Ontogenez 1989; 20: 409–15 (in Russian)
- Liang J, Yao G, Yang L, Hou Y. Dehydroepiandrosterone induces apoptosis of thymocyte through Fas/Fas-L pathway. Int Immunopharmacol 2004; 4: 1467–75
- Yao G, Shang XJ. A comparison of modulation of proliferation of thymocyte by testosterone, dehydroisoandrosterone and androstenedione in vitro. Arch Androl 2005; 51: 257–65
- Yan CH, Jiang XF, Pei X, Dai YR. The in vitro antiapoptotic effect of dehydroepiandrosterone sulfate in mouse thymocytes and its relation to caspase-3/caspase-6. Cell Mol Life Sci 1999; 56: 543–7
- Molinero P, Soutto M, Benot S, Hmadcha A, Guerrero JM. Melatonin is responsible for the nocturnal increase observed in serum and thymus of thymosin alpha1 and thymulin concentrations: observations in rats and humans. J Neuroimmunol 2000; 103: 180–8
- Sainz RM, Mayo RC, Reiter RJ, Antolin I, Esteban MM, Rodriguez C. Melatonin regulates glucocorticoid receptor: an answer to its antiapoptotic action in thymus. FASEB J 1999; 13: 1547–56
- Hoijman E, Rocha Viegas L, Keller Sarmiento MI, Rosenstein RE, Pecci A. Involvement of Bax protein in the prevention of glucocorticoid-induced thymocytes apoptosis by melatonin. Endocrinology 2004; 145: 418–25
- Oner H, Kus I, Oner J, Ogeturk M, Ozan E, Ayar A. Possible effects of melatonin on thymus gland after pinealectomy in rats. Neuro Endocrinol Lett 2004; 25: 115–18
- Pertsov SS. Effect of melatonin on the thymus, adrenal glands, and spleen in rats during acute stress. Bull Exp Biol Med 2006; 141: 292–5
- Presman DM, Hoijman E, Ceballos NR, Galigniana MD, Pecci A. Melatonin inhibits glucocorticoid receptor nuclear translocation in mouse thymocytes. Endocrinology 2006; 147: 5452–9
- Naranjo MC, Guerrero JM, Rubio A, Lardone PJ, Carillo-Vico A, Carrascosa-Salmoral MP, et al. Melatonin biosynthesis in the thymus of humans and rats. Cell Mol Life Sci 2007; 64: 781–90
- Dardenne M, Savino W, Gagnerault MC, Itoh T, Bach JF. Neuroendocrine control of thymic hormonal production. I. Prolactin stimulates in vivo and in vitro the production of thymulin by human and murine thymic epithelial cells. Endocrinology 1989; 125: 3–12
- McNeill WH. Plagues and peoples. Anchor Books, New York 1998
- Maestroni GJ, Covacci V, Conti A. Hematopoietic rescue via T-cell-dependent, endogenous granulocyte-macrophage colony-stimulating factor induced by the pineal neurohormone melatonin in tumor-bearing mice. Cancer Res 1994; 54: 2429–32
- Maestroni GJ. kappa-Opioid receptors in marrow stroma mediate the hematopoietic effects of melatonin-induced opioid cytokines. Ann N Y Acad Sci 1998; 840: 411–19
- Maestroni GJ, Zammaretti F, Pedrinis E. Hematopoietic effect of melatonin involvement of type 1 kappa-opioid receptor on bone marrow macrophages and interleukin-1. J Pineal Res 1999; 27: 145–53
- Srinavasan V, Maestroni G, Cardinali D, Esquifino A, Perumal SP, Miller S. Melatonin, immune function and aging. Immun Ageing 2005; 2: 17
- Tan DX, Manchester LC, Reiter RJ, Qi WB, Karbownik M, Calvo JR. Significance of melatonin in antioxidative defense system: reactions and products. Biol Signals Recept 2000; 9: 137–59
- Tan DX, Manchester LC, Terron MP, Flores LJ, Reiter RJ. One molecule, many derivatives: a never-ending interaction of melatonin with reactive oxygen and nitrogen species?. J Pineal Res 2007; 42: 28–42
- Reiter RJ, Carneiro RC, Oh CS. Melatonin in relation to cellular antioxidative defense mechanisms. Horm Metab Res 1997; 29: 363–72
- Maldonado MD, Murillo-Cabezas F, Terron MP, Flores LJ, Tan DX, Manchester LC, et al. The potential of melatonin in reducing morbidity-mortality after craniocerebral trauma. J Pineal Res 2007; 42: 1–11
- Maldonado MD, Murillo-Cabezas F, Calvo JR, Lardone PJ, Tan DX, Guerrero JM, et al. Melatonin as pharmacologic support in burn patients: a proposed solution to thermal injury-related lymphocytopenia and oxidative damage. Crit Care Med 2007; 35: 1177–85
- Sandyk PR. Possible role of pineal melatonin in the mechanisms of aging. Int J Neurosci 1990; 52: 85–92
- Reiter RJ. Pineal function during aging: attenuation of the melatonin rhythm and its neurobiological consequences. Acta Neurobiol Exp (Wars) 1994; 54(Suppl)31–9
- Ruzsas C, Mess B. Melatonin and aging. A brief survey. Neuro Endocrinol Lett 2000; 21: 17–23
- Kuhn DM, Ruskin B, Lovenberg W. Tryptophan hydroxylase. The role of oxygen, iron, and sulfhydryl groups as determinants of stability and catalytic activity. J Biol Chem 1980; 255: 4137–43
- Johnson M, Hanson GR, Gibb JW. Characterization of acute N-ethyl-3,4-methylenedioxyamphetamine (MDE) action on the central serotonergic system. Biochem Pharmacol 1989; 38: 4333–8
- Stone DM, Johnson M, Hanson GR, Gibb JW. Acute inactivation of tryptophan hydroxylase by amphetamine analogs involves the oxidation of sulfhydryl sites. Eur J Pharmacol 1989; 172: 93–7
- Kuhn DM, Arthur RE, Jr. Inactivation of brain tryptophan hydroxylase by nitric oxide. J Neurochem 1996; 67: 1072–7
- Kuhn DM, Arthur RE, Jr. Inactivation of tryptophan hydroxylase by nitric oxide: enhancement by tetrahydrobiopterin. J Neurochem 1997; 68: 1495–502
- Kuhn DM, Geddes TJ. Peroxynitrite inactivates tryptophan hydroxylase via sulfhydryl oxidation. Coincident nitration of enzyme tyrosyl residues has minimal impact on catalytic activity. J Biol Chem 1999; 274: 29726–32
- Mohan PF, Jacobson MS. Inhibition of macrophage superoxide generation by dehydroepiandrosterone. Am J Med Sci 1993; 306: 10–15
- Padgett DA, Loria RM. In vitro potentiation of lymphocyte activation by dehydroepiandrosterone, androstenediol, and androstenetriol. J Immunol 1994; 153: 1544–52
- Loria RM. Antiglucocorticoid function of androstenetriol. Psychoneuroendocrinology 1997; 22(Suppl 1)S103–S108
- Hernandez-Pando R, De La Luz Streber M, Orozco H, Arriaga K, Pavon L, Al-Nakhli SA, et al. The effects of androstenediol and dehydroepiandrosterone on the course and cytokine profile of tuberculosis in BALB/c mice. Immunology 1998; 95: 234–41
- Loria RM, Padgett DA. Control of the immune response by DHEA and its metabolites. Rinsho Byori 1998; 46: 505–17
- Padgett DA, Loria RM. Endocrine regulation of murine macrophage function: effects of dehydroepiandrosterone, androstenediol, and androstenetriol. J Neuroimmunol 1998; 84: 61–8
- Loria RM, Conrad DH, Huff T, Carter H, Ben-Nathan D. Androstenetriol and androstenediol. Protection against lethal radiation and restoration of immunity after radiation injury. Ann N Y Acad Sci 2000; 917: 860–7
- Whitnall MH, Elliott TB, Harding RA, Inal CE, Landauer MR, Wilhelmsen CL, et al. Androstenediol stimulates myelopoiesis and enhances resistance to infection in gamma-irradiated mice. Int J Immunopharmacol 2000; 22: 1–14
- Loria RM. Immune up-regulation and tumor apoptosis by androstene steroids. Steroids 2002; 67: 953–66
- Chahal HS, Drake WM. The endocrine system and ageing. J Pathol 2007; 211: 173–80
- Ben-Nathan D, Lachmi B, Lustig S, Feuerstein G. Protection by dehydroepiandrosterone in mice infected with viral encephalitis. Arch Virol 1991; 120: 263–71
- Ben-Nathan D, Lustig S, Kobiler D, Danenberg HD, Lupu E, Feuerstein G. Dehydroepiandrosterone protects mice inoculated with West Nile virus and exposed to cold stress. J Med Virol 1992; 38: 159–66
- Loria RM, Padgett DA. Androstenediol regulates systemic resistance against lethal infections in mice. Arch Virol 1992; 127: 103–15
- Carr DJ. Increased levels of IFN-gamma in the trigeminal ganglion correlate with protection against HSV-1-induced encephalitis following subcutaneous administration with androstenediol. J Neuroimmunol 1998; 89: 160–7
- Daigle J, Carr DJ. Androstenediol antagonizes herpes simplex virus type 1-induced encephalitis through the augmentation of type I IFN production. J Immunol 1998; 160: 3060–6
- Padgett DA, Loria RM, Sheridan JF. Endocrine regulation of the immune response to influenza virus infection with a metabolite of DHEA-androstenediol. J Neuroimmunol 1997; 78: 203–11
- Padgett DA, Sheridan JF. Androstenediol (AED) prevents neuroendocrine-mediated suppression of the immune response to an influenza viral infection. J Neuroimmunol 1999; 98: 121–9
- Padgett DA, Loria RM, Sheridan JF. Steroid hormone regulation of antiviral immunity. Ann N Y Acad Sci 2000; 917: 935–43
- Carroll J, Fearnley IM, Shannon RJ, Hirst J, Walker JE. Analysis of the subunit composition of complex I from bovine heart mitochondria. Mol Cell Proteomics 2003; 2: 117–26
- Mansouri A, Muller FL, Liu Y, Ng R, Faulkner J, Hamilton M, et al. Alterations in mitochondrial function, hydrogen peroxide release and oxidative damage in mouse hind-limb skeletal muscle during aging. Mech Ageing Dev 2006; 127: 298–306
- Jornot L, Petersen H, Junod AF. Hydrogen peroxide-induced DNA damage is independent of nuclear calcium but dependent on redox-active ions. Biochem J 1998; 335: 85–94
- Lloyd DR, Carmichael PL, Phillips DH. Comparison of the formation of 8-hydroxy-2′-deoxyguanosine and single- and double-strand breaks in DNA mediated by Fenton reactions. Chem Res Toxicol 1998; 11: 420–7
- Barbouti A, Doulias PT, Zhu BZ, Frei B, Galaris D. Intracellular iron, but not copper, plays a critical role in hydrogen peroxide-induced DNA damage. Free Radic Biol Med 2001; 31: 490–8
- Messina SA, Dawson R, Jr. Attenuation of oxidative damage to DNA by taurine and taurine analogs. Adv Exp Med Biol 2000; 483: 355–67
- Rodriguez B, Sethi AK, Cheruvu VK, Mackay W, Bosch RJ, Kitahata M, et al. Predictive value of plasma HIV RNA level on rate of CD4 T-cell decline in untreated HIV infection. JAMA 2006; 296: 1498–506
- Di Marzio L, Alesse E, Roncaioli P, Muzi P, Moretti S, Marcellini S, et al. Influence of L-carnitine on CD95 cross-lining-induced apoptosis and ceramide generation in human cell lines: correlation with its effects on purified acidic and neutral sphingomyelinases in vitro. Proc Assoc Am Physicians 1997; 109: 154–63
- Di Marzio L, Moretti S, D'Alo S, Zazzeroni F, Marcellini S, Smacchia C, et al. Acetyl-L-carnitine administration increases insulin-like growth factor 1 levels in asymptomatic HIV-1-infected subjects: correlation with its suppressive effect on lymphocyte apoptosis and ceramide generation. Clin Immunol 1999; 92: 103–10
- Christophersen OA, Haug A. Possible roles of oxidative stress, local circulatory failure and nutrition factors in the pathogenesis of hypervirulent influenza: implications for therapy and global emergency preparedness. Microb Ecol Health Dis 2005; 17: 189–99
- Chatterjee S. Sphingolipids in atherosclerosis and vascular biology. Arterioscler Thromb Vasc Biol 1998; 18: 1523–33
- Corda S, Laplace C, Vicaut E, Duranteau J. Rapid reactive oxygen species production by mitochondria in endothelial cells exposed to tumor necrosis factor-alpha is mediated by ceramide. Am J Respir Cell Mol Biol 2001; 24: 762–8
- Suematsu N, Tsutsui H, Wen J, Kang D, Ikeuchi M, Ide T, et al. Oxidative stress mediates tumor necrosis factor-alpha-induced mitochondrial DNA damage and dysfunction in cardiac myocytes. Circulation 2003; 107: 1418–23
- Martin SF, Williams N, Chatterjee S. Lactosylceramide is required in apoptosis induced by N-Smase. Glycoconj J 2006; 23: 147–57
- Garcia-Ruiz C, Colell A, Mari M, Morales A, Fernandez-Checa JC. Direct effect of ceramide on the mitochondrial electron transport chain leads to generation of reactive oxygen species. Role of mitochondrial glutathione. J Biol Chem 1997; 272: 11369–77
- Garcia-Ruiz C, Colell A, Morales A, Calvo M, Enrich C, Fernandez-Checa JC. Trafficking of ganglioside GD3 to mitochondria by tumor necrosis factor-alpha. J Biol Chem 2002; 277: 36443–8
- Garcia-Ruiz C, Colell A, Paris R, Fernandez-Checa JC. Direct interaction of GD3 ganglioside with mitochondria generates reactive oxygen species followed by mitochondrial permeability transition, cytochrome c release, and caspase activation. FASEB J 2000; 14: 847–58
- Goldfeld AE, Doyle C, Maniatis T. Human tumor necrosis factor alpha gene regulation by virus and lipopolysaccharide. Proc Natl Acad Sci U S A 1990; 87: 9769–73
- Yao J, Mackman N, Edgington TS, Fan ST. Lipopolysaccharide induction of the tumor necrosis factor-alpha promoter in human monocytic cells. Regulation by Egr-1, c-Jun, and NF-kappaB transcription factors. J Biol Chem 1997; 272: 17795–801
- Liu H, Sidiropoulos P, Song G, Pagliari LJ, Birrer MJ, Stein B, et al. TNF-alpha gene expression in macrophages: regulation by NF-kappa B is independent of c-Jun or C/EBP beta. J Immunol 2000; 164: 4277–85
- Steer JH, Kroeger KM, Abraham LJ, Joyce DA. Glucocorticoids suppress tumor necrosis factor-alpha expression by human monocytic THP-1 cells by suppressing transactivation through adjacent NF-kappa B and c-Jun-activating transcription factor-2 binding sites in the promoter. J Biol Chem 2000; 275: 18432–40
- Sanceau J, Kaisho T, Hirano T, Wietzerbin J. Triggering of the human interleukin-6 gene by interferon-gamma and tumor necrosis factor-alpha in monocytic cells involves cooperation between interferon regulatory factor-1, NF kappa B, and Sp1 transcription factors. J Biol Chem 1995; 270: 27920–31
- Zhang X, Wu K, Wang D, Yue X, Song D, Zhu Y, et al. Nucleocapsid protein of SARS-CoV activates interleukin-6 expression through cellular transcription factor NF-kappaB. Virology 2007; 365: 324–35
- Yang CM, Chien CS, Hsiao LD, Luo SF, Wang CC. Interleukin-1beta-induced cyclooxygenase-2 expression is mediated through activation of p42/44 and p38 MAPKs, and NF-kappaB pathways in canine tracheal smooth muscle cells. Cell Signal 2002; 14: 899–911
- Luo SF, Wang CC, Chien CS, Hsiao LD, Yang CM. Induction of cyclooxygenase-2 by lipopolysaccharide in canine tracheal smooth muscle cells: involvement of p42/p44 and p38 mitogen-activated protein kinases and nuclear factor-kappaB pathways. Cell Signal 2003; 15: 497–509
- Lin CC, Hsiao LD, Chien CS, Lee CW, Hsieh JT, Yang CM. Tumor necrosis factor-alpha-induced cyclooxygenase-2 expression in human tracheal smooth muscle cells: involvement of p42/p44 and p38 mitogen-activated protein kinases and nuclear factor-kappaB. Cell Signal 2004; 16: 597–607
- Si J, Fu X, Behar J, Wands J, Beer DG, Souza RF, et al. NADPH oxidase NOX5-S mediates acid-induced cyclooxygenase-2 expression via activation of NF-kappa B in Barrett's esophageal adenocarcinoma cells. J Biol Chem 2007; 282: 16244–55
- Leonarduzzi G, Scavazza A, Biasi F, Chiarpotto E, Camandola S, Vogel S, et al. The lipid peroxidation end product 4-hydroxy-2,3-nonenal up-regulates transforming growth factor beta1 expression in the macrophage lineage: a link between oxidative injury and fibrosclerosis. FASEB J 1997; 11: 851–7
- Chiarpotto E, Allasia C, Biasi F, Leonarduzzi G, Ghezzo F, Berta G, et al. Down-modulation of nuclear localisation and pro-fibrogenic effect of 4-hydroxy-2,3-nonenal by thiol- and carbonyl-reagents. Biochim Biophys Acta 2002; 1584: 1–8
- Chiarpotto E, Castello L, Leonarduzzi G, Biasin And F, Poli G. Role of 4-hydroxy-2,3-nonenal in the pathogenesis of fibrosis. Biofactors 2005; 24: 229–36
- Jarnicki AG, Lysaght J, Todryk S, Mills KH. Suppression of antitumor immunity by IL-10 and TGF-beta-producing T cells infiltrating the growing tumor: influence of tumor environment on the induction of CD4+ and CD8+ regulatory T cells. J Immunol 2006; 177: 896–904
- Kriegel MA, Li MO, Sanjabi S, Wan YY, Flavell RA. Transforming growth factor-beta: recent advances on its role in immune tolerance. Curr Rheumatol Rep 2006; 8: 138–44
- Li MQ, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 2006; 25: 455–71
- Kojima M, Morisaki T, Uchiyama A, Doi F, Mibu R, Katano M, et al. Association of enhanced cyclooxygenase-2 expression with possible local immunosuppression in human colorectal carcinomas. Ann Surg Oncol 2001; 8: 458–65
- Harizi H, Juzan M, Pitard V, Moreau JF, Gualde N. Cyclooxygenase-2-issued prostaglandin E2 enhances the production of endogenous IL-10, which down-regulates dendritic cell functions. J Immunol 2002; 168: 2255–63
- Akasaki Y, Liu G, Chung NH, Ehtesham M, Black KL, Yu JS. Induction of a CD4+ T regulatory type 1 response by cyclooxygenase-2-overexpressing glioma. J Immunol 2004; 173: 4352–9
- Landino LM, Crews BC, Gierse JK, Hauser SD, Marnett LJ. Mutational analysis of the role of the distal histidine and glutamine residues of prostaglandin-endoperoxide synthase-2 in peroxidase catalysis, hydroperoxide reduction, and cyclooxygenase activation. J Biol Chem 1997; 272: 21565–74
- Goodwin DC, Rowlinson SW, Marnett LJ. Substitution of tyrosine for the proximal histidine ligand to the heme of prostaglandin endoperoxide synthase 2: implications for the mechanism of cyclooxygenase activation and catalysis. Biochemistry 2000; 39: 5422–32
- Seibold SA, Ball T, Hsi LC, Mills DA, Abeysinghe RD, Micielli R, et al. Histidine 386 and its role in cyclooxygenase and peroxidase catalysis by prostaglandin-endoperoxide H synthases. J Biol Chem 2003; 278: 46163–70
- Rogge CE, Liu W, Wu G, Eang LH, Kulmacz RJ, Tsai AL. Identification of Tyr504 as an alternative tyrosyl radical site in human prostaglandin H synthase-2. Biochemistry 2004; 43: 1560–8
- Rogge CE, Ho B, Liu W, Kulmacz RJ, Tsai AL. Role of Tyr348 in Tyr385 radical dynamics and cyclooxygenase inhibitor interactions in prostaglandin H synthase-2. Biochemistry 2006; 45: 523–32
- Morse DE, Duncan H, Hooker N, Morse A. Hydrogen peroxide induces spawning in mollusks, with activation of prostaglandin endoperoxide synthetase. Science 1977; 196: 298–300
- Seregi A, Serfozo P, Mergl Z. Evidence for the localization of hydrogen peroxide-stimulated cyclooxygenase activity in rat brain mitochondria: a possible coupling with monoamine oxidase. J Neurochem 1983; 40: 407–13
- Hecker G, Utz J, Kupferschmidt RJ, Ullrich V. Low levels of hydrogen peroxide enhance platelet aggregation by cyclooxygenase activation. Eicosanoids 1991; 4: 107–13
- Im JW, Kim HK, Kim ND, Choi JS, Yu BP, Yang HS, et al. Activation of cyclooxygenases by H2O2 and t-butylhydroperoxide in aged rat lung. Biotechnol Lett 2004; 26: 1665–9
- Lands WEM, Rome LH. Inhibition of prostaglandin biosynthesis. Prostaglandins: chemical and biochemical aspects, SMM Karim. MTP Press, LancasterUK 1976; 87–137
- Hemler ME, Lands WE. Evidence for a peroxide-initiated free radical mechanism of prostaglandin biosynthesis. J Biol Chem 1980; 255: 6253–61
- Kulmacz RJ, Lands WE. Requirements for hydroperoxide by the cyclooxygenase and peroxidase activities of prostaglandin H synthase. Prostaglandins 1983; 25: 531–40
- Kulmacz RJ, Wang LH. Comparison of hydroperoxide initiator requirements for the cyclooxygenase activities of prostaglandin H synthase-1 and -2. J Biol Chem 1995; 270: 24019–23
- Kulmacz RJ. Regulation of cyclooxygenase catalysis by hydroperoxides. Biochem Biophys Res Commun 2005; 338: 25–33
- Landino LM, Crews BC, Timmons MD, Morrow JD, Marnett LJ. Peroxynitrite, the coupling product of nitric oxide and superoxide, activates prostaglandin biosynthesis. Proc Natl Acad Sci U S A 1996; 93: 15069–74
- Goodwin DC, Landino LM, Marnett LJ. Effects of nitric oxide and nitric oxide-derived species on prostaglandin endoperoxide synthase and prostaglandin biosynthesis. FASEB J 1999; 13: 1121–36
- Upmacis RK, Deeb RS, Hajjar DP. Regulation of prostaglandin H2 synthase activity by nitrogen oxides. Biochemistry 1999; 38: 12505–13
- Bachschmid M, Schildknecht S, Ullrich V. Redox regulation of vascular prostanoid synthesis by the nitric oxide-superoxide system. Biochem Biophys Res Commun 2005; 338: 536–42
- Schildknecht S, Bachschmid M, Ullrich V. Peroxynitrite provides the peroxide tone for PGHS-2-dependent prostacyclin synthesis in vascular smooth muscle cells. FASEB J 2005; 19: 1169–71
- Kulmacz RJ. Prostaglandin H synthase and hydroperoxides: peroxidase reaction and inactivation kinetics. Arch Biochem Biophys 1986; 249: 273–85
- Wu G, Wei C, Kulmacz RJ, Osawa Y, Tsai AL. A mechanistic study of self-inactivation of the peroxidase activity in prostaglandin H synthase-1. J Biol Chem 1999; 274: 9231–7
- Wu G, Kulmacz RJ, Tsai AL. Cyclooxygenase inactivation kinetics during reaction of prostaglandin H synthase-1 with peroxide. Biochemistry 2003; 42: 13772–7
- Wu G, Rogge CE, Wang JS, Kulmacz RJ, Palmer G, Tsai AL. Oxyferryl heme and not tyrosyl radical is the likely culprit in prostaglandin H synthase-1 peroxidase inactivation. Biochemistry 2007; 46: 534–42
- Hayama M, Inoue R, Akiba S, Sato T. ERK and p38 MAP kinase are involved in arachidonic acid release induced by H2O2 and PDGF in mesangial cells. Am J Physiol Renal Physiol 2002; 282: F485–91
- Coulon L, Calzada C, Moulin P, Vericel E, Lagarde M. Activation of p38 mitogen-activated protein kinase/cytosolic phospholipase A2 cascade in hydroperoxide-stressed platelets. Free Radic Biol Med 2003; 35: 616–25
- Shit S, Atreja SK. Phospholipase A2 activation by hydrogen peroxide during in vitro capacitation of buffalo spermatozoa. Indian J Exp Biol 2004; 42: 486–90
- van Rossum GS, Drummen GP, Verkleij AJ, Post JA, Boonstra J. Activation of cytosolic phospholipase A2 in Her14 fibroblasts by hydrogen peroxide: a p42/44(MAPK)-dependent and phosphorylation-independent mechanism. Biochim Biophys Acta 2004; 1636: 183–95
- Colston JT, de la Rosa SD, Strader JR, Anderson MA, Freeman GL. H2O2 activates Nox4 through PLA2-dependent arachidonic acid production in adult cardiac fibroblasts. FEBS Lett 2005; 579: 2533–40
- Cui XL, Ding Y, Alexander LD, Bao C, Al-Khalili OK, Simonson M, et al. Oxidative signaling in renal epithelium: critical role of cytosolic phospholipase A2 and p38(SAPK). Free Radic Biol Med 2006; 41: 213–21
- Nimmerjahn F, Dudziak D, Dirmeier U, Hobom G, Riedel A, Schlee M, et al. Active NF-kappaB signalling is a prerequisite for influenza virus infection. J Gen Virol 2004; 85: 2347–56
- Wurzer WJ, Ehrhardt C, Pleschka S, Berberich-Siebelt F, Wolff T, Walczak H, et al. NF-kappaB-dependent induction of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) and Fas/FasL is crucial for efficient influenza virus propagation. J Biol Chem 2004; 279: 30931–7
- Abdala-Valencia H, Cook-Mills JM. VCAM-1 signals activate endothelial cell protein kinase Calpha via oxidation. J Immunol 2006; 177: 6379–87
- Wu WS, Tsai RK, Chang CH, Wang S, Wu JR, Chang YX. Reactive oxygen species mediated sustained activation of protein kinase C alpha and extracellular signal-regulated kinase for migration of human hepatoma cell Hepg2. Mol Cancer Res 2006; 4: 747–58
- Lin D, Lobell S, Jewell A, Takemoto DJ. Differential phosphorylation of connexin46 and connexin50 by H2O2 activation of protein kinase Cgamma. Mol Vis 2004; 10: 688–95
- Lin D, Takemoto DJ. Oxidative activation of protein kinase Cgamma through the C1 domain. Effects on gap junctions. J Biol Chem 2005; 280: 13682–93
- Zhang Y, McPherson BC, Liu H, Baman TS, Rock P, Yao Z. H2O2 opens mitochondrial K(ATP) channels and inhibits GABA receptors via protein kinase C-epsilon in cardiomyocytes. Am J Physiol Heart Circ Physiol 2002; 282: H1395–403
- Dina OA, Chen X, Reichling D, Levine JD. Role of protein kinase Cepsilon and protein kinase A in a model of paclitaxel-induced painful peripheral neuropathy in the rat. Neuroscience 2001; 108: 507–15
- Amadesi S, Cottrell GS, Divino L, Chapman K, Grady EF, Bautista F, et al. Protease-activated receptor 2 sensitizes TRPV1 by protein kinase Cepsilon- and A-dependent mechanisms in rats and mice. J Physiol 2006; 575: 555–71
- Mao J, Price DD, Phillips LL, Lu J, Mayer DJ. Increases in protein kinase C gamma immunoreactivity in the spinal cord dorsal horn of rats with painful mononeuropathy. Neurosci Lett 1995; 198: 75–8
- Dina OA, Barletta J, Chen X, Mutero A, Martin A, Messing RO, et al. Key role for the epsilon isoform of protein kinase C in painful alcoholic neuropathy in the rat. J Neurosci 2000; 20: 8614–19
- Olah Z, Karai L, Iadarola MJ. Protein kinase Calpha is required for vanilloid receptor 1 activation. Evidence for multiple signaling pathways. J Biol Chem 2002; 277: 35752–9
- Sweitzer SM, Wong SM, Tjolsen A, Allen CP, Mochly-Rosen D, Kendig JJ. Exaggerated nociceptive responses on morphine withdrawal: roles of protein kinase C epsilon and gamma. Pain 2004; 110: 281–9
- Sweitzer SM, Wong SM, Peters MC, Mochly-Rosen D, Yeomans DC, Kendig JJ. Protein kinase C epsilon and gamma: involvement in formalin-induced nociception in neonatal rats. J Pharmacol Exp Ther 2004; 309: 616–25
- Shumilla JA, Liron T, Mochly-Rosen, Kendig JJ, Sweitzer SM. Ethanol withdrawal-associated allodynia and hyperalgesia: age-dependent regulation by protein kinase C epsilon and gamma isoenzymes. J Pain 2005; 6: 535–49
- Hartung HP, Toyka KV. Activation of macrophages by substance P: induction of oxidative burst and thromboxane release. Eur J Pharmacol 1983; 89: 301–5
- Peck R. Neuropeptides modulating macrophage function. Ann N Y Acad Sci 1987; 496: 264–70
- Serra MC, Bazzoni F, Della Bianca V, Greskowiak M, Rossi F. Activation of human neutrophils by substance P. Effect on oxidative metabolism, exocytosis, cytosolic Ca2+ concentration and inositol phosphate formation. J Immunol 1988; 141: 2118–24
- Perianin A, Snyderman R, Malfroy B. Substance P primes human neutrophil activation: a mechanism for neurological regulation of inflammation. Biochem Biophys Res Commun 1989; 161: 520–4
- Brunelleschi S, Vanni L, Ledda F, Giotti A, Maggi CA, Fantozzi R. Tachykinins activate guinea-pig alveolar macrophages: involvement of NK2 and NK1 receptors. Br J Pharmacol 1990; 100: 417–20
- Brunelleschi S, Tarli S, Giotti A, Fantozzi R. Priming effects of mammalian tachykinins on human neutrophils. Life Sci 1991; 48: PL1–5
- Boichot E, Lagente V, Paubert-Braquet M, Frossard N. Inhaled substance P induces activation of alveolar macrophages and increases airway responses in the guinea-pig. Neuropeptides 1993; 25: 307–13
- Berman AS, Chancellor-Freeland C, Zhu G, Black PH. Substance P primes murine peritoneal macrophages for an augmented proinflammatory cytokine response to lipopolysaccharide. Neuroimmunomodulation 1996; 3: 141–9
- Tanabe T, Otani H, Zeng XT, Mishima K, Ogawa R, Inagaki C. Inhibitory effects of calcitonin gene-related peptide on substance-P-induced superoxide production in human neutrophils. Eur J Pharmacol 1996;314:175–83 [Erratum in: Eur J Pharmacol 1997;321:137–41].
- Tanabe T, Otani H, Bao L, Mikami Y, Yasukura T, Ninomiya T, et al. Intracellular signaling pathway of substance P-induced superoxide production in human neutrophils. Eur J Pharmacol 1996; 299: 187–95
- Brunelleschi S, Nicali R, Lavagno L, Viano I, Pozzi E, Gagliardi L, et al. Tachykinin activation of human monocytes from patients with interstitial lung disease, healthy smokers or healthy volunteers. Neuropeptides 2000; 34: 45–50
- Marriott I, Mason MJ, Elhofy A, Most KL. Substance P activates NF-kappaB independent of elevations in intracellular calcium in murine macrophages and dendritic cells. J Neuroimmunol 2000; 102: 163–71
- Lavagno L, Bordin G, Colangelo D, Viano I, Brunelleschi S. Tachykinin activation of human monocytes from patients with rheumatoid arthritis: in vitro and ex-vivo effects of cyclosporin A. Neuropeptides 2001; 35: 92–9
- Marriott I, Bost KL. Substance P receptor mediated macrophage responses. Adv Exp Med Biol 2001; 493: 247–54
- Delgado AV, McManus AT, Chambers JP. Production of tumor necrosis factor-alpha, interleukin 1-beta, interleukin 2, and interleukin 6 by rat leukocyte subpopulations after exposure to substance P. Neuropeptides 2003; 37: 355–61
- Weinstock JV. The role of substance P, hemokinin and their receptor in governing mucosal inflammation and granulomatous responses. Front Biosci 2004; 9: 1936–43
- Bardelli C, Gunella G, Varsaldi F, Balbo P, Del Boca E, Bernardone IS, et al. Expression of functional NK1 receptors in human alveolar macrophages: superoxide anion production, cytokine release and involvement of NF-kappaB pathway. Br J Pharmacol 2005; 145: 385–96
- Koon HW, Pothoulakis C. Immunomodulatory properties of substance P: the gastrointestinal system as a model. Ann N Y Acad Sci 2006; 1088: 23–40
- Longhurst JC, Dittman LE. Hypoxia, bradykinin, and prostaglandins stimulate ischemically sensitive visceral afferents. Am J Physiol 1987; 253: H556–67
- Martin HA, Basbaum AI, Kwiat GC, Goetzl EJ, Levine JD. Leukotriene and prostaglandin sensitization of cutaneous high-threshold C- and A-delta mechanonociceptors in the hairy skin of rat hindlimbs. Neuroscience 1987; 22: 651–9
- Martin HA, Basbaum AI, Goetzl EJ, Levine JD. Leukotriene B4 decreases the mechanical and thermal thresholds of C-fiber nociceptors in the hairy skin of the rat. J Neurophysiol 1988; 60: 438–45
- Devor M, White DM, Goetzl EJ, Levine JD. Eicosanoids, but not tachykinins, excite C-fiber endings in rat sciatic nerve-end neuromas. Neuroreport 1992; 3: 21–4
- Karla W, Shams H, Orr JA, Scheid P. Effects of the thromboxane A2 mimetic, U46,619, on pulmonary vagal afferents in the cat. Respir Physiol 1992; 87: 383–96
- Rueff A, Dray A. Sensitization of peripheral afferent fibres in the in vitro neonatal rat spinal cord-tail by bradykinin and prostaglandins. Neuroscience 1993; 54: 527–35
- Khasar SG, Ouseph AK, Chou B, Ho T, Green PG, Levine JD. Is there more than one prostaglandin E receptor subtype mediating hyperalgesia in the rat hindpaw?. Neuroscience 1995; 64: 1161–5
- Ouseph AK, Khasar SG, Levine JD. Multiple second messenger systems act sequentially to mediate rolipram-induced prolongation of prostaglandin E2-induced mechanical hyperalgesia in the rat. Neuroscience 1995; 64: 769–76
- Wang JF, Khasar SG, Ahlgren SC, Levine JD. Sensitization of C-fibres by prostaglandin E2 in the rat is inhibited by guanosine 5′-O-(2-thiodiphosphate), 2′,5′-dideoxyadenosine and Walsh inhibitor peptide. Neuroscience 1996; 71: 259–63
- Petho G, Derow A, Reeh PW. Bradykinin-induced nociceptor sensitization to heat is mediated by cyclooxygenase products in isolated rat skin. Eur J Neurosci 2001; 14: 210–18
- Bergren DR. Prostaglandin involvement in lung C-fiber activation by substance P in guinea pigs. J Appl Physiol. 2006; 100: 1918–27
- Frassetto L, Morris RC, Jr, Sellmeyer DE, Todd K, Sebastian A. Diet, evolution and aging – the pathophysiologic effects of the post-agricultural inversion of the potassium-to-sodium and base-to-chloride ratios in the human diet. Eur J Nutr 2001; 40: 200–13
- Gorman Hills A. Acid-base balance. Chemistry, physiology, pathophysiology. Williams & Wilkins, Baltimore 1993
- Maurer M, Riesen W, Muser J, Hulter HN, Krapf R. Neutralization of Western diet inhibits bone resorption independently of K intake and reduces cortisol secretion in humans. Am J Physiol Renal Physiol 2003; 284: F32–40
- Lembeck F, Amann R. The influence of capsaicin sensitive neurons on stress-induced release of ACTH. Brain Res Bull 1986; 16: 541–3
- Amann R, Lembeck F. Stress induced ACTH release in capsaicin treated rats. Br J Pharmacol 1987; 90: 727–31
- Donnerer J, Amann R, Skofitsch G, Lembeck F. Substance P afferents regulate ACTH-corticosterone release. Ann N Y Acad Sci 1991; 632: 296–303
- Watanabe T, Morimoto A, Tan N, Makisumi T, Shimada SG, Nakamori T, et al. ACTH response induced in capsaicin-desensitized rats by intravenous injection of interleukin-1 or prostaglandin E. J Physiol 1994; 475: 139–45
- Franco-Cereceda A, Kallner G, Lundberg JM. Capsazepine-sensitive release of calcitonin gene-related peptide from C-fibre afferents in the guinea-pig heart by low pH and lactic acid. Eur J Pharmacol 1993; 238: 311–16
- Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, et al. The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron 1998; 21: 531–43
- Auberson S, Lacroix JS, Lundberg JM. Modulation of capsaicin-sensitive nerve activation by low pH solutions in guinea-pig lung. Pharmacol Toxicol 2000; 86: 16–23
- Fischer MJ, Reeh PW, Sauer SK. Proton-induced calcitonin gene-related peptide release from rat sciatic nerve axons, in vitro, involving TRPV1. Eur J Neurosci 2003; 18: 803–10
- Geppetti P, Materazzi S, Nicoletti P. The transient receptor potential vanilloid 1: role in airway inflammation and disease. Eur J Pharmacol 2006; 533: 207–14
- van der Merwe MT, Jansson PA, Crowther NJ, Boyd IH, Gray IP, Joffe BI, et al. Lactate and glycerol release from subcutaneous adipose tissue in black and white lean men. J Clin Endocrinol Metab 1999;84:2888–95 [Erratum in: J Clin Endocrinol Metab 2000;85:1670–1].
- Kerckhoffs DA, Arner P, Bolinder J. Lipolysis and lactate production in human skeletal muscle and adipose tissue following glucose ingestion. Clin Sci (Lond) 1998; 94: 71–7
- van der Merwe MT, Crowther NJ, Schlaphoff GP, Boyd IH, Gray IP, Joffe BI, et al. Lactate and glycerol release from the subcutaneous adipose tissue of obese urban women from South Africa; important metabolic implications. J Clin Endocrinol Metab 1998; 83: 4084–91
- Gustafsson J, Eriksson J, Marcus C. Glucose metabolism in human adipose tissue studied by 13C-glucose and microdialysis. Scand J Clin Lab Invest 2007; 67: 155–64
- Kobasa D, Jones SM, Shinya K, Kash JC, Copps J, Ebihara H, et al. Aberrant innate immune response in lethal infection of macaques with the 1918 influenza virus. Nature 2007; 445: 319–23
- Fabris N, Mocchegiani E, Amadio L, Zannotti M, Licastro F, Franceschi C. Thymic hormone deficiency in normal ageing and Down's syndrome: is there a primary failure of the thymus?. Lancet 1984; 1: 983–6
- Mocchegiani E, Fabris N, Travaglini P, Sartorio A, Travaglini P, Sartorio A, et al. Thymic endocrine activity in children with idiopathic growth-hormone deficiency. Int J Neurosci 1991; 59: 151–7
- Mocchegiani E, Paolucci P, Granchi D, Cavalazzi L, Santarelli L, Fabris N. Plasma zinc level and thymic hormone activity in young cancer patients. Blood 1994; 83: 749–57
- Mocchegiani E, Fabris N. Age-related thymus involution: zinc reverses in vitro the thymulin secretion defect. Int J Immunopharmacol 1995; 17: 745–9
- Mocchegiani E, Santarelli L, Muzzioli M, Fabris N. Reversibility of the thymic involution and of age-related peripheral immune dysfunctions by zinc supplementation in old mice. Int J Immunopharmacol 1995; 17: 703–18
- Mocchegiani E, Santarelli L, Costarelli L, Cipriano C, Muti E., Giacconi R, et al. Plasticity of neuroendocrine-thymus interactions during ontogeny and ageing: role of zinc and arginine. Ageing Res Rev 2006; 5: 281–309
- Dardenne M, Boukaiba N, Gagnerault MC, Homo-Delarche F, Chappuis P, Lemonnier D, et al. Restoration of the thymus in aging mice by in vivo zinc supplementation. Clin Immunol Immunopathol 1993; 66: 127–35
- Dardenne M, Savino W, Wade S, Kaiserlian D, Lemonnier D, Bach JF. In vivo and in vitro studies of thymulin in marginally zinc-deficient mice. Eur J Immunol 1984; 14: 454–8
- Prasad AS, Meftah S, Abdallah J, Kaplan J, Brewer GJ, Bach JF, et al. Serum thymulin in human zinc deficiency. J Clin Invest 1988; 82: 1202–10
- Prasad AS. Zinc: an overview. Nutrition 1995;11(1 Suppl):93–9.
- Prasad AS. Zinc and immunity. Mol Cell Biochem 1998; 188: 63–9
- Prasad AS. Zinc: mechanisms of host defense. J Nutr 2007; 137: 1345–9
- Consolini R, Cei B, Cini P, Bottone E, Casarosa L. Circulating thymic hormone activity in young cancer patients. Clin Exp Immunol 1986; 66: 173–80
- Consolini R, Legitimo A, Giorgianni A, Putti MC. Thymic dysfunction in childhood T-acute lymphoblastic leukemia: a possible linkage with a primary thymus involvement. Haematologica 1992; 77: 243–7
- Mocchegiani E, Ciavattini A, Santarelli L, Tibaldi A, Muzzioli M, Bonazzi P, et al. Role of zinc and alpha2 macroglobulin on thymic endocrine activity and on peripheral immune efficiency (natural killer activity and interleukin 2) in cervical carcinoma. Br J Cancer 1999; 79: 244–50
- Mocchegiani E, Perissin L, Santarelli L, Tibaldi A, Zorzet S, Rapozzi V, et al. Melatonin administration in tumor-bearing mice (intact and pinealectomized) in relation to stress, zinc, thymulin and IL-2. Int J Immunopharmacol 1999; 21: 27–46
- Mocchegiani E, Provinciali M, Di Stefano G, Nobilini A, Caramia G, Santarelli L, et al. Role of the low zinc bioavailability on cellular immune effectiveness in cystic fibrosis. Clin Immunol Immunopathol 1995; 75: 214–24
- Franceschi C, Chiricolo M, Licastro F, Zannotti M, Masi M, Mocchegiani E, et al. Oral zinc supplementation in Down's syndrome: restoration of thymic endocrine activity and of some immune defects. J Ment Defic Res 1988; 32: 169–81
- Licastro F, Mocchegiani E, Zannotti M, Arena G, Masi M, Fabris N. Zinc affects the metabolism of thyroid hormones in children with Down's syndrome: normalization of thyroid stimulating hormone and of reversal triiodothyronine plasmic levels by dietary zinc supplementation. Int J Neurosci 1992; 65: 259–68
- Licastro F, Mocchegiani E, Masi M, Fabris N. Modulation of the neuroendocrine system and immune functions by zinc supplementation in children with Down's syndrome. J Trace Elem Electrolytes Health Dis 1993; 7: 237–9
- Licastro F, Chiricolo M, Mocchegiani E, Fabris N, Zannoti M, Beltrandi E, et al. Oral zinc supplementation in Down's syndrome subjects decreased infections and normalized some humoral and cellular immune parameters. J Intellect Disabil Res 1994; 38: 149–62
- Mocchegiani E, Brignola C, Iannone P, Campieri M, Pasquali M, Lanfranchi GA, et al. Levels of zinc and thymulin in plasma from patients with Crohn's disease. J Clin Lab Immunol 1990; 32: 79–84
- Brignola C, Belloli C, De Simone G, Evangelisti A, Parente R, Mancini R, et al. Zinc supplementation restores plasma concentrations of zinc and thymulin in patients with Crohn's disease. Aliment Pharmacol Ther 1993; 7: 275–80
- Mocchegiani E, Veccia S, Ancarani F, Scalise G, Fabris N. Benefit of oral zinc supplementation as an adjunct to zidovudine (AZT) therapy against opportunistic infections in AIDS. Int J Immunopharmacol 1995; 17: 719–27
- Mocchegiani E, Muzzioli M. Therapeutic application of zinc in human immunodeficiency virus against opportunistic infections. J Nutr 2000;130(5S Suppl):1424S–1431S.
- Mocchegiani E, Boemi M, Fumelli P, Fabris N. Zinc-dependent low thymic hormone level in type I diabetes. Diabetes 1989; 38: 932–7
- Travaglini P, Moriondo P, Togni E, Venegoni P, Bochicchio D, Conti A, et al. Effect of oral zinc administration on prolactin and thymulin circulating levels in patients with chronic renal failure. J Clin Endocrinol Metab 1989; 68: 186–90
- Muzzioli M, Mocchegiani E, Bressani N, Bevilacqua P, Fabris N. In vitro restoration by thymulin of NK activity of cells from old mice. Int J Immunopharmacol 1992; 14: 57–61
- Giacconi R, Cipriano C, Muzzioli M, Gasparini N, Orlando F, Mocchegiani E. Interrelationships among brain, endocrine and immune response in ageing and successful ageing: role of metallothionein III isoform. Mech Ageing Dev 2003; 124: 371–8
- Licastro F, Savorani G, Sarti G, Salsi A, Cavazzuti F, Zanichelli L, et al. Zinc and thymic hormone-dependent immunity in normal ageing and in patients with senile dementia of the Alzheimer type. J Neuroimmunol 1990; 27: 201–8
- Licastro F, Davis LJ, Mocchegiani E, Fabris N. Impaired peripheral zinc metabolism in patients with senile dementia of probable Alzheimer's type as shown by low plasma concentrations of thymulin. Biol Trace Elem Res 1996; 51: 55–62
- Mocchegiani E, Verbanac D, Santarelli L, Tibaldi A, Muzzioli M, Radosevic-Stasic B, et al. Zinc and metallothioneins on cellular immune effectiveness during liver regeneration in young and old mice. Life Sci 1997; 61: 1125–45
- Mocchegiani E, Muzzioli M, Cipriano C, Giacconi R. Zinc, T-cell pathways, aging: role of metallothioneins. Mech Ageing Dev 1998; 106: 183–204
- Mocchegiani E, Muzzioli M, Giacconi R. Zinc, metallothioneins, immune responses, survival and ageing. Biogerontology 2000; 1: 133–43
- Mocchegiani E, Giacconi R, Cipriano C, Muzzioli M, Fattoretti P, Bertoni-Freddari C, et al. Zinc-bound metallothioneins as potential biological markers of ageing. Brain Res Bull 2001; 55: 147–53
- Mocchegiani E, Giacconi R, Cipriano C, Gasparini N, Orlando F, Stecconi R, et al. Metallothioneins (I + II) and thyroid-thymus axis efficiency in old mice: role of corticosterone and zinc supply. Mech Ageing Dev 2002; 123: 675–94
- Mocchegiani E, Giacconi R, Cipriano C, Muzzioli M, Gasparini N, Moresi R, et al. MtmRNA gene expression, via IL-6 and glucocorticoids, as potential genetic marker of immunosenescence: lessons from very old mice and humans. Exp Gerontol 2002; 37: 349–57
- Cipriano C, Giacconi R, Muzzioli M, Gasparini N, Orlando F, Corradi A, et al. Metallothionein (I + II) confers, via c-myc, immune plasticity in oldest mice: model of partial hepatectomy/liver regeneration. Mech Ageing Dev 2003; 124: 877–86
- Mocchegiani E, Muzzioli M, Giacconi R, Cipriano C, Gasparini N, Franceschi C, et al. Metallothioneins/PARP-1/IL-6 interplay on natural killer cell activity in elderly: parallelism with nonagenarians and old infected humans. Effect of zinc supply. Mech Ageing Dev 2003; 124: 459–68
- Mocchegiani E, Giacconi R, Cipriano C, Gasparini N, Bernardini G, Malavolta M, et al. The variations during the circadian cycle of liver CD1d-unrestricted NK1.1+TCR gamma/delta+ cells lead to successful ageing. Role of metallothionein/IL-6/gp130/PARP-1 interplay in very old mice. Exp Gerontol 2004; 39: 775–88
- Mocchegiani E, Giacconi R, Cipriano C, Muti E, Gasparini N, Malavolta M. Are zinc-bound metallothionein isoforms (I + II and III) involved in impaired thymulin production and thymic involution during ageing?. Immun Ageing 2004; 1: 5
- Mocchegiani E, Giacconi R, Fattoretti P, Casoli T, Cipriano C, Muti E, et al. Metallothionein isoforms (I + II and III) and interleukin-6 in the hippocampus of old rats: may their concomitant increments lead to neurodegeneration?. Brain Res Bull 2004; 63: 133–42
- Mocchegiani E, Giacconi R, Muti E, Rogo C, Bracci M, Muzzioli M, et al. Zinc, immune plasticity, aging, and successful aging: role of metallothionein. Ann N Y Acad Sci 2004; 1019: 127–34
- Mocchegiani E, Bertoni-Freddari C, Marcellini F, Malavolta M. Brain, aging and neurodegeneration: role of zinc ion availability. Prog Neurobiol 2005; 75: 367–90
- Mocchegiani E, Costarelli L, Giacconi R, Cipriano C, Muti E, Malavolta M. Zinc-binding proteins (metallothionein and alpha-2 macroglobulin) and immunosenescence. Exp Gerontol 2006; 41: 1094–107
- Mocchegiani E, Paolucci E, Balsamo A, Cacciari E, Fabris N. Influence of growth hormone on thymic endocrine activity in humans. Horm Res 1990; 33: 248–55
- Mocchegiani E, Sartorio A, Santarelli L, Ferrero S, Fabris N. Thymulin, zinc and insulin-like growth factor-I (IGF-I) activity before and during recombinant growth hormone (rec-GH) therapy in children and adults with GH deficiency. J Endocrinol Invest 1996; 19: 630–7
- Dardenne M. Role of thymic peptides as transmitters between the neuroendocrine and immune systems. Ann Med 1999; 31(Suppl 2)34–9
- Mocchegiani E, Bulian D, Santarelli L, Tibaldi A, Muzzioli M, Pierpaoli W, et al. The immuno-reconstituting effect of melatonin or pineal grafting and its relation to zinc pool in aging mice. J Neuroimmunol 1994; 53: 189–201
- Mocchegiani E, Bulian D, Santarelli L, Tibaldi A, Muzzioli M, Lesnikov V, et al. The zinc pool is involved in the immune-reconstituting effect of melatonin in pinealectomized mice. J Pharmacol Exp Ther 1996; 277: 1200–8
- Fabris N, Mocchegiani E, Provinciali M. Plasticity of neuro-endocrine-thymus interactions during aging – a minireview. Cell Mol Biol (Noisy-le-grand) 1997; 43: 529–41
- Fabris N, Mocchegiani E, Provinciali M. Plasticity of neuroendocrine-thymus interactions during aging. Exp Gerontol 1997; 32: 415–29
- Mocchegiani E, Santarelli L, Tibaldi A, Muzzioli M, Bulian D, Cipriano K, et al. Presence of links between zinc and melatonin during the circadian cycle in old mice: effects on thymic endocrine activity and on the survival. J Neuroimmunol 1998; 86: 111–22
- Dalton T, Palmiter RD, Andrews GK. Transcriptional induction of the mouse metallothionein-I gene in hydrogen peroxide-treated Hepa cells involves a composite major late transcription factor/antioxidant response element and metal response promoter elements. Nucleic Acids Res 1994; 22: 5016–23
- Ren Y, Smith A. Mechanism of metallothionein gene regulation by heme-hemopexin. Roles of protein kinase C, reactive oxygen species, and cis-acting elements. J Biol Chem 1995; 270: 23988–95
- Dalton TP, Li Q, Bittel D, Liang L, Andrews GK. Oxidative stress activates metal-responsive transcription factor-1 binding activity. Occupancy in vivo of metal response elements in the metallothionein-I gene promoter. J Biol Chem 1996; 271: 26233–41
- Andrews GK. Regulation of metallothionein gene expression by oxidative stress and metal ions. Biochem Pharmacol 2000; 59: 95–104
- McAleer MF, Tuan RS. Metallothionein protects against severe oxidative stress-induced apoptosis of human trophoblastic cells. In Vitro Mol Toxicol 2001; 14: 219–31
- Datta J, Majumder S, Kutay H, Motiwala T, Frankel W, Costa R, et al. Metallothionein expression is suppressed in primary human hepatocellular carcinomas and is mediated through inactivation of CCAAT/enhancer binding protein alpha by phosphatidylinositol 3-kinase signaling cascade. Cancer Res 2007; 67: 2736–46
- Hernandez J, Carrasco J, Belloso E, Giralt M, Bluethmann H, Kee Lee D, et al. Metallothionein induction by restraint stress: role of glucocorticoids and IL-6. Cytokine 2000; 12: 791–6
- Kobayashi K, Himeno S, Satoh M, Kuroda J, Shibata N, Seko Y, et al. Pentavalent vanadium induces hepatic metallothionein through interleukin-6-dependent and -independent mechanisms. Toxicology 2006; 228: 162–70
- Ashino T, Arima Y, Shioda S, Iwakura Y, Numasawa S, Yoshida T. Effect of interleukin-6 neutralization on CYP3A11 and metallothionein-1/2 expressions in arthritic mouse liver. Eur J Pharmacol 2007; 558: 199–207
- Kobayashi K, Kuroda J, Shibata N, Hasegawa T, Seko Y, Satoh M, et al. Induction of metallothionein by manganese is completely dependent on interleukin-6 production. J Pharmacol Exp Ther 2007; 320: 721–7
- Ginn-Pease ME, Whisler RL. Redox signals and NF-kappaB activation in T cells. Free Radic Biol Med 1998; 25: 346–61
- Kabe Y, Ando K, Hirao S, Yoshida M, Handa H. Redox regulation of NF-kappaB activation: distinct redox regulation between the cytoplasm and the nucleus. Antioxid Redox Signal 2005; 7: 395–403
- Cai L, Tsiapalis G, Cherian MG. Protective role of zinc-metallothionein on DNA damage in vitro by ferric nitrilotriacetate (Fe-NTA) and ferric salts. Chem Biol Interact 1998; 115: 141–51
- Iqbal M, Noor R, Mizuno R, Okada S. Protective role of zinc-metallothionein (Zn-MT) in iron nitrilotriacetate (Fe-NTA)-induced renal oxidative damage. Redox Rep 2003; 8: 163–7
- Min KS, Morishita F, Tetsuchikawahara N, Onosaka S. Induction of hepatic and renal metallothionein synthesis by ferric nitrilotriacetate in mice: the role of MT as an antioxidant. Toxicol Appl Pharmacol 2005; 204: 9–17
- Min KS. [The physiological significance of metallothionein in oxidative stress.] Yakugaku Zasshi 2007;127:695–702 (in Japanese).
- Sun Y, Tan M, Duan H, Swaroop M. SAG/ROC/Rbx/Hrt, a zinc RING finger gene family: molecular cloning, biochemical properties, and biological functions. Antioxid Redox Signal 2001; 3: 635–50
- Cragg RA, Phillips SR, Piper JM, Varma JS, Campbell FC, Mathers JC, et al. Homeostatic regulation of zinc transporters in the human small intestine by dietary zinc supplementation. Gut 2005; 54: 469–78
- Mason B. Victor Moritz Goldschmidt: father of modern geochemistry. Special Publication No. 4. Geochemical Society, 1992.
- Goldschmidt VM. Geochemistry. Clarendon Press, Oxford 1954
- Houston MC. The role of mercury and cadmium heavy metals in vascular disease, hypertension, coronary heart disease, and myocardial infarction. Altern Ther Health Med 2007; 13: S128–S133
- Lopez-Alonso M, Prieto F, Miranda M, Castillo C, Hernandez JR, Benedito JL. Intracellular distribution of copper and zinc in the liver of copper-exposed cattle from northwest Spain. Vet J 2005; 170: 332–8
- Sienko NJ, Plane RA. Chemical principles and properties2nd edn. McGraw-Hill, New York 1974
- Underwood EJ. Trace elements in human and animal nutrition4th edn. Academic Press, New York 1977
- Karimbakas J, Langkamp-Henken B, Percival SS. Arrested maturation of granulocytes in copper deficient mice. J Nutr 1998; 128: 1855–60
- Percival SS. Copper and immunity. Am J Clin Nutr 1998;67(5 Suppl):1064S–1068S.
- Lominadze D, Saari JT, Percival SS, Schuschke DA. Proinflammatory effects of copper deficiency on neutrophils and lung endothelial cells. Immunol Cell Biol 2004; 82: 231–8
- Davis GK, Mertz W. Copper. In: Mertz W. Trace elements in human and animal nutrition, 5th edn, Vol 1. New York: Academic Press, 1987:301–64.
- Lentsch AB, Kato A, Saari JT, Schuschke DA. Augmented metalloproteinase activity and acute lung injury in copper-deficient rats. Am J Physiol Lung Cell Mol Physiol 2001; 281: L387–93
- Chowell G, Ammon CE, Hengartner NW, Hyman JM. Estimation of the reproductive number of the Spanish flu epidemic in Geneva, Switzerland. Vaccine 2006; 24: 6747–50
- Chowell G, Ammon CE, Hengartner NW, Hyman JM. Transmission dynamics of the great influenza pandemic of 1918 in Geneva, Switzerland: assessing the effects of hypothetical interventions. J Theor Biol 2006; 241: 193–204